

Журнал неорганической химии, 2022, T. 67, № 6, стр. 817-824
Природа химической связи и структура рентгеновского фотоэлектронного спектра PaO2
Ю. А. Тетерин a, b, *, М. В. Рыжков c, А. Е. Путков a, b, К. И. Маслаков a, А. Ю. Тетерин b, К. Е. Иванов b, С. Н. Калмыков a, В. Г. Петров a
a Московского государственного университета им. М.В. Ломоносова, Химический факультет
119991 Москва, Ленинские горы, 1, Россия
b Национальный исследовательский центр “Курчатовский институт”
123182 Москва, пл. Акад. Курчатова, 1, Россия
c Институт химии твердого тела УрО РАН
620990 Екатеринбург, ул. Первомайская, 91, Россия
* E-mail: Teterin_YA@nrcki.ru
Поступила в редакцию 03.01.2022
После доработки 14.02.2022
Принята к публикации 22.02.2022
- EDN: PJRHEK
- DOI: 10.31857/S0044457X22060289
Аннотация
Релятивистским методом дискретного варьирования рассчитаны плотность состояний и спектр рентгеновской фотоэлектронной спектроскопии валентных электронов в диапазоне энергий связи электронов от 0 до ~50 эВ в PaO2. Построена схема молекулярных орбиталей. Наблюдаются значительные эффекты ковалентности в PaO2, связанные с перекрыванием не только атомных орбиталей Pa6d, но и Pa6p, и Pa5f с орбиталями кислорода. Найдено, что электроны внутренних валентных молекулярных орбиталей ослабляют химическую связь, обусловленную электронами внешних валентных молекулярных орбиталей.
ВВЕДЕНИЕ
Протактиний 231Ра (τ1/2 = 3.28 × 104 лет, α-распад) – природный изотоп, но на практике большое значение имеет и искусственный изотоп 233Ра (τ1/2 = 27 сут, β-распад) – промежуточный продукт в производстве 233U в реакторах-размножителях, работающих на тории. Поскольку имеются доступные количества (~0.5 г) протактиния, то многие его физико-химические свойства и его соединений изучены [1]. Черный РаО2 получается восстановлением Р2О5 водородом при 1550°С c гранецентрированной решеткой ГЦК (СаF2) c a = = 0.5509 нм [2] (a = 0.5505 нм [3]). Методом рентгеновской фотоэлектронной спектроскопии (РФЭС) получен только спектр остовных Pa4f-электронов Pa2O5 [4].
При расчете электронной структуры PaO2 основное внимание уделялось электронам внешних валентных молекулярных орбиталей (ВМО) с энергиями от 0 до ~15 эВ [5, 6] и не рассматривалась область от ~15 до ~50 эВ внутренних валентных МО (ВВМО). Установлено, что структура спектров РФЭС валентных электронов ThO2 и UO2 в диапазоне от 0 до ~50 эВ в основном связана с электронами валентных МО, что подтверждено результатами рентгеновских эмиссионных и конверсионных исследований и релятивистских расчетов [7, 8]. Аналогичные заключения сделаны для AnO2 (An = Np–Bk) [9–14].
В настоящей работе впервые выполнен полностью релятивистский расчет электронного строения РаO2 релятивистским методом дискретного варьирования (РДВ), определена плотность состояний валентных электронов РаO2 и рассчитан спектр рентгеновской фотоэлектронной спектроскопии (РФЭС) в диапазоне энергий связи от 0 до ~50 эВ с целью изучения особенностей химической связи в РаO2 и сравнения полученных результатов со спектральными характеристиками диоксидов других актиноидов.
МЕТОД РАСЧЕТА
Кластер ${\text{PaO}}_{8}^{{12 - }}$ точечной группы симметрии D4h, отражающий ближайшее окружение протактиния в PaO2, представляет объемно-центрированный куб, в центре которого находится протактиний, а в вершинах 8 кислородов с длиной связи rPa–O = 0.2385 нм [2] (0.2384 нм [3]). Расчеты такого кластера впервые проведены в настоящей работе с использованием оригинальной программы, реализующей неэмпирический метод РДВ [15, 16]. Метод РДВ основан на решении уравнения Дирака–Слэтера для 4-компонентных спиноров с обменно-корреляционным потенциалом [17]. Расширенный базис численных атомных орбиталей, полученных при решении уравнения Дирака–Слэтера для изолированных нейтральных атомов, включал, помимо полностью и частично заполненных, вакантные состояния Pa7p1/2, 7p3/2. В настоящей работе ограничились расчетами минимального фрагмента решетки, поскольку, как было показано в работах [10, 11], основные характеристики МО больших кластеров Pu63O216 и Am63O216 оказались близкими к данным, полученным для минимальных кластеров плутония и америция. Необходимо отметить, что указанный в обозначении кластера ${\text{PaO}}_{8}^{{12 - }}$ формальный заряд –12 означал, что в схеме расчета О2р-полоса поддерживалась полностью заполненной. Однако используемая в расчетах минимальных кластеров процедура перенормировки заселенностей внешних орбиталей граничных атомов и псевдопотенциала, моделировавшего влияние окружающего кристалла, обеспечивала положение верхних заполненных уровней в пределах 10 эВ ниже нуля используемой в расчетах энергетической шкалы [9]. Отсутствие muffin-tin – аппроксимации потенциала в методе РДВ является его преимуществом, поскольку нет ограничений на симметрию исследуемого соединения. Любые типы кластеров (в том числе вообще не имеющие симметрии) могут быть рассчитаны с одинаковой точностью. Также результаты расчета, полученные в приближении молекулярные орбитали как линейные комбинации атомных орбиталей, позволяют анализировать роль атомных состояний в электронной структуре, химической связи, спектральных и других свойствах твердофазных соединений.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Схема валентных МО РаO2. Деление валентных МО на ВМО и ВВМО носит условный характер (рис. 1). ВМО в основном образованы внешними валентными (недозаполненными) атомными орбиталями (АО), а ВВМО в большой степени – внутренними валентными (заполненными) АО. Структура спектров РФЭС электронов ВВМО, как правило, хорошо разрешена, что позволяет делать качественные и количественные заключения о строении ближайшего окружения центрального атома в кластере и длине связи от него до ближайших соседей [18].
Рис. 1.
Схема МО РаO2. Стрелками отмечены некоторые разности энергий уровней. Слева приведены рассчитанные значения энергий связи электронов (эВ). Энергетический масштаб не выдержан. В скобках приведены номера групп орбиталей, отмеченных в тексте. Величина ${{\Delta }}E_{{{\text{Pa}}}}^{{\text{T}}}$ = 314.46 эВ [24].

При построении схемы МО РаО2 учитывалось, что экспериментальные значения энергии связи O1s-электронов в AnO2 должны быть равны 529.9 эВ, а разность энергий O1s- и O2s- в атоме кислорода равна 508.3 эВ [9, 10]. Поэтому разность энергий O1s-электронов и квазиатомной $12\gamma _{7}^{ - }$ ВВМО должна равняться 508.3 эВ, а энергия $12\gamma _{7}^{ - }$ ВВМО равна 21.60 эВ, которая отличается от величины 21.17 эВ, приведенной в табл. S1 . Такая величина удовлетворительно согласуется со структурой экспериментальных спектров РФЭС диоксидов других актиноидов и позволяет с малой погрешностью определить энергии связи валентных электронов.
На схеме MO PaO2 вакантные МО приведены в виде штрихов, а занятые МО – сплошных горизонтальных линий (рис. 1). Рассчитанные значения энергий связи электронов валентных МО приведены слева от МО. Над горизонтальными прямыми МО дан состав в %. Справа от МО даны их обозначения (табл. S1 ), а в скобках – номера групп МО. Эти номера необходимы для простоты обсуждения рассматриваемой схемы. Разности энергий валентных и остовных МО приведены на схеме в виде вертикальных прямых со стрелками. Эти разности могут быть измерены экспериментально в случае получения спектров РФЭС валентных и остовных электронов PaO2. Внизу схемы даны значения этих разностей в эВ.
На схеме пунктиром выделены формально “разрыхляющие” $17\gamma _{6}^{ - },$ $13\gamma _{7}^{ - }$ (5) и $16\gamma _{6}^{ - }$ (6) и соответствующие им “связывающие” $15\gamma _{6}^{ - },$ $11\gamma _{7}^{ - }$ (8) и $14\gamma _{6}^{ - }$ (9) ВВМО, а также – “квазиатомные” $12\gamma _{7}^{ - },$ $13\gamma _{7}^{ + },$ $16\gamma _{6}^{ + },$ $12\gamma _{7}^{ + },$ и $15\gamma _{6}^{ + }$ (7) ВВМО, обусловленные в основном O2s АО.
Cхема МО РаO2 позволяет понять природу формирования химической связи в этом диоксиде и может быть использована при интерпретации сложной структуры других его рентгеновских (эмиссионных, поглощения, конверсионных и др.) спектров.
Электронное строение PaO2. Валентная электронная конфигурация основного состояния протактиния – Pa6s26p65f26d17s2, 4K9/2. Эти оболочки протактиния могут принимать участие в образовании МО в его оксидах [18, 19]. Результаты расчета электронного строения PaO2 методом РДВ приведены на рис. 1 и в табл. S1 .
Наибольшее участие в образовании МО принимают Ра6р, 6d, 5f АО. Найдено, что Ра6s АО в малой степени участвует в образовании МО как и Ра7s и 7р AO. Верхнюю заполненную $20\gamma _{7}^{ - }$ МО, содержащую 93% Ра5f AO, можно формально рассматривать как “квазиатомную”, а такие Ра5f-электроны – локализованными вблизи уровня Ферми. Остальные Ра5f-электроны делокализованы в основном в пределах области ВМО. Делокализованные Ра5f-электроны увеличивают ковалентную составляющую химической связи за счет перекрывания Ра5f АО с O2p орбиталями кислорода. Участие Ра5f, 6d и О2р АО в образовании ВМО согласуется с результатами для AnO2 более тяжелых актиноидов [8–13]. При этом ковалентные вклады Ра5f АО в МО O2s-типа отсутствуют.
В РаO2 так же, как для диоксидов других актиноидов, Ра6p АО участвуют в образовании как ВМО, так и ВВМО. В наибольшей степени перекрываются Ра6p3/2 и O2s АО соседних атомов. В результате этого возникают “разрыхляющие” $17\gamma _{6}^{ - },$ $13\gamma _{7}^{ - }$ (5) и $16\gamma _{6}^{ - }$ (6), а также “связывающие” $15\gamma _{6}^{ - },$ $11\gamma _{7}^{ - }$ (8) и $14\gamma _{6}^{ - }$ (9) ВВМО. Эффекты ковалентности связи в РаO2 являются существенными, что обусловлено перекрыванием Ра6d и Ра5f АО с AO лигандов.
Эффективный заряд протактиния в РаO2. Сравнивая валентную атомную конфигурацию Ра6s26p65f26d17s2 и рассчитанную в настоящей работе ионную конфигурацию Ра6s26p65f1.616d1.647s0.247p0.41 для РаO2 можно найти, что эффективный заряд протактиния равен QPa = +1.10e–, что меньше значения (QPa = +4e–) в ионном приближении. Это связано с существенными ковалентными эффектами в диоксиде протактиния. Например, химическая связь в диоксиде протактиния более ионная, чем в диоксиде нептуния, поскольку эффективный заряд нептуния QNp = +0.80e– [9] меньше, чем QPa = = +1.10e– в PaO2. Эти результаты качественно согласуются с данными для диоксидов и других актиноидов [20, 21]. Величина такого небольшого эффективного заряда протактиния в PaO2 также согласуется с данными для химических сдвигов линий актиноидов по отношению к металлам в спектрах РФЭС. Так, для Np 4f7/2-электронов при переходе от металлического Np к NpO2 наблюдается сдвиг, равный ΔEb = 3.9 эВ [9]. Если бы эффективный заряд был равен QPa = +4e–, то это приводило бы к сдвигу в десятки эВ. Например, возникновение вакансии на квазиостовном уровне в CeO2 приводит к сдвигу линий, например, Ce3d-электронов на ~16.0 эВ [22].
Структура спектра РФЭС валентных электронов PaO2. Cпектр РФЭС валентных электронов PaO2 отсутствует в литературе. Для Pa2O5, как отмечалось, имеется только спектр РФЭС остовных Pa4f7/2,5/2-электронов с большой погрешностью [4].
Методика получения теоретических спектров РФЭС диоксидов актиноидов на основе данных расчета электронной структуры описана в работах [9, 10]. В настоящей работе для сравнения теоретического спектра валентных электронов PaO2 со спектрами диоксидов других актиноидов была проведена калибровка по энергии связи электронов. Для того, чтобы сравнивать экспериментальные и теоретические спектры в ряду AnO2 (An = = Th–Cf), их необходимо представить в единой энергетической шкале. В этой шкале энергии для экспериментальной Eb(O1s) и теоретической E($17\gamma _{6}^{ - }$(5)) равны 529.9 и 21.60 эВ соответственно. Поэтому теоретические значения энергий (табл. S1 ) были увеличены по абсолютной величине на 0.48 эВ так, чтобы энергия $17\gamma _{6}^{ - },$ $13\gamma _{7}^{ - }$ (5) МО была равна величине 17.82 эВ, а энергия квазиатомной $12\gamma _{7}^{ - }$ (6) МО, связанной с O2s AO, была равна 21.6 эВ. С учетом состава МО и сечений фотоэффекта [23] были определены теоретические интенсивности спектра РФЭС валентных электронов PaO2 (рис. 2).
Рис. 2.
Гистограмма рассчитанного (РДВ) спектра РФЭС валентных электронов РаO2: черным отмечен вклад Ра5f-электронов; штрихами – вклад Ра6p-электронов; горизонтальными прямыми – вклад Pa6s-электронов в интенсивность. В скобках приведены номера групп орбиталей, отмеченных в тексте.
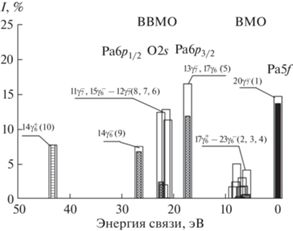
Теоретический спектр РФЭС валентных электронов PaO2 можно условно разделить на две области. Структура, связанная с электронами ВМО, наблюдается в области спектра от 0 до ~15 эВ. Интенсивность этого спектра в большой степени связана с Ра5f- и 6d-электронами, так как сечение фотоэффекта этих электронов существенно больше, чем у Ра7s-, 7p- и O2p-электронов.
Структура, обусловленная электронами ВВМО (от ~15 до 50 эВ), возникает из-за сильного перекрывания Ра6p и O2s АО ближайших атомов. Спектр этой области может быть разделен на пять (5–9) компонент: $17\gamma _{6}^{ - },$ $13\gamma _{7}^{ - }$ (5) и $16\gamma _{6}^{ - }$ (6) – “разрыхляющие” ВВМО; $12\gamma _{7}^{ - },$ $13\gamma _{7}^{ + },$ $12\gamma _{7}^{ + }$ и $16\gamma _{6}^{ + }$ (7) – ВВМО, содержащие квазиатомные O2s AO; $15\gamma _{6}^{ - },$ $11\gamma _{7}^{ - }$ (8) и $14\gamma _{6}^{ - }$ (9) – формально “связывающие” ВВМО.
Эти результаты лежат в основе понимания природы и особенностей химической связи и построения схемы МО для РаO2.
Ковалентный вклад электронов валентных МО в химическую связь в РаО2. Для оценки вклада электронов различных МО в химическую связь РаO2 в приближении Малликена в работе методам РДВ были рассчитаны величины заселенностей перекрывания различных МО [20, 25] (табл. 1). Положительные величины заселенностей характеризуют усиление (связывание) связи, а отрицательные величины – ослабление (разрыхление) связи.
Таблица 1.
Заселенности перекрывания орбиталей в кластере PaO8 (в расчете на одну связь Pa–O, все значения умножены на 103)
| Связь в PaO2 | РДВ | Связь в PaO2 | РДВ |
|---|---|---|---|
| Pa5f5/2–O2p | 25 | Pa6d3/2–O2s | 13 |
| Pa5f7/2–O2p | 47 | Pa6d5/2–O2s | 22 |
| Pa5f5/2–O2s | 4 | $\Sigma _{{{\text{ВМО}}}}^{*}$ | 436 |
| Pa5f7/2–O2s | 8 | ||
| Pa7p1/2–O2p | 17 | Pa6p1/2–O2p | –20 |
| Pa7p3/2–O2p | 24 | Pa6p3/2–O2p | –73 |
| Pa7p1/2–O2s | 14 | Pa6p1/2–O2s | –2 |
| Pa7p3/2–O2s | 22 | Pa6p3/2–O2s | –25 |
| Pa7s–O2p | 23 | Pa6s–O2p | –22 |
| Pa7s–O2s | 21 | Pa6s–O2s | –2 |
| Pa6d3/2–O2p | 80 | $\Sigma _{{{\text{ВВМО}}}}^{*}$ | –144 |
| Pa6d5/2–O2p | 116 | $\Sigma _{{{\text{МО}}}}^{*}$ | 292 |
Суммарный вклад ВМО в заселенность связей РаО2 равен 436 (как и в табл. 1, приводятся значения заселенностей связей, умноженные на 103 для наглядности). Наибольший вклад в усиление связи вносят электроны Ра6d–O2p (196), Ра7p–O2p (41), Ра6d–O2s (35), Ра5f–O2p (72). Электроны ВВМО протактиния разрыхляют связь в РаO2 и их общий вклад в заселенность равен –144. Наибольший вклад в разрыхление такой связи вносят электроны Ра6р–O2p (–93). В совокупности электроны ВВМО (–144) на 33% ослабляют связь, обусловленную электронами ВМО (436). В результате суммарный вклад валентных электронов в связь в РаО2 в единицах заселенностей перекрывания равен 292.
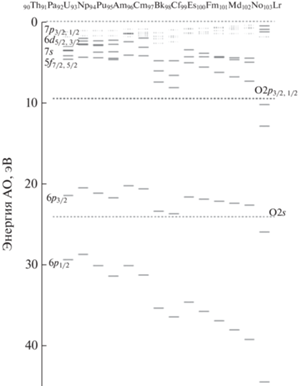
Атомные валентные орбитали актиноидов и кислорода. Энергии E (эВ) валентных АО актиноидов от 90Th до 103Lr, рассчитанные методом РДВ для конфигураций основных состояний An 6s26p65fn6dm7s27p0 (m = 0, 1, 2), за исключением An 6s AO, отражены на hис. 3. Для сравнения приведены энергии АО атома кислорода 8O: E(O2p3/2) = 9.48 эВ, E(O2p1/2) = 9.52 эВ, ΔEsl(O2p) = 0.04 эВ; E(O2s) = 24.06 эВ, где ΔEsl(O2p) – расщепление, связанное со спин-орбитальным взаимодействием O2p-электронов.
Энергии An7s и 7р АО слабо изменяются для всего ряда актиноидов, за исключением Lr, что характеризует их валентный характер. В несколько большей степени наблюдаются изменения для An6d, 5f и 6p3/2 АО, за исключением Lr, что также характеризует их валентный характер и эти АО могут участвовать в образовании МО в диоксидах актиноидов. При этом энергия An6s АО изменяется от 45.90 эВ для Th до 70.68 эВ для Lr. Поскольку энергии O2p3/2,1/2 и O2s уровней равны 9.48, 9.52 и 24.06 эВ соответственно, то не следует ожидать существенного участия An6s АО в образовании МО с АО кислорода. То же можно ожидать для An6p1/2 АО второй половины ряда актиноидов. Эти результаты находятся в согласии с данными энергий связи актиноидов, найденных в релятивистском приближении [24, 26].
Валентные и остовные (An4f) МО AnO2 (An = = Th–Cf). Для сравнения известных экспериментальных и теоретических спектров РФЭС валентных электронов [1–9] необходимо было провести новую одинаковую калибровку их энергий связи. Такая калибровка проведена относительно энергии связи O1s-электронов Eb(O1s) = 529.9 эВ. Поскольку разность энергий ΔEb = Eb(O1s) – Eb(O2s) равна 508.3 эВ для атома [26], то линия электронов квазиатомной $12g_{7}^{ - }$ ВВМО должна наблюдаться при 21.6 эВ. В пределах ошибки измерения (±0.1 эВ) линия O1s-электронов должна наблюдаться при Eb(O1s) = 529.9 эВ в шкале, в которой для калибровочной линии C 1s-электронов насыщенных углеводородов принято значение Eb(С 1s) = 285.0 эВ. Соответствующие экспериментальные значения энергий связи электронов для CfO2 получены в результате оценки с учетом данных РФЭС для Cf2O3 [14] путем увеличения на 1 эВ. Экспериментальные значения энергий связи валентных электронов PaO2 получены интерполяцией, а An4f -электронов Pa(Cf)O2 – экстраполяцией известных экспериментальных величин для AnO2 (An = Th, U–Bk) с использованием уравнений:
где Eb – энергия связи электронов, ΔEsl –величина спин-орбитального расщепления, Z – атомный номер актиноида, а R2 = 0.99991 (R-коэффициент корреляции).В некоторых случаях экспериментальные величины энергий связи заметно отличаются от соответствующих теоретических значений (табл. 2). Это в большой степени связано с погрешностью (трудностью) экспериментального определения энергий связи валентных электронов, в частности электронов $12\gamma _{7}^{ - }$ ВВМО. Несмотря на это, можно заключить, что эти величины находятся в удовлетворительном качественном согласии с соответствующими рассчитанными значениями. Это позволяет предположить, что рассчитанные методом РДВ теоретические спектры РФЭС валентных электронов AnO2 (An = Ра, Es–Lr) будут удовлетворительно отражать структуру экспериментальных спектров. Величины Eb(An 4f7/2) и ΔEsl(An4f) этих диоксидов будут близки реальным значениям. Поскольку погрешность определения по уравнениям (1), (2) этих величин ≤±1.0 эВ, то они будут являться наиболее точными значениями из всех известных ранее величин [27].
Таблица 2.
Рассчитанные (РДВ) и экспериментальные (РФЭС) энергии E* (эВ) валентных и остовных МО AnO2
| zAnO2 | ВМО | ВВМО | ОМО | Источ-ник | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ВЗМО** | $17\gamma _{6}^{ + }$ | ${\text{17}}\gamma _{6}^{ - }{\text{13}}\gamma _{7}^{ - }$ (5) | $16\gamma _{6}^{ - }\left( 7 \right)$ | $12\gamma _{7}^{ - }$ (6) |
${\text{15}}\gamma _{6}^{ - }{\text{11}}\gamma _{7}^{ - }$ (8) | $14\gamma _{6}^{ - }$ (9) | $14\gamma _{6}^{ + }$ (10) | An4f7/2 | ΔEsl(An4f)*** | |||
| 90ThO2 | Расч. | 6.44 | 9.26 | 16.98 | 21.39 | 21.60 | 22.79 | 25.50 | 40.84 | [7] | ||
| Экс. | 4.1 | 9.0 | 16.5 | 21.6 | 21.8 | 23.5 | 25.3 | 41.6 | 334.1 | 9.3 | ||
| 91PaO2 | Расч. | 0.48 | 9.20 | 17.82 | 21.62 | 21.60 | 22.97 | 27.20 | 43.71 | |||
| Интер | – | (9.0)**** | (16.7) | (20.8) | (22.1) | (23.4) | (26.7) | (44.3) | (356.8) | (10.1) | ||
| 92UO2 | Расч. | 1.61 | 9.26 | 18.38 | 21.77 | 21.60 | 23.09 | 28.49 | 45.90 | [8] | ||
| Экс. | 1.1 | 8.9 | 17.2 | 20.0 | 22.3 | 23.3 | 28.0 | 46.9 | 379.7 | 10.8 | ||
| 93NpO2 | Расч. | 3.07 | 9.33 | 18.77 | 21.94 | 21.60 | 23.25 | 29.66 | 47.88 | [9] | ||
| Экс. | 1.9 | 9.1 | 17.2 | 21.0 | 21.9 | 25.7 | 29.1 | 46.9 | 402.8 | 11.7 | ||
| 94PuO2 | Расч. | 4.01 | 9.31 | 19.21 | 21.95 | 21.60 | 23.38 | 31.18 | 50.45 | [10] | ||
| Экс. | 2.4 | 9.6 | 17.7 | 19.9 | 22.2 | 24.3 | 29.9 | 49.9 | 425.9 | 12.7 | ||
| 95AmO2 | Расч. | 4.67 | 9.32 | 19.50 | 22.01 | 21.60 | 23.49 | 32.27 | 52.30 | [11] | ||
| Экс. | 3.1 | 9.7 | 18.2 | 20.3 | 21.9 | 25.0 | 31.7 | 50.7 | 449.1 | 14.0 | ||
| 96CmO2 | Расч. | 4.72 | 9.43 | 19.74 | 22.08 | 21.60 | 23.65 | 33.10 | 53.64 | [12] | ||
| Экс. | 4.7 | 9.4 | 18.2 | 22.5 | 22.5 | 25.3 | 34.6 | – | 472.5 | 14.7 | [14] | |
| 97BkO2 | Расч. | 4.10 | 9.45 | 19.94 | 22.13 | 21.60 | 23.79 | 34.19 | 55.48 | [13] | ||
| Экс. | 4.1 | 9.4 | 18.7 | 22.1 | 22.8 | 25.4 | 34.4 | – | 498.9 | 15.9 | [14] | |
| 98CfO2***** | Расч. | 5.10 | 9.38 | 20.05 | 22.05 | 21.60 | 23.78 | 35.36 | 57.65 | [28] | ||
| Экс. | – | – | 20.0 | – | 21.6 | 25.3 | 35.5 | – | 523.9 | (16.7) | [14] | |
**** В скобках приведены величины, полученные в результате интерполяции и экстраполяции соответствующих экспериментальных данных.
***** Энергии связи для CfO2 оценены с учетом данных для Cf2O3 [14].
ЗАКЛЮЧЕНИЕ
На основе результатов полностью релятивистского расчета электронного строения РаO2 определена плотность состояний валентных электронов и рассчитан теоретический спектр РФЭС этих электронов в диапазоне энергий связи от 0 до ~50 эВ.
Проведен анализ этого спектра с учетом экспериментальных и теоретических данных РФЭС для валентных и остовных электронов AnO2 (An = = Th, U–Cf) и построена схема МО электронов РаO2. Эта схема необходима для понимания особенностей химической связи в РаO2, а также для выяснения общих закономерностей и особенностей формирования химической связи в ряду AnO2 (An = Th–Cf).
Приведены результаты расчета энергий электронов атомных орбиталей актиноидов, из которых следует качественный вывод, что АО An 6p-электронов имеют валентный характер и могут участвовать в образовании МО в диоксидах актиноидов.
На основании результатов сравнительного анализа характеристик теоретических и экспериментальных спектров РФЭС валентных электронов AnO2 (An = Th–Cf) установлено удовлетворительное согласие между такими спектрами. Это позволяет предположить, что рассчитанные методом РДВ спектры РФЭС валентных электронов AnO2 (An = Pa, Es–Lr) также будут отражать сложную структуру их экспериментальных спектров.
Список литературы
Химия актинидов. Т. 1. / Под ред. Кац Дж., Сиборг Г., Морсс Л. Пер. на рус. под ред. Мясоедова Б.Ф., М.: Мир, 1991.
Roberts L.E.J., Walter A.J. // Physico-Chimie du Protactinium / Eds. Bouissieres G., Mixart R. Orsay. 2–8 July 1965. Centre National de la Recherche Scientifique. Paris, 1966. P. 51.
Sellers P.A., Fried S., Elson R.E., Zachariasen W.H. // J. Am. Chem. Soc. 1954. V. 76. № 23. P. 5935. https://doi.org/10.1021/ja01652a011
Krause M.O., Haire R.G., Keski-Rahkonen O., Peterson J.R. // J. Electr. Spectrosc. Relat. Phenom. 1988. V. 47. P. 215. https://doi.org/10.1016/0368-2048(88)85013-8
Prodan I.D., Scuseria G.E., Martin R.L. // Phys. Rev. B. 2007. V. 76. P. 033101. https://doi.org/10.1103/PhysRevB.76.033101
Wen X.-D., Martin R.L., Henderson T.M., Scuseria G.E. // Chem. Rev. 2013. V. 113. P. 1063. https://doi.org/10.1021/cr300374y
Teterin A.Y., Ryzhkov M.V., Teterin Y.A. et al. // Radiochemistry. 2009. V. 51. P. 560. [Тетерин А.Ю., Рыжков М.В., Тетерин Ю.А. и др. // Радиохимия. 2009. Т. 51. № 6. С. 489]. https://doi.org/10.1134/ S1066362209060022
Maslakov K.I., Teterin Yu.A., Ryzhkov M.V. et al. // Int. J. Quantum Chem. 2019. V. 119. № 24. P. e26040. https://doi.org/10.1002/qua.26040
Teterin Yu.A., Teterin A.Yu., Ivanov K.E. et al. // Phys. Rev. B. 2014. V. 89. P. 035102. https://doi.org/10.1103/PhysRevB.89.035102
Teterin Yu.A., Maslakov K.I., Teterin A.Yu. et al. // Phys. Rev. B. 2013. V. 87. P. 245108. https://doi.org/10.1103/PhysRevB.87.245108
Teterin Y.A., Maslakov K.I., Ryzhkov M.V. et al. // Nuclear Technol. Radiation Protection. 2015. V. 30. № 2. P. 83. https://doi.org/10.2298/NTRP1502083T
Putkov A.E., Teterin Y.A., Ryzhkov M.V. et al. // Radiochemistry. 2021. V. 63. № 4. P. 401. [Путков. А.Е., Тетерин Ю.А., Рыжков М.В., и др. // Радиохимия. 2021. Т. 63. № 4. С. 309] https://doi.org/10.1134/S1066362221040020
Putkov A.E., Teterin Y.A., Ryzhkov M.V. et al. // Russ. J. Phys. Chem. A. 2021. V. 95. № 6. P. 1169. [Путков А.Е., Тетерин Ю.А., Рыжков М.В. и др. // Журн. физ. химии. 2021. Т. 95. № 6. С. 908] https://doi.org/10.1134/S0036024421060212
Veal B.W., Lam D.J., Diamond H., Hoekstra H.R. // Phys. Rev. B. 1977. V. 15. № 6 P. 2929. https://doi.org/10.1103/PhysRevB.15.2929
Rosen A., Ellis D.E. // J. Chem. Phys. 1975. V. 62. P. 3039. https://doi.org/10.1063/1.430892
Ellis D.E., Goodman G.L. // Int. J. Quant. Chem. 1984. V. 25. P. 185. https://doi.org/10.1002/qua.560250115
Gunnarsson O., Lundqvist B.I. // Phys. Rev. B. 1976. V. 13. P. 4274. https://doi.org/10.1103/PhysRevB.13.4274
Teterin Yu.A., Gagarin S.G. // Russ. Chem. Rev. 1996. V. 65. P. 825.
Teterin Yu.A., Teterin A.Yu. // Russ. Chem. Rev. 2004. V. 73. P. 541.
Kelly P.J., Brooks M.S., Allen R. // J. Physique. 1979. V. 40. № C4. P. 184. https://doi.org/10.1051/jphyscol:1979458
Gubanov V.A., Rosen A., Ellis D.E. // J. Phys. Chem. Solids. 1979. V. 40. P. 17. https://doi.org/10.1016/0022-3697(79)90090-8
Maslakov K.I., Teterin Yu.A., Ryzhkov M.V. et al. // Phys. Chem. Chem. Phys. 2018. V. 20. № 23. P. 16167. https://doi.org/10.1039/C8CP01442F
Yarzhemsky V.G., Teterin A.Yu., Teterin Yu.A., Trzhaskovskaya M.B. // Nuclear Technol. Radiation Protection. 2012. V. 27. P. 103. https://doi.org/10.2298/NTRP1202103Y
Huang K.N., Aoyagi M., Chen M.N., Crasemann B., Mark H. // At. Data Nucl. Data Tables. 1976. V. 18. P. 243. https://doi.org/10.1016/0092-640X(76)90027-9
Mulliken R.S. // Annu. Rev. Phys. Chem. 1978. V. 29. P. 1. https://doi.org/10.1146/annurev.pc.29.100178.000245
Trzhaskovskaya M.B., Yarzhemsky V.G. // Atom. Data Nucl. Data. 2018. V. 119. P. 99. https://doi.org/10.1016/j.adt.2017.04.003
Sevier K.D. // Atomic Data and Nuclear Data Tables. 1979. V. 24. P. 323. https://doi.org/10.1016/0092-640X(79)90012-3
Путков А.Е., Маслаков К.И., Тетерин Ю.А. и др. // Журн. структ. химии. 2021. V. 62 № 12. P. 1963. https://doi.org/10.26902/JSC_id83848
Дополнительные материалы
- скачать ESM.docx
- Таблица S1.
СПИСОК ЛИТЕРАТУРЫ
Инструменты
Журнал неорганической химии