

Журнал неорганической химии, 2022, T. 67, № 3, стр. 294-325
μ-Нитридо- и μ-карбидодимерные макрогетероциклические комплексы железа и рутения как молекулярная платформа для моделирования окислительных ферментов (обзор)
С. В. Зайцева a, *, С. А. Зданович a, Д. В. Тюрин b, О. И. Койфман a, b
a Институт химии растворов им. Г.А. Крестова РАН
153045 Иваново, ул. Академическая, 1, Россия
b Ивановский государственный химико-технологический университет
153000 Иваново,
Шереметевский пр-т, 7, Россия
* E-mail: svz@isc-ras.ru
Поступила в редакцию 08.09.2021
После доработки 10.11.2021
Принята к публикации 11.11.2021
- EDN: QQMMHR
- DOI: 10.31857/S0044457X22030175
Аннотация
Последние научные публикации демонстрируют новый подход к моделированию катализаторов, имитирующих гемсодержащие ферменты. Он заключается в использовании μ-оксо-, μ-нитридо- и μ-карбидодимерных комплексов переходных металлов в качестве многообещающих миметиков. Высокая каталитическая активность μ-нитридодимерных комплексов железа(III,IV) с макроциклами порфиринового типа, опосредованная образованием высоковалентных оксоформ, имеющих сходство с редокс-сайтом окислительных ферментов, стимулирует интерес к подобного рода соединениям. В данном обзоре особое внимание сконцентрировано на исследованиях, ориентированных на детализацию химических принципов формирования разных видов высокоокисленных оксоформ μ-нитридо- и μ-карбидодимерных комплексов железа(IV) и рутения(IV) и модуляцию их реакционной способности. В работе обобщены данные о спектральных, координационных и редокс-свойствах μ-нитридо- и μ-карбидодимерных комплексов железа(IV) и рутения(IV), проведен их сравнительный анализ и продемонстрирована возможность использования этих соединений в качестве молекулярной платформы для моделирования окислительных ферментов. Отмечено, что биметаллическая конструкция является гибкой, имеет значительный потенциал для дальнейшего развития и точной настройки свойств через включение в структуру различных лигандов, металлов и мостиковых групп.
ВВЕДЕНИЕ
Химия тетрапирролов — это область, которая во многом обусловлена биологическими процессами. Эти уникальные молекулы выполняют жизнеобеспечивающие функции в биологических системах. Важная роль отводится природным тетрапиррольным макроциклам и их комплексам с металлами, которые находят широкое применение в химии, биологии и медицине. Особый интерес вызывают гемовые оксидоредуктазы. Природа использует гемзависимые ферменты, такие как каталаза, пероксидаза и ферменты цитохрома P450, чтобы активировать различные редокс- процессы с чрезвычайно высокой эффективностью и селективностью в мягких условиях [1]. Хорошо известно, что биоинспирация и биомиметика – это одни из стратегий в области химии тетрапирролов. Их реализация напрямую зависит от обширных фундаментальных исследований свойств искусственных соединений порфиринового ряда. К настоящему времени выявлены ключевые химические принципы, лежащие в основе эффективности природных катализаторов аэробного окисления. На основании этих знаний достигнуты определенные успехи в разработке синтетических катализаторов на основе тетрапиррольных макроциклов, моделирующих простетическую группу фермента. Постоянно растущее структурное многообразие этих соединений, существующее благодаря направленной модификации периферии и координационного центра [2–14], а также высокая стабильность и доступность этих комплексов создают перспективы расширения спектра их рецепторных, каталитических и биологических функций для практического применения. Тетрапиррольные макроциклические комплексы переменных металлов можно использовать для модуляции молекулярных или клеточных функций [15]. В настоящее время они широко изучаются в биомедицине для фотодинамической терапии [16–20], антимикробной и противовирусной фототерапии [21–24], для катализа [25–27], вмешательства в биохимические пути [28, 29]. Кроме того, соединения порфиринового типа используются для собирания световой энергии с целью ее преобразования в химическую [30–32], в фотовольтаике [33] и электрохимии [34, 35], в супрамолекулярной химии [36]. Следует отметить, что одной из важнейших проблем современной бионеорганической химии является создание функциональных моделей, способных выступать в качестве биокатализаторов или ингибиторов ферментативных реакций. В рамках этой проблемы ставятся и решаются актуальные задачи, связанные с синтезом новых соединений для направленных каталитических превращений различных субстратов, расширением представлений о деталях механизма окислительной биотрансформации экзо- и эндогенных веществ, выявлением факторов, позволяющих модулировать реакционную способность металлсодержащих редокс-сайтов окислительных ферментов путем разработки искусственных молекулярных платформ на основе тетрапиррольных макроциклических комплексов переходных металлов. Эти соединения хорошо зарекомендовали себя в качестве миметиков активных центров при моделировании разных стадий биотрансформации или окислительной деградации ксенобиотиков. Такой подход предполагает формирование базы модельных комплексов, обладающих изменяемым редокс-состоянием, модулируемой каталитической активностью и субстратной специфичностью. Он позволяет получить информацию, которая может быть полезна для интерпретации свойств ферментов и их действия на молекулярном уровне. Биомиметические исследования и их результаты имеют важное значение при усовершенствовании старых или разработке новых фармакотерапевтических методов для решения проблем этиологии и патогенеза, а также для создания доступных и селективных катализаторов, работающих в умеренных энергосберегающих условиях экологически чистым способом.
В активном центре многих окислительных ферментов содержится железо. Одной из причин важной роли железа в биологии является возможность тонкой настройки его химических свойств благодаря различным координационным состояниям. Природные ферменты со сходными железосодержащими активными центрами катализируют широкий спектр химических реакций, таких как десатурация, окислительная циклизация, монооксигенация и диоксигенация, гидропероксидирование, гидроксилирование и эпоксидирование [37–39]. Несмотря на различие в конечном результате этих процессов, ключевые стадии каталитического механизма для многих из них удивительно похожи и связаны с образованием оксоферрильной активной частицы (FeIV=O) в качестве ключевого промежуточного соединения [40, 41]. Большое разнообразие в реакционной способности ферментов со сходными активными центрами делает этот класс отличной мишенью для биомиметических исследований.
Для реакций с участием гемомонооксигеназ активным интермедиатом являются катион-радикальные формы оксогема высоковалентного железа(IV) (FeIV=O) [42]. Каталитические свойства этих высоковалентных оксоформ железа во многом определяются природой экваториальных и аксиальных лигандов, связанных с металлом. Несмотря на обширные данные по структуре и активности высоковалентных интермедиатов, вопрос о способе и характере разрыва связи O–O в гидропероксокомплексе Fe–OOH, механизме переноса атома кислорода, возможности управления редокс-состоянием и реакционной способностью оксоферрильных частиц остается нерешенным [43–45]. Причина существования неоднозначности суждений тесно связана с тем, что реакционная способность миметиков, наблюдаемая в модельных реакциях “пероксошунта”, окислительной биотрансформации или других каталитических стадиях редокс-процесса, сильно зависит от разных факторов, которые, в свою очередь, определяются и контролируются выбранными условиями реакции [46]. Подробно изучить эти факторы можно путем расширения диапазона синтетических соединений с прогнозируемыми каталитическими свойствами и установления детализации механизма редокс-превращений с их участием.
В настоящее время исследуются и уже используются в качестве металлических центров в модельных ферментных системах синтетические моноядерные комплексы, включающие катионы железа, меди, рутения, никеля и марганца [47–49].
В поисках эффективных катализаторов сложных процессов окисления была предложена новая стратегия биоинспирации с использованием N‑ и С‑мостиковых комплексов железа и рутения с макроциклами порфиринового типа. Эта молекулярная платформа сочетает в себе структурные особенности активных центров двух самых мощных окисляющих ферментов: сайта дижелеза, как в метанмонооксигеназе, и порфиринового лиганда, как в P450. Она характеризуется низкой трансформационной подвижностью, ион металла благодаря N‑ или C‑мостиковому фрагменту прочно связан с макроциклическим лигандом и имеет фиксированную высокую степень окисления. Возможность делокализации заряда на двух металлических центрах, двух макроциклических лигандах и μ-мостике этих соединений делает предсказуемой активацию пероксида водорода и надкислот с последующей генерацией высоковалентных оксоформ, обладающих как каталазной, так и пероксидазной активностью. Наличие двух редокс-активаторов позволяет расширить диапазон способов настройки и контроля реакционной способности такой биомиметической системы. Для μ-нитридо- и μ-карбидодимерных комплексов железа(IV) и рутения(IV) в реакциях с перекисями возможно индуцирование или регулирование их активности, а также симультанного проявления каталазных и пероксидазных свойств [50–54] посредством аксиальной координации различных биоактивных молекул.
В рамках химического аспекта моделирования редокс-центров окислительных ферментов необходимо проводить систематические исследования спектральных, координационных, окислительно-восстановительных и каталитических свойств димерных тетрапиррольных макроциклических комплексов переходных металлов с различной модификацией, определять взаимосвязь структура–активность и детализировать химические принципы окислительных реакций, имитирующие разные стадии каталитического цикла в биологических системах.
ОСНОВНЫЕ ПРИНЦИПЫ СИНТЕЗА И НЕКОТОРЫЕ ОТЛИЧИТЕЛЬНЫЕ ХАРАКТЕРИСТИКИ μ-НИТРИДО- И μ-КАРБИДОДИМЕРНЫХ КОМПЛЕКСОВ С МАКРОЦИКЛАМИ ПОРФИРИНОВОГО ТИПА
Биядерные порфириновые и фталоцианиновые комплексы, имеющие в своем составе кислородный, азотный и углеродный мостиковый фрагмент, интенсивно исследуются в течение последних десятилетий. Интерес к этим макроциклическим биядерным комплексам возобновился после открытия интересных с практической точки зрения каталитических свойств μ-оксо- и μ-нитридожелезных фталоцианинов и порфиринов в окислении метана, бензола и алкилароматических соединений, в образовании связи C–С и окислительном дегалогенировании ароматических углеводородов, включая поли- и перфторированные молекулы [55–60]. Так, в работе впервые показано, что µ-карбидодимерный комплекс фталоцианината рутения проявляет высокую каталитическую активность в реакции переноса карбенов [61]. Фталоцианиновые μ-оксокомплексы железа, в отличие от μ‑нитридо и μ‑карбидодимерных комплексов, являются нестабильными и легко подвергаются мономеризации. Поэтому целесообразнее использовать последние соединения в качестве объектов биомиметического исследования.
Согласно данным [62–96 ], синтез биядерных комплексов с общей формулой [M1P1(μ-X)M2P2], где М1 и М2 – ионы переходных металлов (М1 = М2 для гомоядерных комплексов, М1 ≠ М2 для гетероядерных комплексов), P1, P2 – порфириновый, фталоцианиновый или порфиразиновый дианион (Р1 = P2 для гомолептичеких комплексов, Р1 ≠ P2 для гетеролептических комплексов), Х – атом кислорода, азота или углерода, связывающий две структурно-эквивалентные или неэквивалентные части, можно описать в соответствии со схемой 1 :
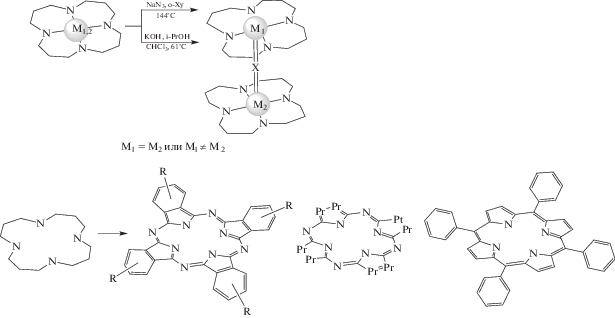
Схема 1 .
Наиболее доступными в ряду μ-Х-биядерных соединений являются µ‑оксодимерные комплексы. Их можно получить также при окислении мономерных порфиринатов железа кислородом воздуха или простым хроматографированием, пропуская через колонку, заполненную основным или нейтральным оксидом алюминия [62, 63, 72, 75]. Первые μ-нитридодимерные комплексы получены и спектрально охарактеризованы около 40 лет назад [79, 80]. Существенный вклад в разработку методов синтеза μ‑нитридодимерных комплексов внесли работы Эрколани [64–68, 70, 73]. В этих работах представлен синтез и спектральные данные различных типов μ‑нитридодимерных комплексов, в том числе систем с гетеролептическими лигандами ТРРFe–N=FeРс (TPP – тетрафенилпорфирин, Pc – замещенный фталоцианин), а также с разными металлами: ТРРFe=N–RuРс, ТРРMn=N–FeРс и Рс(tBu)4Mn=N–FeРс(tBu)4 [44, 45, 48, 49, 95]. μ-Нитридодимерный комплекс фталоцианината железа (μ-N(FeIII,IVPc)2), в котором металл формально имеет степени окисления +3 и +4, легко окисляется в присутствии слабых окислителей, например тетрацианохинодиметана или йода, до μ-N(FeIVPc)2+ [54]. Использование сильных окислителей, таких как бром, азотная кислота или трифторуксусная кислота, на воздухе приводит к двухэлектронному переносу, происходящему на атоме железа и макроциклическом лиганде [86]. В последнее время появилось большое количество работ, посвященных синтезу и изучению физико-химических свойств данного класса соединений [50–54, 76, 81–91]. Впервые о теоретически возможном существовании μ‑карбидодимерных комплексов железа с макроциклическими лигандами – стабильными диамагнитными соединениями с линейной структурой M=С=M – заявил Роальд Хоффманн (Roald Hoffmann) в работе [92]. В 1981 г. Дэниелом Мэнсуем (Daniel Mansuy) был получен первый μ-карбидодимерный комплекс тетрафенилпорфирината железа(IV) (TPPFeIV=C=FeIVTPP) путем взаимодействия мономерного FeTPP, растворенного в смеси дихлорметан–хлороформ (9 : 1), с тетрайодметаном в присутствии избытка железного порошка или с тетрайодметаном в бензоле в присутствии избытка дитионита натрия или бораната натрия [93]. μ‑Карбидодимерный фталоцианинат железа (μ‑С(FeIVPc)2) был получен в результате взаимодействия мономерного фталоцианината железа с тетрайодметаном в хлорнафталине в присутствии дитионита натрия или железного порошка [95, 96]. В настоящее время синтезирован μ‑карбидодимерный октасульфофталоцианинат железа (μ‑С(FeIVPc(SO3H)8)2) [97]. Выход комплекса μ‑С(FeIVPc(SO3H)8)2 составил 40%. Фталоцианинаты рутения обычно получают конденсацией фталонитрилов в присутствии солей или комплексов рутения [66]. Прямое внедрение рутения в предварительно сформированный макроцикл используется в основном для металлирования алкилзамещенных фталоцианинов додекакарбонилтрирутением (Ru3(CO)12) в кипящем бензонитриле с образованием комплексов с двумя аксиально координированными молекулами растворителя бензонитрила [98]. В работе [61] впервые показано, что µ‑карбидодимерный комплекс фталоцианината рутения можно получить по реакции 4,5‑октабутоксизамещенного фталоцианина с Ru3(CO)12 в кипящем о-дихлорбензоле по схеме 2 :
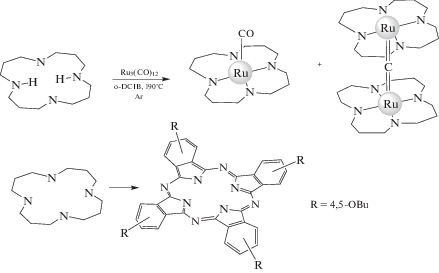
Схема 2 .
Синтез μ-карбидодимерных комплексов с макроциклическими лигандами (μ‑С(MP)2, где M = Fe, Ru; P = Pc, TPP, октаэтилпорфирин (OEP)) можно разделить на три основные стадии [69]:
1) реакция легкодоступного мономерного предшественника, например (Cl)FeP (P = Pc, TPP, OEP), с гидроксидом калия в кипящем изопропаноле в анаэробных условиях:
(1)
$\begin{gathered} 2{{\left[ {{{{\text{M}}}^{{{\text{III}}}}}{{{\left( {{\text{OH}}} \right)}}_{{\text{2}}}}{\text{P}}} \right]}^{--}} + {\text{2O}}{{{\text{H}}}^{--}} \to 2{{\left[ {{{{\text{M}}}^{{{\text{II}}}}}{{{\left( {{\text{OH}}} \right)}}_{{\text{2}}}}{\text{P}}} \right]}^{{2--}}} \to \\ \to \,\,2{{\left[ {{{{\text{M}}}^{{\text{I}}}}{\text{P}}} \right]}^{--}} + 1{\text{/}}2{{{\text{O}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} + 2{\text{O}}{{{\text{H}}}^{--}};~~~ \\ \end{gathered} $2) окисление восстановленной частицы MIP– дихлоркарбеном, образующимся in situ при взаимодействии хлороформа с гидроксидом калия, с выделением соответствующего дихлоркарбенового комплекса:
(2)
$2{{\left[ {{{{\text{M}}}^{{\text{I}}}}{\text{P}}} \right]}^{--}} + {\text{CC}}{{{\text{l}}}_{2}} \to \left[ {{{{\text{M}}}^{{{\text{II}}}}}\left( {{\text{CC}}{{{\text{l}}}^{{\text{2}}}}} \right){\text{P}}} \right] + {{\left[ {{{{\text{M}}}^{{\text{I}}}}{\text{P}}} \right]}^{{2--}}};$3) конденсация in situ карбенового комплекса с восстановленными частицами, приводящая к образованию μ‑карбидодимерного комплекса:
(3)
$\begin{gathered} \left[ {{{{\text{M}}}^{{{\text{II}}}}}\left( {{\text{CC}}{{{\text{l}}}_{{\text{2}}}}} \right){\text{P}}} \right] + {{\left[ {{{{\text{M}}}^{{\text{I}}}}{\text{P}}} \right]}^{{2--}}} \to \\ \to \left[ {{{{\left( {{\text{MP}}} \right)}}_{2}}(\mu {\text{ - C}})} \right] + 2{\text{C}}{{{\text{l}}}^{--}}, \\ \end{gathered} $(4)
$\begin{gathered} \left[ {{{{\text{M}}}^{{{\text{II}}}}}\left( {{\text{CC}}{{{\text{l}}}_{{\text{2}}}}} \right){\text{P}}} \right] + 2{{\left[ {{{{\text{M}}}^{{\text{I}}}}{\text{P}}} \right]}^{--}} \to \\ \to \left[ {{{{\left( {{\text{MP}}} \right)}}_{2}}(\mu {\text{ - C}})} \right] + \left[ {{{{\text{M}}}^{{{\text{II}}}}}{\text{P}}]{\text{ }} + {\text{ }}2{\text{C}}{{{\text{l}}}^{--}}} \right]; \\ \end{gathered} $4) очистка и выделение μ-карбидодимерного комплекса.
В соответствии с предложенным механизмом синтеза μ‑карбидодимерных комплексов к настоящему времени получен целый ряд биядерных соединений.
На основании результатов многолетних исследований, связанных с синтезом биметаллических μ‑Х‑димерных комплексов с макроциклами порфиринового типа [62–98 ] можно выделить несколько основных способов структурной вариации молекулярной платформы на основе биметаллических комплексов железа и рутения (схема 3 ):
– модификация макроциклического лиганда за счет введения различных электронодонорных или электроноакцепторных заместителей;
– изменение типа макроциклического каркаса путем замены лиганда или интеграцией двух разных макроциклов (порфирин, фталоцианин, порфиразин и др.);
– варьирование катиона переходного металла (Fe, Mn, Ru, Cr, V, Os и др.);
– варьирование мостикового атома с образованием, например, структурных единиц FeIII–O–FeIII, FeIII–N=FeIV и FeIV=C=FeIV для μ‑оксо-, μ‑нитридо- и μ‑карбидодимерных комплексов соответственно.
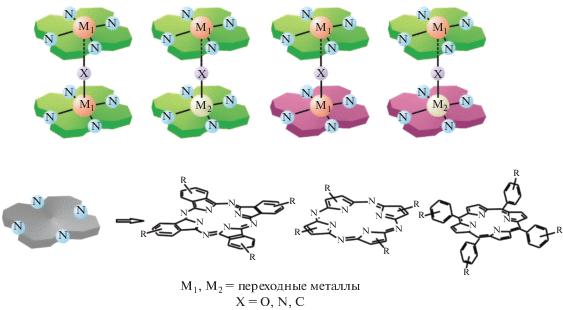
Схема 3 . Возможные структуры μ-Х-димерных биметаллических макроциклических комплексов.
За счет структурных модификаций можно регулировать такие свойства соединений, как перенос электронов, проводимость, редокс-потенциал, координирующая и реакционная способность. Их адресная настройка имеет большой потенциал практического применения.
μ‑Х‑димерные биметаллические комплексы железа и рутения μ‑X(МP)2 (M = Fe, Ru, P – фталоцианин, октапропилпорфиразин (OPrTAP), тетрафенилпорфирин; X = C, N, O), представляющие собой многообещающую молекулярную платформу для окислительного катализа, были исследованы методами Uv-vis-, ИК- и ЯМР-спектроскопии, рентгеновского поглощения вблизи краевой структуры (XANES), масс-спектрометрии, а также с помощью теоретических расчетов [50–54, 61–106 ].
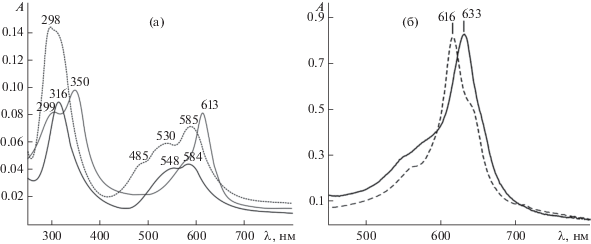
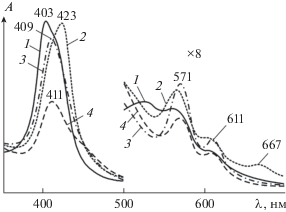
Общий вид ЭСП μ‑нитридо- и μ‑карбидодимерных комплексов железа и рутения с макроциклами порфиринового типа соответствует мономерным предшественникам, за исключением гипсохромного сдвига В‑ и Q‑полос и их уширения для соединений с N‑, C‑мостиками вследствие экситонного взаимодействия двух макроциклов [50–54, 69, 78, 86, 100–104]. Для μ‑карбидодимерных комплексов железа гипсохромный сдвиг в Q‑области выражен в меньшей степени. В ЭСП μ‑карбидодимерных комплексов рутения с фталоцианиновым лигандом не наблюдается низкоинтенсивных Q-полос в области 580–600 нм по сравнению с мономерными аналогами [61, 99]. Uv‑vis‑спектры μ‑Х‑димерных биметаллических макроциклических комплексов в растворе демонстрируют характерную интенсивную Q‑полосу поглощения в диапазоне 500–680 нм (табл. S1 ). В ЭСП соединений μ‑оксодимерного 2,3,7,8,12,13,17,18-октапропилтетраазапорфирината железа(III) (µ-О(FeIIIOPrTAP)2), μ-нитридодимерного 2,3,7,8,12,13,17,18-октапропилтетраазапорфирината железа(III, IV) (µ-N(FeIII,IVOPrTAP)2) и μ-карбидодимерного 2,3,7,8,12,13,17,18-октапропилтетраазапорфирината железа(IV) (µ-С(FeIVOPrTAP)2) присутствуют типичные полосы поглощения в области от 500 до 650 нм. Следует отметить, что В‑ и Q‑полосы в ЭСП комплекса µ-О(FeIIIOPrTAP)2 заметно смещены в красную область по сравнению с полосами поглощения µ-N(FeIII,IVOPrTAP)2 и µ-С(FeIVOPrTAP)2. Наблюдаемый красный сдвиг для μ‑оксодимера предполагает более слабое взаимодействие между двумя порфиразиновыми фрагментами, чем в μ‑нитридо- и μ‑карбидокомплексах. Это предположение согласуется с более короткими межплоскостными расстояниями в µ‑N(FeIII,IVOPrTAP)2 и µ‑С(FeIVOPrTAP)2 (табл. S2 ) [78]. Более гипсохромно сдвинуты В‑ и Q‑полосы в ЭСП μ-карбидодимерного тетра-трет-бутилзамещенного фталоцианината рутения(IV) (μ-С(RuIVPc(tBu)4)2) по сравнению с аналогичным комплексом железа (рис. 1). Форма и положение Q-полос в ЭСП µ-карбидодимеров фталоцианината рутения зависят от природы заместителей. Так, трет-бутилзамещенный комплекс имеет относительно узкую Q-полосу при 616–619 нм [99, 100], а для комплексов с окта- и гексабутоксизамещенными лигандами наблюдаются более широкие Q‑полосы с максимумами при 607 и 615 нм соответственно. Авторы [99] показали, что форма полосы не связана с агрегацией, а является результатом электронных и конформационных изменений при замещении макроцикла. Аналогичным образом можно охарактеризовать спектральное поведение для µ-нитридодимерных фталоцианинатов железа [54, 76, 86, 90, 95, 102–104, 106]. Введение гибких линейных алкоксигрупп на периферии макроцикла приводит к сильному уширению Q‑полос димерных комплексов, тогда как комплексы с объемными разветвленными алкоксигруппами в “сэндвичевых” комплексах обычно демонстрируют узкие, хорошо разрешенные Q‑полосы. Такое спектральное поведение, вероятно, связано с разницей в степени вращения макроциклических лигандов [107].
Рис. 1.
ЭСП µ-О(FeIIIOPrTAP)2 (серая сплошная линия), µ-N(FeIII,IVOPrTAP)2 (черная сплошная линия) и µ‑С(FeIVOPrTAP)2 (черная пунктирная линия), с = 2 × 10–5 моль/л, в CH2Cl2 (а); ЭСП комплексов μ‑С(FeIVPc(tBu)4)2 (черная сплошная линия) и μ-С(RuIVPc(tBu)4)2 (черная пунктирная линия), с = 10–6 моль/л, в бензоле (б).
В ИК-спектрах μ-нитридо- и μ-карбидодимерных комплексов железа с макроциклами порфиринового типа наблюдается характерная полоса в области 900–1000 см–1, соответствующая асимметричным валентным колебаниям фрагмента Fe–X–Fe. Для рутениевых μ-карбидодимерных комплексов полоса колебаний νas(Ru=C=Ru) смещается в высокочастотную область более 1000 см–1 (табл. S1 ). В μ-карбидодимерном гетеролептическом комплексе железа эта полоса может расщепляться, вероятно, из-за различной прочности связывания двух частей мостиковой системы Fe=С=Fe, которая определяется окружением атома металла [100]. Кроме того, наличие четырех или восьми трет-бутильных групп хорошо проявляется в области алифатических C–H валентных колебаний при 2950 см–1, в то время как наблюдается постепенное уменьшение интенсивности фенильных C–H валентных колебаний в диапазоне 1600–700 см–1 при переходе от незамещенного гомолептического μ-карбидодимерного комплекса железа с фталоцианином к замещенному аналогу. Масс-спектры μ-нитридо- и μ-карбидодимерных комплексов демонстрируют заметные молекулярные пики, соответствующие ожидаемым значениям молекулярного иона (рис. S1 ) [50–54, 61–109 ].
Для соединений µ-О(FeIIIOPrTAP)2, µ-N(FeIII,IVOPrTAP)2 и µ-С(FeIVOPrTAP)2 выполнен подробный сравнительный анализ структурных особенностей (табл. S2–S4 [78]). Многие тенденции, установленные в ходе анализа структуры этих соединений, характерны и для µ-Х-димерных комплексов железа с порфириновыми и фталоцианиновыми лигандами [76, 86, 103–106].
Данные рентгеновской дифракции (ST1-ST2, SP1-SP2) показывают, что атомы железа μ-оксодимера соединены почти симметричным μ-оксомостиком с расстояниями Fe(1)–O(1) и Fe(2)–O(1) 1.760 и 1.750 Å соответственно, образуя угол FeOFe 158.52° (табл. S2 ). Атомы железа расположены в центре плоскости порфиразина с расстояниями Fe(1)–Npyr и Fe(2)–Npyr 1.934–1.936 и 1.936–1.946 Å соответственно. Порфиразиновые макроциклы почти плоские. Отклонения пиррольных атомов азота от Fe(1) и Fe(2) N4-плоскостей составляют ±0.061 и ±0.032 Å соответственно.
Максимальные отклонения мезоатомов азота от плоскости N4 составляют 0.071–0.075 Å. Выход атомов Fe(1) и Fe(2) из плоскости макроциклов к кислородному мостику равен 0.336 и 0.367 Å соответственно. Двугранный угол между двумя порфиразиновыми плоскостями, обусловленный единицей FeOFe, составляет 19.3°. Макроциклы комплекса µ-О(FeIIIOPrTAP)2 смещены относительно друг друга и образуют конформационный угол 24.2°.
Замена кислородного мостика на азот сопровождается уменьшением длин связей Fe(1)–Npyr и Fe(2)–Npyr до 1.914–1.929 и 1.915–1.932 Å соответственно. Повышается планарность макроциклов, максимальные отклонения пиррольных и мезоатомов азота составляют ±0.011 и ±0.063 Å соответственно. Связи Fe–μ-N в структурной единице Fe–Х–Fe короче по сравнению со связями Fe–O. Угол фрагмента FeNFe увеличивается до 168.5°, а двугранный угол между порфиразиновыми плоскостями уменьшается до 11.5° (табл. S2 ). Однако относительная ориентация макроциклов в димерных молекулах соразмерна углам кручения 24.2° и 26.2° для µ-О(FeIIIOPrTAP)2 и µ-N(FeIII,IVOPrTAP)2 соответственно. Смещение атомов железа из плоскостей порфиразина в комплексе µ-N(FeIII,IVOPrTAP)2 уменьшается на 0.053 и 0.056 Å по сравнению с комплексом µ-О(FeIIIOPrTAP)2.
При переходе к углеродному мостику наблюдается существенное отличие молекулярной геометрии соединений µ-С(FeIVOPrTAP)2 и µ-О(FeIIIOPrTAP)2. Значительно уменьшается расстояние Fe–Npyr (до 1.910–1.915 Å). Длины связей Fe–C короче на 0.079–0.091 Å, а фрагмент Fe=C=Fe имеет угол 175.10° и приближается к линейной конфигурации. Такая структурная трансформация приводит к резкому уменьшению двугранного угла до 8.2°. Плоскости макроциклов становятся почти параллельными. Пиррольные и мезоатомы азота выступают из плоскости порфиразина на 0.011 и 0.082 Å соответственно. Выход атома железа из координационной плоскости в сторону С-мостика меньше, чем в соединениях µ-О(FeIIIOPrTAP)2 и µ‑N(FeIII,IVOPrTAP)2 (табл. S2 ).
С увеличением степени окисления железа и кислотности по Льюису величина связи Fe–Npyr, смещение атомов Fe из плоскости макроцикла и двугранный угол между двумя плоскостями уменьшаются в ряду µ-О(FeIIIOPrTAP)2 > > µ-N(FeIII,IVOPrTAP)2 > µ-С(FeIVOPrTAP)2. Аналогично изменяются данные параметры и для µ-димерных комплексов железа с фталоцианином [110]. Однако изменение связи в мостиковом фрагменте Fe–X–Fe не соответствует приведенной выше тенденции (табл. S2 ). Несмотря на более высокую степень окисления катиона железа (Fe4+) и формальную двойную связь Fe=C в µ-С(FeIVOPrTAP)2 по сравнению с µ-N(FeIII,IVOPrTAP)2, где степень окисления Fe3,5+ и порядок связи 1.5, длина связи Fe–C больше, чем Fe–N. Подобный факт наблюдался и в ряду μ-O‑, μ-N‑ и μ-C‑димерных фталоцианинов железа [100], а для димерных комплексов с тетрафенилпорфириновым макроциклом эти связи равны [105]. Это можно объяснить снижением порядка связи Fe=C, которое возникает из-за высокого вклада обратной дативной π-связи. Структурная единица Fe–X–Fe становится более линейной при переходе от μ-O‑ к μ-C‑димерному комплексу, о чем свидетельствует увеличение угла от 158° до 175° и от 174° до 180° для порфиразиновых и порфириновых комплексов соответственно (табл. S2, S3 ). Природа мостикового атома не оказывает существенного влияния на конформационные углы во всех трех димерах. Межплоскостное расстояние, определяемое как сумма двух длин связей между Fe–X и двумя отклонениями катионов железа от плоскости, уменьшается от 4.213 Å в случае соединения µ-О(FeIIIOPrTAP)2 до 3.891 и 3.766 Å для µ-N(FeIII,IVOPrTAP)2 и µ‑С(FeIVOPrTAP)2 соответственно [78], что указывает на более сильное π–π‑взаимодействие макроциклов в этих комплексах.
Следует отметить влияние природы макроцикла на наиболее важные структурные параметры µ-димерных комплексов (табл. S3, S4 ). Анализ рентгеноструктурных данных показывает, что все соединения демонстрируют довольно короткие расстояния Fe–Fe (от 3.28 до 3.39 Å), что предполагает сильное π–π-взаимодействие между ароматическими макроциклическими кольцами. Причиной роста расстояния Fe–Fe для представленных комплексов является стерический фактор, обусловленный арильными или алкильными заместителями в макроциклах. Величина угла мостикового фрагмента увеличивается в ряду: μ-N(FeIII,IVОЕP)2 < μ-N(FeIII,IVTPP)2 < μ-N(FeIII,IVPctBu)2 ≤ μ-N(FeIVPctBu)2Br2 = μ-N(FeIIIРсFeIVТРP) < < μ-N(FeIVPc)2Br2 (табл. S4 ). Линейной структурной единицей Fe–N–Fe обладает димерный комплекс на основе фталоцианина.
Сравнение структурных параметров μ-О-, μ-N- и μ-С-димерных порфиразиновых комплексов железа с μ-димерными порфириновыми и фталоцианиновыми аналогами показывает, что природа макроциклического лиганда не оказывает значимого влияния на длину связи Fe–X (табл. S3 ) [76, 78, 102, 104, 105]. Длины связей Fe–O, Fe–N и Fe–C для димерных комплексов порфиринового типа изменяются в пределах 1.75–1.76, 1.64–1.68 и 1.66–1.68 Å соответственно. Однако смещение атомов железа из плоскости макроцикла значительно больше в порфириновых комплексах, вероятно, из-за наличия объемных мезофенильных групп. Порфиразиновые лиганды обеспечивают большую гибкость во взаимном расположении макроциклов в димерах. Угол FeXFe для димеров с порфириновым макроциклом на 5°–16° больше, чем у порфиразиновых аналогов, что также объясняется влиянием стерического фактора, вызванного объемными арильными заместителями (табл. S3 ).
ЭПР-исследования µ-О(FeIIIOPrTAP)2, µ-N(FeIII,IVOPrTAP)2 и µ-С(FeIVOPrTAP)2 показали, что µ-О- и µ-С-комплексы не дают сигнала, тогда как μ-нитридодимерный комплекс показывает сильный сигнал, характерный для низкоспинового железа. Спектры ЭПР µ-N(FeIII,IVOPrTAP)2 демонстрируют хорошо разрешенную сверхтонкую структуру за счет наличия мостикового атома 14N [76, 78]. Присутствие пиридина в составе комплекса µ-N(FeIII,IVOPrTAP)2 приводит к исчезновению в спектрах ЭПР сверхтонкого расщепления. Координация основания сопровождается образованием аддукта с кислородом, и в ЭПР-спектре наблюдаются сигналы g1 = 2.062, g2 = 2.007, g3 = 2.000 без сверхтонкой структуры 14N [78].
Степень окисления железа и его координационное окружение претерпевают важные изменения в каталитическом цикле, поэтому использование спектроскопии XANES полезно для идентификации активных интермедиатов. Высоковалентное состояние железа в µ-Х-димерных комплексах порфиринового типа хорошо иллюстрируют спектры XANES. Положение и интенсивность FeK XANES предкраевого пика зависят от электронной структуры, геометрии железосодержащих частиц, координационного числа металла, симметрии и природы лиганда. Частицы, содержащие высоковалентное железо, в отличие от их низковалентных аналогов, характеризуются более интенсивными предкраевыми пиками из-за наличия большого числа вакантных d-орбиталей [78]. В ряду µ-димерных октапропилпорфиразинов µ-О < µ-N < µ-С отмечен рост предкраевой энергии и интенсивности предкраевого пика (рис. 2), что говорит об увеличении степени окисления железа в этих соединениях.
Рис. 2.
Спектры XANES комплексов µ-О(FeIIIOPrTAP)2, µ-N(FeIII,IVOPrTAP)2 и µ-С(FeIVOPrTAP)2. Предкраевая энергия указана над кривыми.
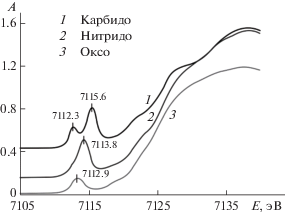
Подобная картина наблюдается в спектрах XANES для µ-нитридодимерных комплексов железа с фталоцианиновыми лигандами [105], у которых также подтверждается высоковалентное состояние атомов железа. Положение и интенсивность предкраевого пика увеличиваются при переходе от мономера (Cl)FeIIIPc к димерным аналогам с мостиковым фрагментом FeIII–N–FeIV и FeIV–N–FeIV с 7113.7 до 7113.8 и 7114.7 эВ соответственно. Введение заместителей в макроцикл сопровождается еще большим ростом интенсивности (от 1.16 эВ для мономера FeIII до 1.6 эВ для µ-димера FeIV) и смещением пика в область высокой энергии в ряду (Cl)FeIIIPc(tBu)4 (7112.8 эВ) < µ-N(FeIII,IVPc(tBu)4)2 (7114.1 эВ) < µ-N(FeIVPc(tBu)4)2 (7115.2 эВ). Это свидетельствует о вкладе электронодонорных заместителей в стабилизацию высоковалентного состояния атома железа.
Следует отметить разное электрохимическое поведение μ-оксо-, μ-нитридо- и μ-карбидодимерных комплексов железа. На примере соединений с порфиразином рассмотрим влияние природы μ-Х-мостика на редокс-потенциалы и структуру окисленных частиц. Методом циклической вольтамперометрии и вольтамперометрии на вращающихся дисковых электродах в CH2Cl2 получены редокс-потенциалы для µ-О(FeIIIOPrTAP)2, µ-N(FeIII,IVOPrTAP)2 и µ-С(FeIVOPrTAP)2 (табл. 1) [78]. Все потенциалы относятся к полуволновому потенциалу ферроцена (Fc/Fc+).
Таблица 1.
Редокс-потенциалы комплексов µ-О(FeIIIOPrTAP)2, µ-N(FeIII,IVOPrTAP)2 и µ-С(FeIVOPrTAP)2
| Комплекс | Стадия 1 | Стадия 2 | ||||
|---|---|---|---|---|---|---|
| Epa, эВ | Epc, эВ | E1/2, эВ | Epa, эВ | Epc, эВ | E1/2, эВ | |
| μ-O | 0.240 | 0.070 | 0.155 | 0.860 | – | – |
| μ-N | –0.052 | –0.174 | –0.113 | 0.640 | 0.520 | 0.580 |
| μ-C | 0.356 | 0.252 | 0.304 | 0.656 | 0.538 | 0.597 |
Электрохимические исследования показали, что в случае комплекса µ‑N(FeIII,IVOPrTAP)2 два обратимых окисления могут быть связаны как с окислением металлоцентра ($E_{{1/2}}^{1}$), так и с окислением лиганда до π-катион-радикала ($E_{{1/2}}^{2}$) (табл. 1). В результате одноэлектронного окисления μ-N-димера образуются диамагнитные комплексы с двумя эквивалентными FeIV-центрами. Окисление металла происходит при более низких потенциалах, чем отрыв электрона от макроцикла.
Данные табл. 1 показывают, что в случае µ-С(FeIVOPrTAP)2 протекают два последовательных обратимых одноэлектронных процесса окисления при +0.304 и +0.597 В. Оба процесса представляют собой обратимый одноэлектронный перенос и связаны с окислением порфиразиновой части до π-дикатион-радикала (OPrTAP+•)FeIV=C=FeIV(OPrTAP+•).
Электрохимическое поведение μ-карбидодимерных комплексов железа с тетрафенилпорфиновым, октаэтилпорфириновым и фталоцианиновым лигандами описано в работах [53, 69, 89, 101, 103]. Все соединения демонстрируют две одноэлектронные волны окисления, связанные с последовательным отщеплением электрона на макроциклическом лиганде. В более ранних работах имеются данные для соединений μ-N(FeTPP)2 и μ-С(FeTPP)2, которые подвергались четырехстадийному обратимому одноэлектронному окислению в дихлорметане [55, 108]. Авторы связывают эти четыре процесса с последовательным образованием положительно заряженных комплексов (1+, 2+, 3+, 4+) без каких-либо дополнительных подробностей о локализации зарядов.
КООРДИНАЦИОННЫЕ СВОЙСТВА µ-НИТРИДО И µ-КАРБИДОДИМЕРНЫХ КОМПЛЕКСОВ ЖЕЛЕЗА(IV) И РУТЕНИЯ(IV) В РЕАКЦИЯХ С БИОЛОГИЧЕСКИ АКТИВНЫМИ ОСНОВАНИЯМИ
В биологических системах простетическая группа гемсодержащего фермента функционирует в составе сложного белково-пигментного комплекса. Поэтому окружение координационного центра, электронная и конформационная структура боковых цепей гемма и природа связи с белковой молекулой определяют его селективность и реакционную способность. Аксиальное лигандирование порфириновых комплексов, участвующих в каталитических циклах, существенно влияет на протонное состояние активных центров, определяет их основность и тип высоковалентного интермедиата [109]. Даже небольшие изменения, происходящие с центральным атомом металла, структурой лиганда и внутренней координационной сферой, приводят к ощутимым различиям в реакционной способности порфиринового комплекса.
Моделирование феррильных частиц природных окислительных ферментов связано как с различными аспектами дизайна искусственных молекулярных платформ, так и с изучением их свойств. Аксиальное лигандирование соединений порфиринового типа, с одной стороны, является одним из видов модификации макроцикла, а с другой – определяет субстратную специфичность активного сайта миметика. Исследования координирующей способности комплексов в зависимости от природы макроциклического лиганда, субстрата и склонности атома металла к различным координационным состояниям дают возможность получить фундаментальное химическое представление о структуре ферментов, прочности связи металл–субстрат в редокс-центре, влиянии аксиального лигандирования порфиринового сегмента на его реакционную способность. Результаты таких исследований могут быть полезны при разработке способов инициирования и контроля генерации различных видов высокоактивных интермедиатов.
В последнее время изучение µ-нитридо- и µ-карбидодимерных комплексов железа и рутения не ограничивается синтезом и спектральными свойствами. В литературе представлено несколько работ, посвященных исследованию координирующей способности µ-нитридо- и μ-карбидодимерных комплексов железа(IV) по отношению к различным субстратам [51, 54, 103, 110, 111]. Авторами [103] была показана более высокая устойчивость µ-карбидодимерного комплекса железа с незамещенным фталоцианином μ-C(FePc)2 к мономеризации в растворах N-оснований (пиридина (Py) и 1-метилимидазола (1-MeIm)) за счет координации последних на металлоцентрах димера по сравнению с порфириновыми комплексами. Гексакоординированная структура координационных центров молекул μ-C((1-MeIm)FePc)2 подтверждена данными рентгеноструктурного анализа [103]. Способность μ-C(FePc)2 аксиально координировать различные субстраты была использована в работе [111] при исследовании этого соединения в качестве материала для солнечных элементов и высокочувствительного сенсора на NO2. Подробно образование аксиальных комплексов µ-нитридо- и μ‑карбидодимеров железа(IV) с азотсодержащими органическими основаниями исследовано в работах [51, 54, 110]. Интерес к этим субстратам связан с тем, что большинство из них являются связующим звеном между белковой и простетической группой и входят в состав активного сайта фермента, проявляют бактерицидные, противоопухолевые, противоишемические, антиаллергические, гербицидные свойства и могут служить прекурсорами лекарственных препаратов. Эти молекулы легко образуют комплексы с ионами переходных металлов [112–115].
Известно, что UV-vis-спектры являются наиболее распространенным методом изучения взаимодействия макроциклических тетрапиррольных комплексов металлов с различными субстратами. В UV-vis-спектрах Q-диапазон, в отличие от В-диапазона при λ = 290–340 нм, особенно для металлофталоцианинов и металлопорфиразинов, существенно зависит от заместителей в макроцикле и природы атома-комплексообразователя. Кроме того, изменение положения или интенсивности Q-полос чувствительно к присутствию различных соединений во внутренней координационной сфере и может указывать на сильное взаимодействие между электронными состояниями хромофора и молекулой субстрата.
Координационное связывание μ-нитридо- и μ-карбидодимерными октапропилпорфиразином, тетрафенилпорфирином и тетра-трет-бутилфталоцианином железа(IV) (μ-С(FeIVOPrTAP)2, μ-С(FeIVTPP)2 и μ-C(FeIVPc(tBu)4)2, cкомпл = 10–6 моль/л) различных N-оснований (L = диэтиламин (Et2N), имидазол (Im), 1-метилимидазол (1-MeIm), 2-метилимидазол (2-MeIm), пиридин (Py)) изучено спектральными методами в широком диапазоне концентраций (${{c}_{{{\text{E}}{{{\text{t}}}_{{\text{2}}}}{\text{NH}}}}}$ = 1.36 × 10–4–4.8 × 10–1 моль/л, c1-MeIm = = 6.4 ×10–5–2.5 × 10–1 моль/л, c2-MeIm = 2.01 × 10–5 – – 2.9 × 10–2 моль/л, cIm = 9.5 × 10–5– 2.5 × 10–2 моль/л, cPy = 7.77 × 10–4–5.2 × 10–1 моль/л) [51, 54, 110]. Исследуемая обратимая реакция сопровождается четким спектральным откликом на изменение состава комплекса, который различается в зависимости от природы макроциклического лиганда и субстрата (рис. 3, 4) [51, 54, 110].
Рис. 3.
Изменение ЭСП μ-карбидодимерных комплексов железа с макроциклами порфиринового типа при взаимодействии с 1-метилимидазолом (с1-MeIm = 1.76 × × 10–4–3.0 × 10–1 моль/л) в бензоле: μ-С(FeIVOPrTAP)2 (с = 6.4 × 10–6 моль/л), μ-C(FeIVPc(tBu)4)2 (с = 6.84 × × 10–6 моль/л), μ-С(FeIVTPP)2 (с = 8.2 × 10–6 моль/л) и соответствующая кривая титрования.
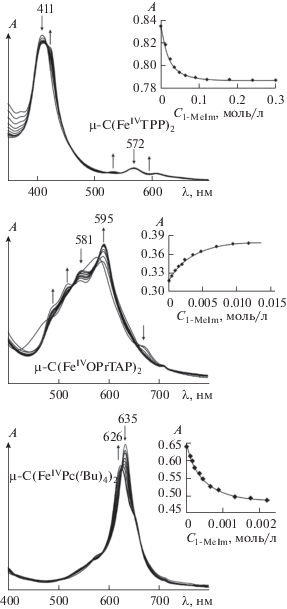
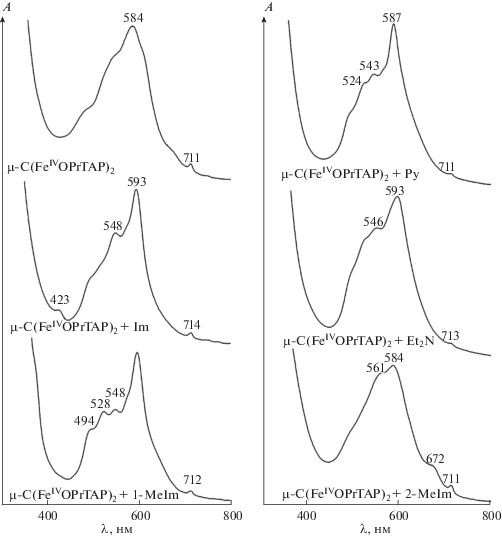
Изменение интенсивности и смещение Q-полос с сохранением изобестических точек в электронном спектре поглощения комплексов μ-С(FeIVOPrTAP)2, μ-C(FeIVPc(tBu)4)2 и μ-С(FeIVTPP)2 свидетельствуют об образовании донорно-акцепторного комплекса [51, 54, 110]. Подобная закономерность наблюдается и для мономерных аналогов [115, 116]. Величина смещения полосы при λmax в ЭСП составляет 7–14, 5–9 и 1–10 нм для μ-С(FeIVOPrTAP)2, μ-C(FeIVPc(tBu)4)2 и μ-С(FeIVTPP)2 соответственно [51, 110].
ЭСП μ-С(FeIVOPrTAP)2 с N-основаниями, в отличие от исходного соединения, характеризуются наличием в длинноволновой области между полосой Соре при λ = 320–322 нм и интенсивной Q-полосой при λ = 587–595 нм ряда менее интенсивных, но четко выраженных полос переноса заряда (рис. 4) [51]. Их появление зависит от электронной плотности на атоме железа [75], которая меняется в результате образования связи металл–субстрат. Наиболее интенсивные полосы переноса заряда наблюдаются у комплекса с 1-MeIm. Это качественно характеризует более прочное связывание 1-метилимидазола по сравнению с другими основаниями.
В отличие от μ-карбидодимерных комплексов, взаимодействие μ-нитридодимерного тетра-трет-бутилфталоцианината железа(IV) (μ-N(FeIVPc(tBu)4)2Cl) с N-основаниями происходит во времени. Трансформация ЭСП заключается в смещении полосы Qmax на 2–4 нм и увеличении ее интенсивности (рис. 5). Принимая во внимание сходство спектральных изменений при координации N-оснований, подробно рассмотрим взаимодействие μ-N(FeIVPc(tBu)4)2Cl с имидазолом.
Рис. 5.
Изменения во времени ЭСП μ-N(FeIVPc(tBu)4)2Cl (1) в бензоле в присутствии имидазола при 298 K: μ‑N((Im)FeIVPc(tBu)4)2 (2), μ-N((Im)FeIII,IVPc(tBu)4)2 (3) (c = 4.13 × 10–6 моль/л, cIm = 5.77 × 10–4 моль/л).
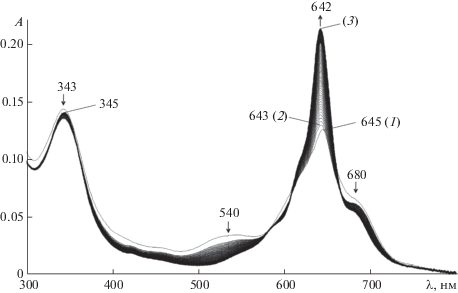
Добавление Im к бензольному раствору μ-N(FeIVPc(tBu)4)2Cl сопровождается мгновенным гипсохромным смещением на 2 нм полосы при λ = 645 нм. Далее наблюдается рост ее интенсивности со сдвигом 642 нм и одновременным снижением интенсивности широких полос при λ = = 540 и 680 нм. Конечный спектр продукта реакции аналогичен спектру μ-нитридодимерного фталоцианина с мостиковым фрагментом FeIII–N=FeIV [117]. Наличие и сохранение изобестических точек в процессе трансформации ЭСП указывает на то, что в ходе координации дополнительные промежуточные продукты не накапливаются в спектроскопически значимых концентрациях. Таким образом, координационное связывание субстрата с комплексом μ-N(FeIVPc(tBu)4)2Cl приводит к одноэлектронному восстановлению последнего с образованием соединения μ-N((L)FeIII,IVPc(tBu)4)2 в соответствии с уравнениями [54]:
Вторая стадия представляет собой медленное одноэлектронное восстановление одного металлического центра FeIV в FeIII. Процесс характеризуется константой скорости (kv, л2/(моль2с)), представляющей собой произведение величин k2 и Kу. Авторы [54] показали, что скорость и характер процесса зависят от природы лиганда. Получен ряд, в котором с увеличением значений рKа субстрата растет константа скорости одноэлектронного восстановления: 2-MeIm (рKа = 5.89, kv = 6.54) < < Im (рKа = 6.65, kv = 16.98) < 1-MeIm (рKа = 7.33, kv = 20.18) < Et2N (рKа = 10.93, kv = 46.77). Доказательством представленной корреляции является то, что присоединение диметилформамида с pKa ≈ 2 вообще не приводит к генерации восстановленных частиц с активным мостиковым фрагментом FeIII–N=FeIV [54].
В ИК-спектрах соединений μ-С(FeIVOPrTAP)2, μ-C(FeIVPc(tBu)4)2, μ-С(FeIVTPP)2 и μ-N(FeIVPc(tBu)4)2Cl полоса ν(Fe–Х–Fe), относящаяся к асимметричным колебаниям μ‑мостика и находящаяся в диапазоне 935–990 см–1, чувствительна к наличию аксиальных лигандов (рис. S2 ) [51, 54, 110]. Донорно-акцепторное взаимодействие μ-димерных комплексов с N-основаниями почти всегда сопровождается сдвигом этой полосы в низкочастотную область спектра на 2–87 см–1. Самые сильные сдвиги наблюдаются у μ‑N(FeIVPc(tBu)4)2Cl [54]. Присутствие 2-метилимидазола в рабочем растворе μ‑С(FeIVOPrTAP)2, μ-C(FeIVPc(tBu)4)2 и μ-С(FeIVTPP)2 вызывает высокочастотный сдвиг полосы колебаний связей Fe=C=Fe до 24 см–1. Для μ-С(FeIVTPP)2 с N-основаниями в ИК‑спектре наблюдаются сигналы ν(Fe–NL) при 411–419 см–1. В области колебаний макроциклического кольца (350–1500 см–1) спектры аддуктов аналогичны спектрам исходных соединений, однако большинство полос смещено. Подобные смещения сигналов, в первую очередь сдвиг полосы колебаний ν(Fe–X–Fe), свидетельствуют об изменении положения атома железа относительно координационной плоскости и указывают на его взаимодействие с субстратом. Похожая картина наблюдалась ранее при использовании ИК-спектров для исследования аксиального лигандирования μ‑оксомостиковых структур μ-O(FePc)2 [118] и μ-O(FeTPTAP)2 (TPTAP – тетрафенилтетраазапорфирин-дианион) [119].
При образовании донорно-акцепторной связи металла с N-основанием димерная структура комплекса сохраняется, об этом свидетельствуют интенсивные молекулярные пики соответствующих ионов в масс-спектре (рис. S3 ). Данная тенденция характерна для всех соединений, рассмотренных в работах [51, 54, 110]. Результаты масс-спектрометрии показывают, что продуктом реакци μ-N(FeIVPc(tBu)4)2, μ‑С(FeIVOPrTAP)2, μ-C(FeIVPc(tBu)4)2 и μ-С(FeIVTPP)2 с азотсодержащими основаниями является донорно-акцепторный комплекс состава 1 : 2, за исключением координации молекулы 2-MeIm μ-С-димерными соединениями.
Степень субстратной специфичности любого комплекса существенно зависит от реализации координационных взаимодействий в системе рецептор–субстрат и определяется ее устойчивостью и составом. Наряду с масс-спектрометрией для установления состава лигандированного комплекса используются методы спектрофотометрического титрования, в том числе метод молярных отношений и ограниченно-логарифмический [118, 119]. Представленные в работах [51, 54, 110] исследования по определению количества присоединившихся субстратов также говорят о том, что два металлических центра μ-димерных комплексов легко координируют по одному N-основанию со сравнительно высокой прочностью (рис. 6, табл. 2).
Рис. 6.
Зависимости lg[(Ap – A0)/(А∞ – Ap)] от lgcL для реакций μ-димерных комплексов с N-основаниями (L) в бензоле при 295–298 K: а – μ-N(FeIVPc(tBu)4)2Cl; б – μ-C(FeIVPc(tBu)4)2; А0, Ар, А∞ – оптические плотности растворов на рабочей длине волны для димеров железа, равновесной смеси и молекулярного комплекса соответственно.
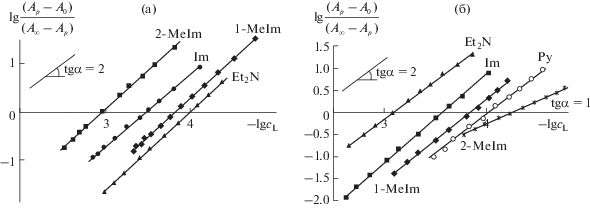
Таблица 2.
Величины констант равновесия реакций μ-нитридо- и μ-карбидодимерных комплексов железа(IV) с азотсодержащими основаниями, показателя основности субстратов и значения частот колебаний мостикового фрагмента
| Комплекс | pKa | lg Kр | Δλ, нм | νas(М–X–М), см–1 | Ссылка |
|---|---|---|---|---|---|
| μ-С((Et2NH)FeIVPc(tBu)4)2 | 10.93 | 6.23 | 7 | [115] | |
| μ-С((1-MeIm)FeIVPc(tBu)4)2 | 7.33 | 7.67 | 10 | [115] | |
| μ-С((Im)FeIVPc(tBu)4)2 | 6.65 | 7.16 | 9 | [115] | |
| μ-С(FeIVPc(tBu)4)2(2-MeIm)* | 5.89 | 4.56 | 9 | [115] | |
| μ-С((Py)FeIVPc(tBu)4)2 | 5.29 | 6.51 | 5 | [115] | |
| μ-С((Et2NH)FeIVTPP)2 | 10.93 | 2.50 | 3 | [51] | |
| μ-С((1-MeIm)FeIVTPP)2 | 7.30 | 3.93 | 16 | 906 | [51] |
| μ-С((Im)FeIVTPP)2 | 6.65 | 3.31 | 8 | 936 | [51] |
| μ-С(FeIVTPP)2(2-MeIm)* | 5.89 | 2.33 | 1 | [51] | |
| μ-С((Py)FeIVTPP)2 | 5.29 | 0.69 | 2 | [51] | |
| μ-С((Et2NH)FeIVOPrTAP)2 | 10.93 | 3.15 | 13 | 969 | [51] |
| μ-С((1-MeIm)FeIVOPrTAP)2 | 7.30 | 5.35 | 14 | [51] | |
| μ-С((Im)FeIVOPrTAP)2 | 6.65 | 4.88 | 13 | 937 | [51] |
| μ-С(FeIVOPrTAP)(2-MeIm)* | 5.89 | 4.00 | 6 | 995 | [51] |
| μ-С((Py)FeIVOPrTAP)2 | 5.29 | 2.88 | 7 | [51] | |
| μ-N((Et2NH)FeIVPc(tBu)4)2 | 10.93 | 4.31 | 4 | 851 | [54] |
| μ-N((1-MeIm)FeIVPc(tBu)4)2 | 7.33 | 3.93 | 2 | 921 | [54] |
| μ-N((Im)FeIVPc(tBu)4)2 | 6.65 | 3.76 | 3 | 896 | [54] |
| μ-N((2-MeIm)FeIVPc(tBu)4)2 | 5.89 | 3.03 | 2 | [54] |
Анализ величин констант равновесного процесса показывает, что субстратная специфичность μ-карбидодимерных соединений железа по отношению к азотсодержащим основаниям растет в ряду: μ-С(FeIVTPP)2 < μ-С(FeIVOPrTAP)2 < μ-С(FeIVPc(tBu)4)2 (табл. 2) [51, 110]. Полученная тенденция связана с влиянием основности макроциклического лиганда на эффективный положительный заряд металлического центра. Чем ниже pKa макроцикла, тем прочнее связь рецептор–субстрат. Уменьшение основности макроциклических лигандов в ряду: H2TPP > H2TAP > H2Pc > H2Pc(tBu)4 объясняет представленную выше тенденцию. Также имеет место конформационный фактор. Деформация макроцикла снижает его ароматичность и повышает основность. Комплекс μ-N(FeIVPc(tBu)4)2Cl не входит в установленную зависимость. Причина в том, что процесс координации субстрата сопровождается одноэлектронным восстановлением одного из атомов железа.
Устойчивость донорно-акцепторных комплексов μ-димеров [51, 54, 110] линейно коррелирует с основностью (pKa) субстрата (табл. 2). При составе комплекса 1 : 2 значения Kр растут в ряду N-оснований: Py < 2-MeIm < Im < 1-MeIm < Et2NH.
Структурные единицы FeIV–N=FeIII и FeIV–N=FeIV редокс-подвижны и могут легко одноэлектронно окисляться и восстанавливаться в присутствии слабых окислителей и органических оснований. Эту способность можно использовать как перспективный способ переключения свойств или регулирования реакционной активностью.
Быстрое установление равновесия координационного связывания μ-карбидодимерными комплексами N-содержащих субстратов, заметный спектральный отклик на образование связи рецептор–субстрат, а также умеренно высокая субстратная специфичность соединения, зависящая от природы макроцикла и основания, дают хороший прогноз на связывание подобными комплексами биологически активных молекул, имеющих в своем составе сами N-основания или их производные. Степень субстратной специфичности комплексов-миметиков позволяет оценить основность различных форм активного сайта фермента, образующихся в каталитическом цикле, и предсказать их активность в окислительно-восстановительных реакциях [120–122].
ХИМИЧЕСКАЯ ГЕНЕРАЦИЯ ВЫСОКОВАЛЕНТНЫХ ОКСОФОРМ μ-НИТРИДО И μ-КАРБИДОДИМЕРНЫХ КОМПЛЕКСОВ ЖЕЛЕЗА(IV) И РУТЕНИЯ(IV) С ЛИГАНДАМИ ПОРФИРИНОВОГО ТИПА В РЕАКЦИИ С ПЕРОКСИДАМИ
Детальное понимание механизмов каталитического окисления и выявление факторов, позволяющих моделировать активность металлсодержащих редокс-сайтов окислительных ферментов, являются одной из актуальных проблем современной химии. Решить эту проблему пытаются путем разработки искусственных молекулярных платформ, имитирующих активные центры, и моделирования разных стадий биотрансформации или окислительной деградации эндо- или экзогенных веществ.
Имеющиеся в литературе данные по моделированию природных ферментов гемовых оксидоредуктаз однозначно указывают на то, что проявление ферментоподобной каталитической активности связано с наличием высоковалентных оксоформ FeIV. Эти интермедиаты генерируются за счет разрыва связи О–О в алкилпероксо- или гидропероксокомплексах металлов с тетрапиррольными макроциклическими лигандами. Прямое изучение этих частиц является очень сложной задачей из-за их высокой реакционной способности и крайней нестабильности. Поэтому используют биомиметические исследования с применением синтетических моделей каталаз и пероксидаз на основе порфиринов и их аналогов с целью генерации активных форм. Полученные результаты дают ценную информацию о фундаментальных свойствах форм гема и влиянии различных факторов, определяющих механизм реакции и хемоселективность реакционноспособных промежуточных продуктов в реакциях оксигенирования [120–125]. В последнее время появились публикации, в которых показано, что димерные металлопорфирины зарекомендовали себя как более активные и стабильные катализаторы по сравнению с мономерными аналогами, а μ-нитридо- и μ-карбидодимерные порфириновые и фталоцианиновые комплексы железа и рутения оказались эффективными катализаторами окисления многих органических субстратов в мягких условиях, в том числе метана и бензола [61, 76–78, 84–87, 99, 102, 104, 106, 117, 124].
μ-Нитридо- и μ-карбидодимерные комплексы высоковалентного железа и рутения, имеющие в своем составе фрагмент М–Х–М, могут рассматриваться в качестве источника высокоокисленных форм (π-катион-радикальные или МV-формы [50, 52, 53, 86, 89–91, 101–104, 108, 125–127]), обладающих как каталазной, так и пероксидазной активностью. Кроме того, для μ-карбидодимерных комплексов железа и рутения возможно регулирование их каталитической активности в реакциях с перекисями путем аксиальной координации биологически активных молекул, а также симультанное проявление каталазных и пероксидазных свойств [53, 89–91, 101, 128, 129].
Реакция комплексов μ-N(FeIVPc(tBu)4)2, μ-С(FeIVTPP)2, μ-С(FeIVOPrTAP)2, μ-C(FeIVPc(tBu)4)2, μ-С(FeIVPc(HSO3)8)2 (μ-карбидодимерного октасульфофталоцианината железа(IV)) и μ-C(RuIVPc(tBu)4)2 (с = 6.4 × 10–6–8.2 × 10–6 моль/л) с органическими пероксидами (перекись дикумола (ПДК), трет-бутилпероксид (ТБП), трет-бутилгидропероксид (ТБГП)) изучена в широком диапазоне их концентраций (сПДК = 2.4 × 10–2–8.0 × 10–1 моль/л, сТБП = = 2.7 × 10–5–1.15 моль/л, сТБГП = 2.7 × 10–5–‒ 1.1 × 10–1 моль/л) [50, 52, 53, 89–91, 97, 101]. Она протекает во времени и сопровождается изменением ЭСП (табл. S5 ) с сохранением четко выраженных изобестических точек.
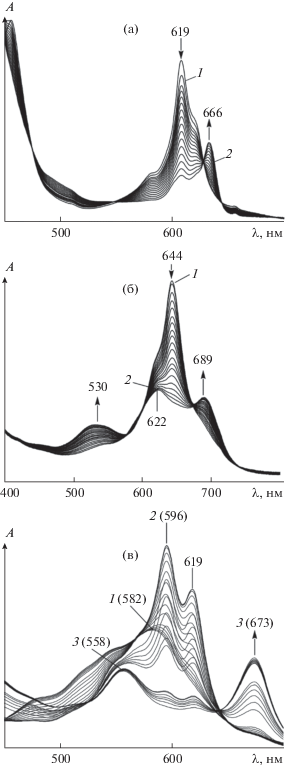
Активация пероксидов в присутствии μ-N(FeIVPc(tBu)4)2, μ-С(FeIVTPP)2, μ‑С(FeIVOPrTAP)2, μ-C(FeIVPc(tBu)4)2, μ-С(FeIVPc(HSO3)8)2 и μ-C(RuIVPc(tBu)4)2 приводит к появлению в ЭСП этих соединений широкой Q-полосы в области 627–690 нм, характерной для катион-радикальных форм (рис. 7) [50, 52, 53, 89–91, 97, 101].
Рис. 7.
Изменение ЭСП μ-Х-димерных комплексов в ходе реакции с пероксидами: а – μ-C(RuIVPc(tBu)4)2 (c = 1.95 × 10−6 моль/л) с ТБГП (c = 2.74 × 10−2 моль/л); б – μ-N(FeIVPc(tBu)4)Cl2 с ТБП (c = 1.1 × 10–2 моль/л) (1 – исходный комплекс, 2 – π-катион-радикальная форма); в) μ-C(FeOPrTAP)2 (с = 6.4 × 10–6 моль/л) c ТБГП (с = 2.27 × 10–4 моль/л) (1 – исходный комплекс, 2 – π-дикатион-радикальная форма, 3 – π-катион-радикальная форма) в бензоле при 295–298 K.
В зависимости от природы пероксида меняется состав координационного узла димера. Подтверждением этому служит появление в ИК-спектрах бензольных растворов μ-N(FeIVPc(tBu)4)2, μ-C(FeOPrTAP)2 и μ-C(FeIVPc(tBu)4)2 с ПДК или ТБП полосы колебаний ν(Fe–O) в Fe–OR в области 530–585 см–1 (табл. 3). Такие изменения говорят о наличии в реакционной смеси соединения с кислородсодержащим лигандом OR в аксиальном положении. Присутствие ТБГП приводит к возникновению в ИК-спектре комплексов полосы с частотой 841–883 см–1, характеризующей колебание ν(Fe=O) (для сравнения в пероксидазе ν(Fe=O) = 787 см–1, в (O)FeTPP ν(Fe=O) = 852 см–1 [130]). Общими чертами спектральных изменений в ИК-спектрах димерных комплексов являются также низкочастотные сдвиги колебаний ν(М–Х–М) и появление новых полос в области 1290–1366 см–1, относящихся к колебаниям связей Cα–Cβ и Cα–N в пиррольных кольцах макроцикла с π-катион-радикальным характером. Сильное поглощение в области 1200–1500 см–1, характеризующее π-катион-радикальную форму, наблюдалось также для мономерных порфириноподобных комплексов и μ-нитридодимерного FeIII,IV [131, 132]. Образование π-катион-радикала подтверждается и масс-спектрометрией. Высокоокисленные формы комплексов сохраняют свою биядерную структуру, о чем свидетельствуют интенсивные молекулярные пики соответствующих ионов в масс-спектре (рис. S4 ) [50, 52, 53, 89–91, 97, 101].
Таблица 3.
Основные частоты колебаний (ν, см–1) в ИК-спектрах бензольных растворов μ-нитридо- и μ-карбидодимерных комплексов железа и рутения c органическими пероксидами
| Комплекс | Отнесение полос | ||||
|---|---|---|---|---|---|
| ν(Fe=C=Fe) | ν(Fe=O) | ν(Cα–Cβ), ν(Cα–N) в катион-радикальной форме |
ν(Fe–O) | ссылка | |
| μ-С(FeIVTPP)2 + ТБГП | 940 | 864 | 1290 | [53] | |
| μ-C(FeIVOPrTAP)2 + ПДК | 855 | 581 | [50] | ||
| μ-C(FeIVOPrTAP)2 + ТБП | 874 | 534 | [50] | ||
| μ-C(FeIVOPrTAP)2 + ТБГП | 924 | 845 | 1366 | [89] | |
| μ-С(FeIVPc(tBu)4)2 + ТБП | 920 | 564 | [52] | ||
| μ-С(FeIVPc(tBu)4)2 + ТБГП | 917 | 841 | 1353 | [52] | |
| μ-С(FeIVPc(SO3H)8)2 + ТБГП | 940* | 854* | 1363* | [97] | |
| μ-N(FeIVPc(tBu)4)2 + ТБП | 901 | 535 | [90] | ||
| μ-N(FeIVPc(tBu)4)2 + ТБГП | 909 | 883 | [91] | ||
| (FeIVPc(tBu)4)N(FeIIIPc(tBu)4) + ТБГП | 935** | 833** | 1350** | [131] | |
| μ-С(RuIVPc(tBu)4)2 + ТБГП | 911 | 843 | 1363 | [101] | |
| (O)FeTPP | 852* | [131] | |||
| Пероксидаза | 787* | [131] | |||
| Оксигемоглобин | 567 | [131] | |||
В работах [50, 52, 53, 89–91, 97, 101] представлены кинетические параметры (табл. 4) и механизм генерации активных окси- и оксоформ Х-мостиковых структур через быстрое образование донорно-акцепторного алкилпероксокомплекса с последующим гомолитическим разрывом связи О–О (схема 4 ). Спектральные характеристики служат надежным доказательством представленного механизма, а продукты и интермедиаты надежно идентифицированы. Подробно химические принципы образования активных высокоокисленных частиц (схема 4 ) иллюстрируют спектральные изменения, наблюдаемые при активации ТБГП комплексом μ-С(FeIVTPP)2. В ЭСП комплекса μ‑С(FeIVTPP)2 зафиксированы три серии постепенно изменяющихся кривых с собственным набором изобестических точек, соответствующие трехстадийной реакции (рис. 8). Первая стадия относится к координации молекулы пероксида, которая в предлагаемых условиях происходит практически мгновенно. В ЭСП μ-С(FeIVTPP)2 поглощение при 667 нм (рис. 8а) может указывать на присутствие координированного BuOO–. Длинноволновая полоса типична для образования связи M–O–O и наблюдалась при спектральных исследованиях ряда макроциклических комплексов металлов, имеющих в качестве аксиального лиганда кислород или пероксид [133]. Вторая стадия относится к разрыву связи О–О и образованию короткоживущей частицы с радикалом на аксиальном кислороде O•–FeIVTPP=C=FeIVTPP с константой скорости 3.1 × 10–3 л/(моль с) (рис. 8б). Третья стадия – это образование π-катион-радикальной оксоформы с Q-полосой при λ = 627 нм (kv = 3.9 × × 10–4 л/(моль с)) (рис. 8в).
Таблица 4.
Константы образования высоковалентных окси- и оксоформ μ-Х-димерных комплексов железа и рутения с макроциклами порфиринового типа [50, 51, 53, 89–91, 97, 101]
| Концентрация комплекса, моль/л | Концентрация пероксида, моль/л | kv, л/(моль с) |
|---|---|---|
| ${{с}_{{\mu - {\text{N}}~{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{Pc}}{{{{{{\text{(}}}^{t}}{\text{Bu}})}}_{4}})}}_{2}}}}}$ = 8.9 × 10–6 | сПДК = 2.3 × 10–4–2.3 × 10–2 | 3.2 × 10–4 |
| cТБП = 1.1 × 10–4–1.1 × 10–2 | 4.4 × 10–2 | |
| cТБГП = 2.3 × 10–4–2.3 × 10–2 | 1.95 × 10–1 | |
| ${{с}_{{\mu - {\text{C}}~{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{OPrTAP}})}}_{2}}}}}$ = 6.4 × 10–6 | сПДК = 2.4 × 10–1–8.0 × 10–1 | 7.4 × 10–4 |
| cТБП = 1.1 × 10–4– 1.15 | 4.6 × 10–3 | |
| cТБГП = 2.27 × 10–4–4.2 × 10–2 | 1.86 × 10–2 | |
| ${{с}_{{\mu - {\text{C}}~{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{Pc}}{{{{{{\text{(}}}^{t}}{\text{Bu)}}}}_{{\text{4}}}})}}_{2}}}}}$ = 5.2 × 10–6 | сТБП = 2 × 10–3–1.1 × 10–1 | 1.55 × 10–2 |
| cТБГП = 2.3 × 10–3–2.18 × 10–2 | 6.16 × 10–2 | |
| ${{с}_{{\mu - {\text{C}}~{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{TPP}})}}_{2}}}}}$ = 8.9 × 10–6 | cТБГП = 2.3 × 10–3–2.18 × 10–2 | 3.9 × 10–4 |
| ${{с}_{{\mu - {\text{C}}~{{{((\operatorname{Im} ){\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{TPP}})}}_{2}}}}}$= 5.6 × 10–6 | cТБГП = 2.3 × 10–3–2.18 × 10–2 | 1.28 × 10–2 |
| ${{с}_{{\mu - {\text{C}}~{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{Pc(S}}{{{\text{O}}}_{3}}{\text{H}}{{{\text{)}}}_{{\text{8}}}})}}_{2}}}}}$ = 5.5 × 10–6 | cТБГП = 2.5 × 10–5–2.5 × 10–3 | 5.37 × 10–1 |
| ${{с}_{{\mu - {\text{C}}{{{({\text{R}}{{{\text{u}}}^{{{\text{IV}}}}}{\text{Pc}}{{{{{{\text{(}}}^{t}}{\text{Bu}})}}_{4}})}}_{2}}}}}$ = 5.2 × 10–6 | cТБГП = 2.74 × 10–3–2.62 × 10–1 | 3.2 × 10–2 |
Рис. 8.
Изменение ЭСП μ-карбидодимерного тетрафенилпорфирината железа(IV) (с(FeТРР)2C = = 9.1 × 10–6 моль/л) в реакции с ТБГП (сТБГП = = 2.27 × 10–3 моль/л (а, б, в), сТБГП = 2.27 × 10–5 моль/л (на вставке показана координация во времени)) при 298 K: a – образование донорно-акцепторного комплекса; б – разрыв связи О–О и образование радикала на аксиальном кислороде; в – образование π-катион-радикальной оксоформы.
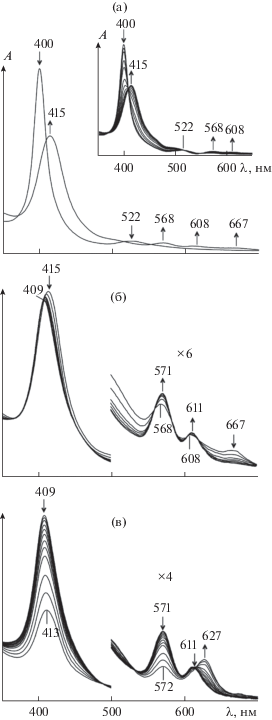
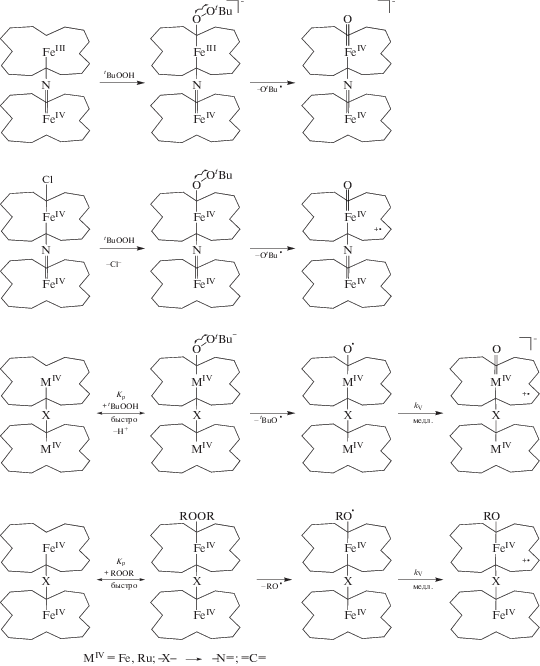
Схема 4 . Механизм генерации π-катион-радикальных форм μ-Х-димерных комплексов железа и рутения с макроциклами порфиринового типа под действием пероксидов.
Возможность спектрального наблюдения образования донорно-акцепторных комплексов с молекулой пероксида или с радикалом на аксиальном кислороде в составе биядерной частицы можно реализовать при изменении условий процесса, например, при уменьшении концентрации пероксида, замене растворителя, понижении температуры. Например, в реакции с μ-С(FeIVTPP)2 или μ-N(FeIVPc(tBu)4)2 снижение концентрации ТБГП до 2.7 × 10–5 моль/л или ПДК до 2.7 × 10–2 моль/л позволяет спектрально зафиксировать во времени присоединение молекулы пероксида в аксиальное положение комплекса (вставка на рис. 8a) и определить константу скорости этой стадии (kv = = 2.8 л/(моль с) или 6.87 × 10–1 л/(моль с) соответственно) [53, 90].
Подтверждением предлагаемого механизма (схема 4 ) и доказательством аксиальной координации молекулы пероксида на катионе металла служат также ИК- и масс-спектры μ-димерных комплексов, зарегистрированные до и сразу после добавления пероксидов [50, 52, 53, 89–91, 97, 101, 129]. Для большинства μ-Х-мостиковых биядерных структур наблюдаются низкочастотные сдвиги асимметричных валентных колебаний мостикового фрагмента ν(М–Х–М), чувствительного к присутствию аксиальных лигандов (рис. S5 ). Кроме того, появляются новые полосы в области 520–600 и 1105–1155 см–1, которые, безусловно, принадлежат колебаниям ν(Fe–O) и ν(O–O) соответственно (567, 1107 и 1155 см–1 для ν(Fe–O) и ν(O–O) в оксогемоглобине [130]). В области колебаний макроцикла (350–1500 см–1) спектр аналогичен спектру исходного соединения, однако имеет место смещение большинства полос, как и в случае координации N-оснований. Дальнейшее выдерживание раствора приводит к исчезновению в ИК-спектре сигналов, характеризующих соединение состава μ-Х(MIVP)2(ROOR') (ROOR' – молекула пероксида, R' = Н, R), увеличению сдвига колебаний ν(М–Х–М) и появлению полос, соответствующих связям MIV=O, Cα–Cβ и Cα–N пиррола макроцикла в π-катион-радикальной форме, которые свидетельствуют об образовании высокоокисленной частицы (рис. S5d, S5e , табл. 3).
Для некоторых μ-Х-димерных комплексов железа обнаружен новый молекулярный пик в масс-спектре, записанном сразу после добавления пероксида, который соответствует ионной форме донорно-акцепторного комплекса (рис. S6 ).
Общей закономерностью для μ-С-димерных комплексов железа и рутения является их схожее электрохимическое поведение в процессе окисления, исследованное с помощью циклической вольтамперометрии в анодном диапазоне [52, 53, 89, 97, 109]. Вольтамперограмма показывает два процесса одноэлектронного переноса в области от +0.304 до +1.02 эВ (табл. 1, 5). Первый потенциал окисления относится к снятию электрона с одного из двух макроциклов, что подтверждается его отсутствием на поляризационной кривой у системы μ-С-димерный комплекс–ТБГП (с = 2 × 10–3 моль/л) из-за химического образования π-катион-радикала. Второе одноэлектронное окисление комплекса дают дважды окисленные частицы, которые могут быть либо π-дикатион-радикалом с положительной локализацией заряда на двух окисленных макроциклах, либо сильно окисленными частицами, несущими радикал на мостиковом атоме углерода [102] и одном из макроциклов. Гипотетическая геометрическая и электронная структура такого комплекса была рассчитана для μ-C(FePc)2 методом DFT [102]. Однако экспериментальные данные, подтверждающие предложенную структуру, отсутствуют.
Соединение μ-С(FeIVTPP)2 имеет два редокс-потенциала (+0.72 и +1.02 эВ), которые соответствуют двум процессам переноса электрона. При добавлении к раствору μ-С(FeIVTPP)2 трет-бутилгидропероксида фиксируется только один полуволновой потенциал +0.76 эВ, отнесенный ко второму окислению. Похожая картина наблюдается и для μ-С(RuIVPc(tBu)4)2 (табл. 5). Тот факт, что первые потенциалы окисления комплексов μ-С(FeIVTPP)2 и μ-С(RuIVPc(tBu)4)2, а также вторые потенциалы этих же комплексов только в присутствии ТБГП близки, указывает на возможность протекания электрохимической реакции на втором макроцикле уже после появления π-катион-радикальной формы. Образование стабильного дикатиона для порфириноподобного соединения не является неожиданностью с учетом предыдущих исследований димерных порфириновых комплексов тория, урана и цинка. Дикатионные формы этих соединений были выделены в твердом виде с хорошим выходом и охарактеризованы рентгеноструктурным анализом, различными физико-химическими методами и расчетами методом DFT [132, 134, 135].
Таблица 5.
Потенциалы полуволн окисления соединений μ-С(FeIVTPP)2и μ-С(RuIVPc(tBu)4)2 и их комплексов с имидазолом [53, 101]
| Комплекс | Растворитель | E1/2, В | |
|---|---|---|---|
| 0/+ | +/2+ | ||
| μ-С(FeIVTPP)2 | ДМФА | 0.72 | 1.02 |
| CH2Cl2 | 0.67 | 1.00 | |
| PhCN | 0.67 | 1.02 | |
| Py [108] | 0.52 | 1.03 | |
| DMF/2 ммоль ТБГП | – | 0.76 | |
| μ-С((Im)FeIVTPP)2 | DMF | 0.45 | 0.74 |
| DMF/2 ммоль ТБГП | – | – | |
| μ-С(RuIVPc(tBu)4)2 | DMF | 0.53 | 0.74 |
| DMF/2 ммоль ТБГП | – | 0.73 | |
| μ-С((Im)RuIVPc(tBu)4)2 | DMF | 0.83 | |
| DMF/2 ммоль ТБГП | 0.86 | ||
| μ-С(RuIVPc)2 [80] | Py/CH2Cl2 | 0.55 | 1.0 |
Дикатион-радикальная форма была спектрально зафиксирована для μ-C(FeIVOPrTAP)2 в реакции с ТБГП (рис. 7) [89]. Процесс окисления сопровождался трансформацией полосы исходного комплекса при λ = 582 нм и накоплением интермедиата π-дикатион-радикала с оптической плотностью при λ = 596 и 619 нм (рис. 7, кривые 1 и 2). Затем во времени они исчезали и появлялись новые широкие полосы при λ = 558 и 673 нм (рис. 8), характерные для π-катион-радикальных частиц OPrTAPFe=C=Fe(O)OPrTAP+•. В ИК-спектрах μ-C(FeIVOPrTAP)2, зарегистрированных по истечении некоторого времени после добавления ТБГП, обнаружены два сигнала колебаний связей Сα–Сβ и Сα–N в пирролах макроцикла с π-катион-радикальным характером при 1365 и 1448 см–1 [89].
Согласно электрохимическим исследованиям μ-C(FeIVOPrTAP)2 [78], окисление при +0.304 и +0.597 эВ (табл. 1) связано с обратимым отводом одного электрона c порфиразиновых макроциклов до образования дикатион-радикала. Возможность более легкого двухэлектронного окисления μ-C(FeIVOPrTAP)2 определяется низким значением второго окислительного потенциала (+0.597 эВ). Действительно, его можно сравнить со значениями первого полуволнового потенциала для комплексов μ-C(FeIVTPP)2 (+0.67 и +1.02 эВ) и μ-C(RuIVPc(tBu)4)2 (+0.53 и +0.74 эВ). Очевидно, что он менее анодный (на 0.073 эВ) в случае первого комплекса и более анодный (на 0.069 эВ) в случае второго [53, 101]. Представленные спектральные характеристики в сочетании с электрохимическими данными [78, 89] надежно характеризуют соединение OPrTAP+•Fe=C=Fe(O)OPrTAP+• как двухэлектронно-окисленную разновидность оксоформ, которая образуется по схеме 5 :
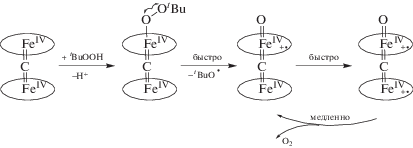
Схема 5 .
Химическое восстановление дикатион-радикала в катион-радикальную форму (рис. 7, спектральные кривые 2, 3) связано с его высокой активностью, проявляющейся в окислении пероксида. Подобную реакционную способность проявляют также π-катион-радикалы μ-нитридо- и μ-карбидодимерных комплексов железа и рутения с фталоцианиновым лигандом [52, 97, 101]. Выделение О2 наблюдалось и в реакции с π-катион- радикалом комплекса μ-N(FeIII,IVPc(tBu)4)2, где пероксиды участвовали в качестве восстановителей [131].
Для высокоокисленной оксоформы комплекса μ-C(FeIVTPP)2 не наблюдается восстановления во времени [53]. Однако участие в активации ТБГП лигандированной молекулы μ-C((Im)FeIVTPP)2 сопровождается выделением кислорода. Это свидетельствует о наличии в растворе активной оксоформы, отличной от π-катион-радикала μ-C(FeIVTPP)2, способной окислять пероксид и быстро восстанавливаться в нейтральную форму (kred = 2.66 × × 10–1 c–1). Процесс активации ТБГП с участием μ-C((Im)FeIVTPP)2 сопровождается аналогичными изменениями в ЭСП комплекса для ранее изученной реакции окисления μ-C(FeIVTPP)2 (рис. 8). Однако конечный спектр не соответствует катион-радикальной оксоформе, отсутствует характерное поглощение при λ = 630 нм (рис. 9, спектральная кривая 4). Авторы [53] относят его к восстановленной форме (μ-C(FeIVTPP)2(Im)(HOR)) активной частицы лигандированного комплекса, которая спектрально не регистрируется. Идентифицировать данную частицу сложно, но с большой вероятностью можно утверждать, что аксиальная группа, имеющая π- и π*-орбитали, способные перекрываться с π-, π*-орбиталями oксoжелеза(IV), делает такие оксочастицы более реакционноспособными. Подобный факт отмечался при изучении μ-нитридодимерных макроциклических соединений железа(III, IV) [124].
Рис. 9.
Изменение ЭСП μ-C((Im)FeIVTPP)2 (с = 5.6 × × 10–6 моль/л) в бензоле в присутствии ТБГП (с = = 2.14 × 10–3 моль/л) при 298 K: кривые 1–4 соответствуют μ-C((Im)FeIVTPP)2, μ-C(FeIVTPP)2(Im)(OOR), (O•)(FeIVTPP)C(FeIVTPP)(Im) и μ-C(FeIVTPP)2(Im)(HOR) соответственно.
Электрохимическое исследование μ-C((Im)FeIVTPP)2 показало наличие двух окислительных потенциалов (табл. 5), что подтверждает возможность двойного окисления. При переходе от μ-C(FeIVTPP)2 к имидазол-лигандированному комплексу в растворах ДМФА наблюдались значительные сдвиги потенциалов полуволн (~0.3 эВ). Это согласуется с большей стабильностью однократно окисленного μ‑C((Im)FeIVTPP) по сравнению с μ-C(FeIVTPP)2 из-за делокализации положительного заряда по всей молекуле. Отсутствие сигналов на вольтамперных кривых для системы μ-C((Im)FeIVTPP)/ДМФА/2мМ ТБГП свидетельствует о наличии в исследуемом растворе сильно окисленных частиц, которые не могут больше подвергаться окислению. В целом для μ-C((Im)FeIVTPP), как и для μ-C(FeIVTPP)2, наблюдается по два одноэлектронных переноса, сосредоточенных в макроциклах [89, 136]. В результате этих реакций в качестве высоковалентных разновидностей может образовываться π-дикатон-радикальная форма или оксокомплекс с положительной локализацией заряда на π-системе одного из макроциклических лигандов и радикала на мостиковом атоме углерода [50, 52, 53, 89, 91, 97, 101, 102]. Из-за сильных экситонных взаимодействий соседних порфириновых лигандов в димерных соединениях железа и рутения первый макроцикл окисляется легче, а второй требует более высоких потенциалов [53, 100, 101].
Следует отметить, что для μ-оксо- и μ-нитридодимерных комплексов железа(III) и железа(III, IV) соответственно образование высокоокисленных частиц с π-катион-радикалом на макроцикле в реакциях с пероксидом водорода идет посредством гетеролитического разрыва связи О–О в гидропероксокомплексе, а в реакции с ТБГП наблюдается гомолитический разрыв с одноэлектронным окислением одного из металлических центров с FeIII до FeIV и появлением нейтральной гидроксо- или оксоформы биядерной структуры [76–78, 86, 104, 117] (схема 2 ). Дикатион-радикальных форм, полученных химическим окислением, для этих соединений не наблюдается. Как показывают исследования, структура высокоокисленных частиц μ-оксо- и μ-нитридодимерных комплексов с МIII и MIII,IV напрямую зависит от типа разрыва связи О–О, который, в свою очередь, определяется не только природой пероксида, но и его концентрацией, свойствами растворителя, наличием аксиального лиганда, рН среды для водных и буферных растворов [137]. В случае же μ-нитридо- и μ-карбидодимерных комплексов с MIV любой тип разрыва связи О–О приводит к образованию высокоактивной заряженной оксоформы.
Анализ кинетических параметров реакции μ-Х-димерных комплексов с пероксидами (табл. 4) показывает, что самая высокая скорость генерации активных форм наблюдается у комплекса μ-C(FeIVPc(SO3H)8)2. Это связано c влиянием периферийных электроноакцепторных заместителей на донорно-акцепторную связь металл–пероксид. Сульфогруппы, оттягивая на себя электронную плотность с макроцикла, увеличивают эффективный положительный заряд на атоме железа, что способствует более быстрому прохождению реакции координации молекулы пероксида и образованию связи Fe–O в алкилпероксокомплексе. На более быстрое и легкое расщепление связи О–О влияет также способность водного боратного буфера к межмолекулярному водородному связыванию с координированной молекулой пероксида.
В работе [88] расчетным методом DFT показано, что в гетеролептическом (Pc, Pc(tBu)4) μ-карбидодимерном комплексе железа одноэлектронное окисление должно с большей вероятностью проходить на трет-бутилзамещенном фталоцианине, который более богат электронами, чем незамещенный аналог. В таком случае следовало ожидать более высокой скорости реакции окисления с участием μ-С(RuIV(tBuPc)4)2, однако данные, представленные в табл. 4, позволяют говорить о росте скорости реакции окисления в ряду: μ-С(FeIVTPP)2 < < μ‑С(FeIVOPrTAP)2 < μ-С(RuIVPc(tBu)4)2 < < μ‑С(FeIV(tBuPc)4)2 < μ-C(FeIVPc(SO3H)8)2. Это, вероятно, связано с тем, что стадия координации и последующий разрыв связи О–О вносят гораздо больший вклад в скорость процесса генерации высокоокисленных частиц, чем перенос электрона с макроцикла на радикальный кислород. Полученная тенденция соответствует снижению основности и увеличению координирующей способности комплексов при переходе от μ-С(FeIVTPP)2 к μ-C(FeIVPc(SO3H)8)2. При сравнении величин констант скорости окисления μ-С(FeIV(tBuPc)4)2 и μ-N(FeIV(tBuPc)4)2Cl (табл. 4) наблюдается влияние природы мостика и наличия ацидолиганда. Важным фактом является регенерирование катион-радикальных форм μ-Х-димерных комплексов железа в присутствии имидазола или 1-метилимидазола в нейтральную лигандированную форму [89, 90]. Состав восстановленного комплекса доказан методами Uv-vis-спектроскопии и масс-спектрометрии. Например, сигнал молекулярного пика m/z = 1557.8 указывает на наличие в реакционной смеси μ-С(FeIVOPrTAP)2–ТБП после добавления имидазола ионной формы соединения μ-С((Im)FeIVOPrTAP)2 (рис. S6b ).
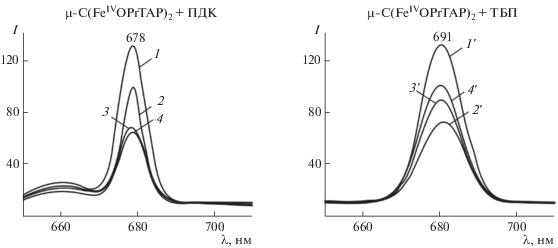
При рассмотрении редокс-свойств карбидодимерных комплексов следует учесть, что катион-радикальные формы μ-карбидодимерного октапропилтетраазапорфирината железа(IV), полученные в реакции с ПДК и ТБП в бензоле, образуют агрегированные формы [50]. Агрегация оксичастиц (OR)FeIVOPrTAP+•CFeIVOPrTAP связана с их меньшей растворимостью по сравнению с нейтральной молекулой. В случае ПДК имеют место H-агрегаты, а в присутствии ТБГП – J-агрегированные формы комплекса, которые различаются фотоактивными свойствами (рис. 10). Добавление имидазола в раствор, содержащий агрегаты, приводит к их разрушению посредством аксиальной координации N-основания с последующим восстановлением π-катион-радикала до нейтральной молекулы состава μ-C(FeIVOPrTAP)2(Im)n [50].
Рис. 10.
Спектры флуоресценции μ-C(FeIVOPrTAP)2 при добавлении ПДК (λex = 673 нм, сПДК = 0.52 моль/л, ${{с}_{{{\text{C}}{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{OPrTAP}})}}_{2}}}}}$ = 6.4 × 10–6 моль/л) (1–4) и ТБП (λex = 675 нм, сТБП = 1.15 × 10–2 моль/л, ${{с}_{{{\text{C}}{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{OPrTAP}})}}_{2}}}}}$ = 8.2 × 10–6 моль/л) (1'–4'). 1 и 1' – молекулярные формы; 2 и 2' – катион-радикалы; 3 и 4 – H-агрегаты; 3' и 4' – J-агрегаты.
Процесс активации органических пероксидов μ-Х-димерными комплексами железа и рутения с последующим образованием таких активных интермедиатов, как высоковалентные алкил- и гидропероксо-комплексы (миметики Сompound 0), π-ди- и π-катион-радикальные оксоформы (миметики Сompound 1), может рассматриваться как одна из модельных стадий каталитического цикла редокс-процессов, протекающих в биологических системах. Использование N-основания в качестве аксиального лиганда способствует генерации высокоокисленных видов оксоформ μ-Х-димерных комплексов, отличных от π-катион-радикальных частиц, способных окислять пероксид с выделением кислорода. Установление химических принципов образования различных видов высокоокисленных частиц, оценка их активности и определение способов ее модуляции представляет интерес для создания синтетических редокс-регуляторов.
ОКИСЛИТЕЛЬНАЯ СПОСОБНОСТЬ ВЫСОКОАКТИВНЫХ ОКСОФОРМ μ-КАРБИДОДИМЕРНЫХ КОМПЛЕКСОВ ЖЕЛЕЗА И РУТЕНИЯ С МАКРОЦИКЛАМИ ПОРФИРИНОВОГО ТИПА
Каталитические свойства тетрапиррольных макроциклических комплексов железа и рутения, которые они проявляют в различных реакциях окисления, позволяют рассматривать их в качестве моделей активных центров природных ферментов. Сравнение реакционной способности моно- и биядерных структур порфиринового типа в условиях однократного оборота каталитического цикла показало, что, например, μ-нитридодимерные комплексы железа(III, IV) являются гораздо более сильными окислителями. Двухъядерные оксочастицы при окислении этилбензола и адамантана соответственно в 26 и 130 раз активнее, чем одноядерные аналоги.
Комплексы μ-нитридожелеза(III,IV) способны реагировать с перфторированными ароматическими соединениями в присутствии пероксидов, в то время как самые сильные окисляющие ферменты не могут [104]. μ-Карбидодимерные комплексы железа и рутения, имеющие в своем составе фрагмент МIV=C=МIV, где за счет карбидного мостика стабилизируется высокая степень окисления металла, могут рассматриваться в качестве источника высокоокисленных катион- или дикатион-радикальных форм и, как следствие, обладать хорошей каталитической активностью. Эти димеры кажутся весьма актуальными в контексте проблемы создания синтетических биокатализаторов, но в сравнении с μ-нитридо- и μ-оксоаналогами, имеющими мостиковую структуру МIV=N–МIII и МIII–O–МIII, остаются малоизученными. Высоковалентная оксоформа μ‑N(FeIII,IVTPP)2 проявляет сильную реакционную способность при окислении ряда алканов [104], что позволяет предсказывать перспективность использования μ-нитридодимеров железа в катализе и стимулировать интерес к исследованиям этих соединений. Действительно, μ-нитридо- и μ-карбидокомплексы, содержащие в своей структуре два металлических высоковалентных центра МIV, могут выступать как основа для генерации сильно окисляющих оксоразновидностей и рассматриваться в качестве потенциальной молекулярной платформы для создания миметиков. Идентификация оксоформ, механизмы, приводящие к их образованию, и оценка реакционной способности в окислительных процессах являются предметом интенсивных исследований.
На сегодняшний день μ-оксо- и μ-нитридомакроциклические комплексы уже нашли широкое каталитическое применение в различных синтетических процессах, а именно в селективном окислении прочных связей C–H в алканах, бензоле, алкилароматических и других соединениях в мягких условиях [76, 84–87, 102, 106, 136]. Недавние исследования μ-карбидодимеров железа(IV) и рутения(IV) продемонстрировали каталитическую активность в реакциях циклопропанирования ароматических олефинов, введения карбена в связи N–H ароматических или алифатических аминов [61], окисления β-каротина [89, 101, 129], ликопина [52], морина [138], метилового оранжевого [97]. Способность генерировать высоковалентные частицы во время реакций с пероксидами делает μ-карбидодимерные соединения перспективными мощными окислителями [50, 52, 53, 61, 89, 101, 129, 138]. В присутствии комплекса μ-С(FeIVOPrTAP)2 протекает реакция окисления циклогексана, характеризующаяся высоким числом оборотов катализатора (табл. 6) [78]. Основными продуктами реакции являются кетоны. Первичный кинетический изотопный эффект, величина которого составляет 5.4, указывает на то, что основной маршрут реакции включает в себя взаимодействие образовавшегося оксокомплекса с субстратом, тогда как в случае радикальных реакций изотопный эффект не превышает 2.
Таблица 6.
Окисление углеводородов трет-бутилгидропероксидом в присутствии комплекса μ-С(FeIVOPrTAP)2. Продукты реакции идентифицированы методом хромато-масс-спектрометрии
| Субстрат | Продукты | Число оборотов катализатора |
|---|---|---|
| Циклогексан | Циклогексанол (0.423 моль/л) Циклогеканон (1.68 моль/л) |
11 493 |
| Тетралин | 1-Тетралон (3.2 моль/л) 1-Гидрокситетралин (0.11 моль/л) |
15 143 |
| Изопропилбензол | Кумол (2.1 моль/л) | 6000 |
Кинетические исследования каталитической активности μ-карбидодимерных комплексов железа и рутения в реакции окисления органических соединений пероксидами представлены в работах [52, 53, 89, 97, 101, 129]. В качестве модельных процессов, которые могут имитировать отдельные стадии каталитического цикла цитохрома р450 или пероксидазы, выбраны реакции окисления β-каротина, ликопина, морина и метилового оранжевого.
В зависимости от природы окислителя и среды время протекания конверсии этих соединений различно. На примере окисления β-каротина высоковалентными оксоформами μ-карбидодимерных комплексов железа и рутения можно подробно рассмотреть химические принципы этого процесса и факторы, его регулирующие.
При воздействии азо-бис-изобутиронитрила окисление β-каротина занимает 72 ч в хлороформе [139], 24 ч в гексане [140] и 2 ч бензоле [141] в реакции с азо-бис(2,4-диметилвалеронитрилом) и 2 ч под действием озона в 2,4-нитрофенилгидразине [142]. Каталитическая роль мономерных металлопорфиринов в окислении β-каротина представлена в единичных работах: тетра-дихлорофенилпорфирин рутения(II) и карбонил(5,10,15,20-тетра-2,4,6-триметилфенилпорфирин) рутения(II) были использованы в качестве соокислителей в реакции разложения каротина с ТБГП в гексане [143] и мета-хлорнадбензойной кислотой в бензоле [144] соответственно. Без описания механизма ранее были представлены исследования, которые показали, что порфириновые комплексы рутения вступают в реакцию в виде активной оксоформы O=RuIV [145]. В работах [52, 53, 89, 97, 101, 129] также отмечается прямое участие оксочастиц μ-карбидодимерных комплексов железа и рутения в окислении субстратов. В отсутствие димерных комплексов реакция субстрата с пероксидом не идет или протекает крайне медленно (конверсия или окислительная деградация за 24–48 ч составляет 5–15%). Добавление комплекса в раствор ТБГП и субстрата приводит к исчезновению полос поглощения последнего в ЭСП. Это доказывает, что система комплекс–ТБГП опосредует процесс окисления за счет образования оксоформ. Реакционная способность μ-Х-димерных комплексов и возможность ее регулирования связаны с видами активных частиц, которые они формируют при окислении, и с факторами, влияющими на это формирование. Наличие двух редокс-центров и углеродного мостика, способных к переносу электрона, определяет существование дикатион-радикальных форм у μ-С-мостиковых соединений, полученных химическим путем. Как уже отмечалось выше, природа сильно окисленных мономерных комплексов порфиринов железа и рутения, а также их димерных аналогов чрезвычайно чувствительна к природе аксиальной координации [53, 89, 124, 129]. Использование аксиально лигандированных димерных комплексов для генерации активных интермедиатов может приводить к образованию высокоактивных частиц, окисленных не только по макроциклу, μ-С-мостику, но и по металлическому центру [101]. Однако их изучение и идентификация затруднены из-за короткого времени жизни. Поэтому отнесение таких видов остается под вопросом.
Высокую активность и разницу в реакционной способности окислительных форм μ-Х-димеров железа(IV) и рутения(IV) демонстрируют данные табл. 7, 8. Кинетические параметры показывают, что процессы окисления β-каротина идут с разным порядком, поэтому их корректное сравнение представляет сложность. В случае окисления β-каротина оксоформами μ-C(FeOPrTAP)2 и μ-С(RuIV(tBuPc)4)2 порядок по субстрату n = 0, а в реакции с μ-С(FeIVTPP)2 и μ-С(FeIV(tBuPc)4)2n = 1. Наиболее высокой окислительной способностью отличаются оксоформы лигандированных N-основанием димерных комплексов железа и рутения. Окислительное разложение β-каротина представляет собой многоступенчатый процесс, который может происходить через образование переходных моно- и бирадикалов, апокаротеналей, эпоксидов и других частиц [89, 129, 140–143]. Поэтому нулевой порядок можно объяснить участием в лимитирующей стадии реакции продуктов первичной конверсии β-каротина, а не исходного соединения. Значительной реакционной способностью отличаются дикатион- радикальные формы и высокоокисленные лигандированные частицы (рис. 11). Например, в условиях одновременного вовлечения реагентов в реакционной смеси μ‑C(FeOPrTAP)2–ТБГП–β-каротин наряду с π-катион-радикальной формой образуется более мощный окислитель – дикатион- радикал комплекса μ-C(FeOPrTAP)2, который значительно увеличивает скорость реакции. Прямое окисление β-каротина в присутствии дикатион-радикальной частицы или высокоокисленной оксоформы лигандированного комплекса μ-C((Im)FeOPrTAP)2 происходит в 18 или 36 раз быстрее, чем с участием π-катион-радикала (рис. 11, табл. 7, 8). Параллельно эти два вида активных интермедиатов окисляют пероксид с выделением О2 [89, 97, 101, 129]. Данный факт позволяет объяснить падение скорости разложения β-каротина при использовании оксоформ лигандированных μ-С((L)RuIVPc(tBu)4)2. Для аксиальных комплексов μС(RuIV(tBuPc)4)2 с N-основанием возможно прохождение металлоцентрированного одноэлектронного химического окисления с образованием оксоформы, содержащей RuV=O. Эта высокоактивная частица является изоэлектронным аналогом π-катион-радикала димерного фталоцианината RuIV=O только с более высокой энергией. Наряду с тем, что природа последнего не оставляет сомнений, отнесение высокоокисленного лигандированного соединения μ-С((L)RuIV(tBuPc)4)2 остается под вопросом и может только предполагаться. В ЭСП системы μ-С((L)RuIV(tBuPc)4)2–ТБГП–β-каротин не наблюдалось полосы в области 600–700 нм, характерной для катион-радикала порфирина или фталоцианина (666 нм для (O)RuIVPc+•(tBu)4СRuIVPc(tBu)4), что может свидетельствовать об участии в реакции такой разновидности, как оксочастицы с RuV [101]. Электрохимическое исследование μ-С((Im)RuIV(tBuPc)4)2 показало значительный сдвиг потенциалов полуволн (E1/2 = +0.83 эВ, табл. 5), что также указывает на образование высокоокисленной формы.
Таблица 7.
Кинетические параметры реакции окисления* морина, ликопина, каротина и метилового оранжевого (МО) катион-радикальными оксоформами μ-карбидодимерных комплексов железа и рутения [52, 89, 97, 101, 129]
| μ-C(FeIVOPrTAP)2${{с}_{{\mu {\text{ - }}{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{OPrTAP}})}}_{2}}}}}$ = 4.60 × 10–6 моль/л | |
|---|---|
| смор= (5.50–20.0) × 10–5 моль/л | kv = 4.80 × 10–1 л/(моль с), nмор = 1** |
| скар = (1.64–8.05) × 10–4 моль/л | kv = 2.60 × 10–2 с–1, nкар = 0*** |
| скар = (1.64–8.05) × 10–4 моль/л | kv = 0.09 × 10–2 с–1, nкар = 0 |
| μ-C(FeIVOPrTAP)2${{с}_{{\mu {\text{ - }}{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{OPrTAP}})}}_{2}}}}}$ = 4.60 × 10–6 моль/л, cIm = 10–2 моль/л | |
| скар = (1.64–8.05) × 10–4 моль/л | kv = 5.70 × 10–2 с–1, nкар = 0 |
| μ-С(FeIVPc(tBu)4)2${{с}_{{\mu {\text{ - }}{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{Pc}}{{{{{{\text{(}}}^{t}}{\text{Bu)}}}}_{4}})}}_{2}}}}}$ = 6.50 × 10–6 моль/л | |
| сликоп = (4.77–12.30) ×. 10–5 моль/л | kv = 15.53 л/(моль с), nликоп = 1 |
| μ-С(FeIVTPP)2${{с}_{{\mu {\text{ - }}{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{TPP}})}}_{2}}}}}$ = 8.90 × 10–6 моль/л | |
| скар = (4.32–9.57) × 10–4 моль/л | kv = 3.30 л/(моль с), nкар = 1 |
| μ-С((Im)FeIVTPP)2${{с}_{{\mu {\text{ - }}{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{TPP}})}}_{2}}}}}$ = 8.9 × 10–6 моль/л, cIm = 10–3 моль/л | |
| скар = (4.32–9.57) × 10–4 моль/л | kv = 10.30 л/(моль с), nкар = 1 |
| μ-С(RuIVPc(tBu)4)2${{с}_{{\mu {\text{ - }}{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{Pc}}{{{{{{\text{(}}}^{t}}{\text{Bu)}}}}_{4}})}}_{2}}}}}$ =1.95 × 10–6 моль/л | |
| скар = (2.69–8.09) × 10–4 моль/л | kv = 2.04 × 10–2 с–1, nкар = 0 |
| μ-С((Im)(RuIVPc(tBu)4)2${{с}_{{\mu {\text{ - C}}{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{Pc}}{{{{{{\text{(}}}^{t}}{\text{Bu)}}}}_{4}})}}_{2}}}}}$ = 1.95 × 10–6 моль/л, cIm = 2.08 × 10–3 моль/л | |
| скар = (2.69–8.05) × 10–4 | kv = 2.65 × 10–3 с–1, nкар = 0 |
| μ-С((1-MeIm)(RuIVPc(tBu)4)2${{с}_{{\mu {\text{ - C}}{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{Pc}}{{{{{{\text{(}}}^{t}}{\text{Bu)}}}}_{4}})}}_{2}}}}}$ = 1.95 × 10–6 моль/л, c1-MeIm = 2.02 × 10–3 моль/л | |
| скар = (2.69–8.05) × 10–4 моль/л | kv = 3.33 × 10–3 с–1, nкар = 0 |
| μ-С((BzIm)(RuIVPc(tBu)4)2${{с}_{{\mu {\text{ - C}}{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{Pc}}{{{{{{\text{(}}}^{t}}{\text{Bu)}}}}_{4}})}}_{2}}}}}$ =1.95 × 10–6 моль/л, cBzIm = 2.17 × 10–3 моль/л | |
| скар = (2.69–8.05) × 10–4 моль/л | kv = 9.54 × 10–3 с–1, nкар = 0 |
| μ-C(FeIVPc(SO3H)8)2${{с}_{{\mu {\text{ - C}}{{{({\text{F}}{{{\text{e}}}^{{{\text{IV}}}}}{\text{Pc(S}}{{{\text{O}}}_{3}}{\text{H}}{{{\text{)}}}_{8}})}}_{2}}}}}$ = 6.50 × 10–6 моль/л | |
| сМО = (2.0–9.0) × 10–5 моль/л | kv = 1.58 × 10–1 л/(моль с), nMO = 1, моль/л |
Таблица 8.
Окисление β-каротина высокоактивными оксоформами μ-карбидодимерных комплексов железа и рутения [89, 101]
| Окислитель | Время реакции | Конверсия, % |
|---|---|---|
| μ-C(FeOPrTAP)2–ТБГП одновременное добавление | 4 мин | 100 |
| Дикатион-радикальная форма μ-C(FeOPrTAP)2 |
2 мин | 100 |
| Катион-радикальная форма μ-C(FeOPrTAP)2 |
36 мин | 100 |
| Высокоокисленная форма μ-C((Im)FeOPrTAP)2 |
1 мин | 100 |
| Катион-радикальная форма μ-С(RuIV(tBuPc)4)2 |
2 мин 40 с | 99 |
| Высокоокисленная форма μ-С((Im)RuIV(tBuPc)4)2 |
18 | 99 |
| Высокоокисленная форма μ-С((1-MeIm)RuIV(tBuPc)4)2 |
18 | 99 |
| Высокоокисленная форма μ-С((BzIm)RuIV(tBuPc)4)2 |
6 мин | 99 |
| RuII(TDFPP)–tBuOOH [146] | 24 ч | 100 |
| ТБГП | 5–24 ч | 4–5 |
Рис. 11.
Изменение ЭСП β-каротина (c = 1.64 × × 10–4 моль/л) при окислении его разными видами высоковалентных оксоформ комплекса μ-C(FeIVOPrTAP)2: а – τ, с = 0 (1), 120 (2); б – τ, с = = 0 (1), 2160 (2); в – высокоокисленная частица лигандированного комплекса μ-C((Im)FeIVOPrTAP)2, τ, с = = 0 (1), 60 (2).
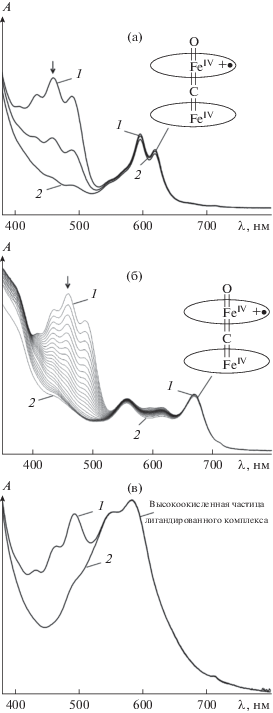
Известно, что высокореакционные формы с редокс-центром RuV=O способны параллельно окислять как используемый субстрат, так и органический пероксид. Одноядерные порфириновые комплексы рутения(V) уже были предложены в качестве метастабильных интермедиатов в каталитическом окислении алканов, дифенилметанола и циклогексена [144, 146, 147]. Использование богатого электронами аксиального лиганда может не только менять место локализации заряда, но и стабилизировать такие активные частицы для спектроскопического обнаружения.
Следует также отметить низкую степень деградации оксоформ (5–10%) в процессе окисления при рециклировании. При полном разложении субстрата, в частности β-каротина, с участием различных высокоокисленных активных частиц в ЭСП реакционной смеси наблюдаются полосы этих видов (рис. 11) [52, 89, 97, 101, 129], что указывает на присутствие данных частиц в растворе после реакции. Это возможно вследствие быстрого повторного окисления восстановленных форм, которые не фиксируются спектрально. Активность высокоокисленных видов подтверждается дальнейшей реакцией разложения субстрата без добавок пероксида. Каталитические свойства частиц при рециклировании не снижаются, а константа скорости процесса практически не меняется в течение шести–десяти циклов (рис. 12).
Рис. 12.
Конверсия β-каротина (c = 1.64 × 10–4 моль/л) при его окислении различными формами μ-C(FeIVOPrTAP)2: а – π-дикатион-радикалом; б – π-катион-радикалом; в – высокоокисленной формой лигандированного комплекса μ-C((Im)FeIVOPrTAP)2, с периодическим добавлением субстрата.
На основании спектральных изменений с учетом полученных кинетических параметров и идентифицированных интермедиатов авторы [52, 89, 97, 101, 129] предположили, что реакция окислительной трансформации субстрата протекает по схеме 6 :
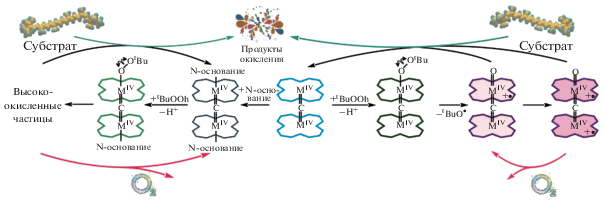
Схема 6 .
ЗАКЛЮЧЕНИЕ
Обобщенные в работе исследования спектральных, координационных и редокс-свойств μ-нитридо- и μ-карбидодимерных комплексов железа(IV) и рутения(IV), а также приведенные возможные механизмы генерации активных оксоформ разных видов с высокой степенью идентификации интермедиатов и продуктов реакции показывают, что благодаря особой электронной структуре эти комплексы способны образовывать частицы, подобные активным сайтам окислительных ферментов, характеризующиеся высокой активностью. Использование аксиального лигандирования и варьирования природы аксиального лиганда можно рассматривать как способ формирования редокс-центров разной природы и регулирования реакционной способности μ-Х-димерных комплексов.
Таким образом, генерирование μ-нитридо- и μ-карбидодимерными комплексами железа(IV) и рутения(IV) в реакции с пероксидами разных видов высокоокисленных частиц и симультанное проявление этими формами каталазных и пероксидазных свойств способствуют использованию таких комплексов в качестве биомиметической молекулярной платформы. Благодаря простоте и гибкости структуры одноатомных мостиковых макроциклических комплексов металлов возможна ее дальнейшая разработка. Для конструирования макроциклической единицы и металлического центра могут быть использованы фталоцианины, порфиразины, порфирины, возможно, корролазины, корролы с разным сочетанием в составе одной молекулы и различные переходные металлы с мостиковыми группами. Модификация периферии и координационного центра, использование гомо- и гетерометаллических центров в гомолептической или гетеролептической молекуле служат инструментом, который может эффективно регулировать и настраивать свойства этих димерных комплексов, открывая новые возможности применения. Эта биметаллическая платформа имеет значительный потенциал для дальнейшего развития, что, несомненно, должно стимулировать к ней интерес с целью создания искусственных биокатализаторов.
Список литературы
Poulos T.L. // Chem. Rev. 2014. V. 114. № 7. P. 3919. https://doi.org/10.1021/cr400415k
Senge M.O., MacGowan S.A., O’Brien J.M. // Chem. Commun. 2015. V. 51. № 96. P. 17031. https://doi.org/10.1039/C5CC06254C
MacGowan S.A., Senge M.O. // Inorg. Chem. 2013. V. 52. № 21. P. 1228. https://doi.org/10.1021/ic4016228
Silva A.M.G., Tomé A.C., Neves M.G.P.M.S. et al. // Eur. J. Org. Chem. 2002. V. 67. № 3. P. 726. https://doi.org/10.1021/jo0106703
Akhigbe J., Samankumara L.P., Brückner C. // Tetrahedron Lett. 2012. V. 53. № 27. P. 3524. https://doi.org/10.1016/j.tetlet.2012.04.137
Boerner L.J.K., Dye D.F., Köpke T. et al. // Coord. Chem. Rev. 2013. V. 257. № 2. P. 599. https://doi.org/10.1016/j.ccr.2012.07.009
Brückner C., Akhigbe J., Samankumara L. // Handbook of Porphyrin Science / Eds. Kadish K.M., Smith K.M., Guilard R. N.Y.: River Edge, 2014. V. 31. P. 1.
Callot H.J. // Dalton Trans. 2008. P. 6346. https://doi.org/10.1039/B807066K
Szyszko B., Latos-Grazynski L. // Chem. Soc. Rev. 2015. V. 44. № 11. P. 3588. https://doi.org/10.1039/C4CS00398E
Costa L.D., Costa J.I.T., Tomé A.C. // Molecules. 2016. V. 21. № 3. P. 320. https://doi.org/10.3390/molecules21030320
Simonova O.R., Zdanovich S.A., Zaitseva S.V. et al. // Russ. J. Inorg. Chem. 2020. V. 65. № 7. P. 1006. [Симонова О.Р., Зданович С.А., Зайцева С.В. и др. // Журн. неорган. химии. 2020. Т. 65. № 7. С. 922.]https://doi.org/10.1134/S0036023620070207
Simonova O.P., Zaitseva S.V., Koifman O.I. // Russ. J. Gen. Chem. 2014. V. 84. № 12. P. 2429. [Симонова О.Р., Зайцева С.В., Койфман О.И. // Журн. общей химии. 2014. Т. 84. № 12. С. 2012.]https://doi.org/10.1134/S1070363214120135
Simonova O.R., Zaitseva S.V., Koifman O.I. // Russ. J. Inorg. Chem. 2012. V. 57. № 6. P. 903. [Симонова О.Р., Зайцева С.В., Койфман О.И. // Журн. неорган. химии. 2012. Т. 57. № 6. С. 976.]https://doi.org/10.1134/S0036023612060204
Zaitseva S.V., Simonova O.R., Zdanovich S.A. et al. // Russ. J. Inorg. Chem. 2010. V. 55. № 6. P. 959. [Зайцева С.В., Симонова О.Р., Зданович С.А. и др. // Журн. неорган. химии. 2010. Т. 55. № 6. С. 1026. https://doi.org/10.1134/S0036023610060215]
Shimizu T., Lengalova A., Martinek V. et al. // Chem. Soc. Rev. 2019. V. 48. № 24. P. 5624. https://doi.org/10.1039/C9CS00268E
Wen X., Li Y., Hamblin M.R. // Photodiagn. Photodyn. Ther. 2017. V. 19. P. 140. https://doi.org/10.1016/j.pdpdt.2017.06.010
Hisamatsu Y., Umezawa N., Yagi H. et al. // Chem. Sci. 2018. V. 9. № 38. P. 7455. https://doi.org/10.1039/C8SC02133C
Sun M., Xu L., Bahng J.H. et al. // Nat. Commun. 2017. V. 8. P. 1847. https://doi.org/10.1038/s41467-017-01337-2
Zhdanova K.A., Savelyeva I.O., Mironov A.F. et al. // Dyes and Pigments. 2020. V. 181. P. 108561. https://doi.org/10.1016/j.dyepig.2020.108561
Grin M.A., Pogorilyy V.A., Noev A.N. et al. // Macroheterocycles. 2018. V. 11. № 1. P. 89. [Грин М.А., Погорилый В.А., Ноев А.Н. и др. // Макрогетероциклы. 2018. Т. 11. № 1. С. 89.]https://doi.org/10.6060/mhc180176p
Harris F., Pierpoint L. // Med. Res. Rev. 2012. V. 32. № 6. P. 1292. https://doi.org/10.1002/med.20251
Wiehe A., O’Brien J.M., Senge M.O. // Photochem. Photobiol. Sci. 2019. V. 18. № 11. P. 2565. https://doi.org/10.1039/C9PP00211A
Kustov A.V., Kustova T.V., Berezin D.B. et al. // Dyes and Pigments. 2020. V. 173. P. 107948. https://doi.org/10.1016/j.dyepig.2019.107948
Kaigorodova E.Y., Mamardashvili G.M., Mamardashvili N.Z. // Russ. J. Inorg. Chem. 2018. V. 63. № 9. P. 1192. [Кайгородова Е.Ю., Мамардашвили Г.М., Мамардашвили Н.Ж. // Журн. неорган. химии. 2018. Т. 63. № 9. С. 1167. https://doi.org/10.1134/S0044457X18090064]
Brandenberg O.F., Fasan R., Arnold F.H. // Curr. Opin. Biotechnol. 2017. V. 47. P. 102. https://doi.org/10.1016/j.copbio.2017.06.005
Zhang R.K., Huang X., Arnold F.H. // Curr. Opin. Chem. Biol. 2019. V. 49. P. 67. https://doi.org/10.1016/j.cbpa.2018.10.004
Kato R., Kobayashi Y., Akiyama M. et al. // Dalton Trans. 2013. V. 42. № 45. P. 15889. https://doi.org/10.1039/C3DT51418H
Ozaki S.-I., Nakahara A., Sato T. // Chem. Lett. 2011. V. 40. № 4. P. 362. https://doi.org/10.1246/cl.2011.362
Lu Y., Sousa A., Franco R. et al. // Biochem. 2002. V. 41. № 26. P. 8253. https://doi.org/10.1021/bi025569m
Alderman N., Danos L., Fang L. et al. // Chem. Commun. 2017. V. 53. № 89. P. 12120. https://doi.org/10.1039/C7CC04767C
Oohora K., Mashima T., Ohkubo K. et al. // Chem. Commun. 2015. V. 51. № 55. P. 11138. https://doi.org/10.1039/C5CC02680F
Lepeshkevich S.V., Parkhats M.V., Stasheuski A.S. et al. // J. Phys. Chem. A. 2014. V. 118. № 4. P. 1864. https://doi.org/10.1021/jp411302d
Stuzhin P.A., Mikhailov M.S., Travkin V.V. et al. // Macroheterocycles. 2012. V. 5. № 2. P. 162. [Стужин П.А., Михайлов М.С., Травкин В.В. и др. // Макрогетероциклы. 2012. Т. 5. № 2. С. 162. https://doi.org/10.6060/mhc2012.120573p]
Metzger T.S., Tel-Vered R., Willner I. // Small. 2016. V. 12. № 12. P. 1605. https://doi.org/10.1002/smll.201503077
Lalaoui N., Le Goff A., Holzinger M. et al. // Chem. Eur. J. 2015. V. 21. № 47. P. 16868. https://doi.org/10.1002/chem.201502377
Fry H.C., Garcia J.M., Medina M.J. et al. // J. Am. Chem. Soc. 2012. V. 134. № 36. P. 14646. https://doi.org/10.1021/ja304674d
Costas M., Mehn M.P., Jensen M.P. et al. // Chem. Rev. 2004. V. 104. № 2. P. 939. https://doi.org/10.1021/cr020628n
Koehntop K.D., Emerson J.P., Que Jr.L. // J. Biol. Inorg. Chem. 2005. V. 10. P. 87. https://doi.org/10.1007/s00775-005-0624-x
Bruijnincx P.C.A., van Koten G., Gebbink R.J.M.K. // Chem. Soc. Rev. 2008. V. 37. № 12. P. 2716. https://doi.org/10.1039/B707179P
Krebs C., Galonic F.D., Walsh C.T. et al. // Acc. Chem. Res. 2007. V. 40. № 7. P. 484. https://doi.org/10.1021/ar700066p
Solomon E.I., Wong S.D., Liu L.V. et al. // Curr. Opin. Chem. Biol. 2009. V. 13. № 1. P. 99. https://doi.org/10.1016/j.cbpa.2009.02.011
Rittle J., Green M.T. // Science. 2010. V. 330. № 6006. P. 933. https://doi.org/10.1126/science.1193478
Wong J.S., Sun Y.-J., Choi S.K. et al. // Chem. Eur. J. 2006. V. 12. № 1. P. 130. https://doi.org/10.1002/chem.200500128
Shaik S., Hirao H., Kumar D. // Acc. Chem. Res. 2007. V. 40. № 7. P. 532. https://doi.org/10.1021/ar600042c
Hessenauer-Ilicheva N., Franke A., Meyer D. et al. // Chem. Eur. J. 2009. V. 15. № 12. P. 2941. https://doi.org/10.1002/chem.200801423
Wolak M., van Eldik R. // Chem. Eur. J. 2007. V. 13. № 17. P. 4873. https://doi.org/10.1002/chem.200601148
Li N., Wanga Y., Wua C. et al. // Appl. Surf. Sci. 2018. V. 434. P. 1112. https://doi.org/10.1016/j.apsusc.2017.11.048
Rosser T.E., Reisner E. // ACS Catal. 2017. V. 7. № 5. P. 3131. https://doi.org/10.1021/acscatal.7b00326
Sengupta K., Chatterjee S., Dey A. // ACS Catal. 2016. V. 6. № 3. P. 1382. https://doi.org/10.1021/acscatal.5b02668
Simonova O.R., Zaitseva S.V., Tyulyaeva E.Y. et al. // Russ. J. Inorg. Chem. 2017. V. 62. № 4. P. 508. https://doi.org/10.1134/S0036023617040179
Zaitseva S.V., Zdanovich S.A., Kudrik E.V. et al. // Russ. J. Inorg. Chem. 2017. V. 62. № 9. P. 1257. [Зайцева С.В., Зданович С.А., Кудрик Е.В. и др. // Журн. неорган. химии. 2017. Т. 62. № 9. С. 1265.]https://doi.org/10.1134/S0036023617090194
Simonova O.R., Zaitseva S.V., Zdanovich S.A. et al. // Macroheterocycles. 2018. V. 11. № 1. P. 29. https://doi.org/10.6060/mhc180173s
Zaitseva S.V., Tyulyaeva E.Yu., Simonova O.R. et al. // J. Coord. Chem. 2018. V. 71. № 16–18. P. 2995. https://doi.org/10.1080/00958972.2018.1506109
Zaitseva S.V., Zdanovich S.A., Tyulyaeva E.Y. et al. // J. Porph. Phthal. 2016. V. 20. № 5. P. 639. https://doi.org/10.1142/S1088424616500474
Kadish K.M., Bottomley L.A., Brace J.G. et al. // J. Am. Chem. Soc. 1980. V. 102. № 13. P. 4341. https://doi.org/10.1021/ja00533a008
Schick G.A., Bocian D.F. // J. Am. Chem. Soc. 1983. V. 105. № 7. P. 1830. https://doi.org/10.1021/ja00345a025
Bocian D.F., Findsen E.W., Hofmann Jr. J.A. et al. // Inorg. Chem. 1984. V. 23. № 7. P. 800. https://doi.org/10.1021/ic00175a002
Floris B., Donzello M. P., Ercolani C. // The Porphyrin Handbook / Eds. Kadish K.M., Smith K.M., Guilard R. San Diego: Elsevier Science, 2003. V. 18. P. 1.
Kudrik E.V., Albrieux F., Afanasiev P. et al. // J. Porph. Phthal. 2013. V. 17. № 8–9. P. 791. https://doi.org/10.1142/S1088424613500417
Li M., Scheidt W.R. // J. Porph. Phthal. 2014. V. 18. № 5. P. 380. https://doi.org/10.1142/S1088424614500096
Kroitor A.P., Cailler L.P., Martynov A.G. et al. // Dalton Trans. 2017. V. 46. № 45. P. 15651. https://doi.org/10.1039/C7DT03703A
Chin D.-H., La Mar G.N., Balch A.L. // J. Am. Chem. Soc. 1980. V. 102. № 13. P. 4344. https://doi.org/10.1021/ja00533a009
Balch A.L., Chan Y.W., Cheng R.J. et al. // J. Am. Chem. Soc. 1984. V. 106. № 25. P. 7779. https://doi.org/10.1021/ja00337a022
Bottomley L.A., Gorce J.-N., Goedken V.L. et al. // Inorg. Chem. 1985. V. 24. № 23. P. 3733. https://doi.org/10.1021/ic00217a008
Ercolani C., Gardini M., Pennesi G. et al. // Inorg. Chem. 1988. V. 27. № 2. P. 422. https://doi.org/10.1021/ic00275a036
Rossi G., Gardini M., Pennesi C. et al. // J. Chem. Soc., Dalton Trans. 1989. № 1. P. 193. https://doi.org/10.1039/DT9890000193
Ercolani C., Hewage S., Heucher R. et al. // Inorg. Chem. 1993. V. 32. № 13. P. 2975. https://doi.org/10.1021/ic00065a031
Ercolani C., Jubb J., Pennesi G. et al. // Inorg. Chem. 1995. V. 34. № 10. P. 2535. https://doi.org/10.1021/ic00114a010
Kienast A., Galich L., Murray K.S.H. et al. // J. Porph. Phthal. 1997. V. 1. № 2. P. 141. https://doi.org/10.1002/(SICI)1099-1409(199704)1:2-%3C141::AID-JPP18%3E3.0.CO;2-M
Donzello M.P., Ercolani C., Kadish K.M. et al. // Inorg. Chem. 1998. V. 37. № 15. P. 3682. https://doi.org/10.1021/ic971585+
Sorokin A.B., Tuel A. // Catal. Today. 2000. V. 57. № 1–2. P. 45. https://doi.org/10.1016/S0920-5861(99)00312-0
Ercolani C., Monacelli F. // J. Porph. Phthal. 2001. V. 5. № 9. P. 668. https://doi.org/10.1002/jpp.376
Donzello M.P., Ercolani C., Russo U. et al. // Inorg. Chem. 2001. V. 40. № 13. P. 2963. https://doi.org/10.1021/ic000874m
Stuzhin P.A., Latos-Grazynski L., Jezierski A. // Trans. Met. Chem. 1989. V. 14. № 5. P. 341. https://doi.org/10.1007/BF01032506
Stuzhin P.A. // Macroheterocycles. 2009. V. 2. № 2. P. 114.
Isci U., Afanasiev P., Millet J.-M.M. et al. // Dalton Trans. 2009. № 36. P. 7410. https://doi.org/10.1039/B902592H
Sorokin A.B., Kudrik E.V. // Catal. Today. 2011. V. 159. № 1. P. 37. https://doi.org/10.1016/j.cattod.2010.06.020
Colomban C., Kudrik E.V., Tyurin D.V. et al. // Dalton Trans. 2015. V. 44. № 5. 2240. P. https://doi.org/10.1039/C4DT03207A
Summerville D.A., Cohen I.A. // J. Am. Chem. Soc. 1976. V. 98. № 7. P. 1747. https://doi.org/10.1021/ja00423a019
Goedken V.L., Ercolani C. // J. Chem. Soc., Chem. Commun. 1984. № 6. P. 378. https://doi.org/10.1039/C39840000378
Kennedy B.J., Murray K.S., Homborg H. et al. // Inorg. Chim. Acta. 1987. V. 134. № 1. P. 19. https://doi.org/10.1016/S0020-1693(00)84449-7
Moubaraki P.B., Benlian D., Baldy A. et al. // Acta Crystallogr. 1989. V. C45. P. 393. https://doi.org/10.1107/S0108270188011886
Stuzhin P.A., Hamdush M., Homborg H. // Mendeleev Commun. 1997. V. 7. № 5. P. 196. https://doi.org/10.1070/MC1997v007n05ABEH000819
Kudrik E.V., Afanasiev P., Sorokin A.B. // Macroheterocycles. 2010. V. 3. № 1. P. 19.
Kudrik E.V., Afanasiev P., Bouchu D. et al. // J. Porph. Phthal. 2011. V. 15. № 7–8. P. 583. https://doi.org/10.1142/S1088424611003495
Stuzhin P.A., Ivanova S.S., Dereven’kov I. et al. // Macroheterocycles. 2012. V. 5. № 2. P. 175. https://doi.org/10.6060/mhc2012.120360s
Colomban C., Kudrik E.V., Sorokin A.B. // J. Porph. Phthal. 2017. V. 21. № 4–6. P. 345. https://doi.org/10.1142/S1088424617500274
İşci Ü., Faponle A.S., Afanasiev P. et al. // Chem. Sci. 2015. V. 6. № 8. P. 5063. https://doi.org/10.1039/C5SC01811K
Zaitseva S.V., Tyulyaeva E.Y., Zdanovich S.A. et al. // J. Mol. Liq. 2019. V. 287. P. 111023. https://doi.org/10.1016/j.molliq.2019.111023
Zaitseva S.V., Simonova O.R., Zdanovich S.A. et al. // Macroheterocycles. 2014. V. 7. № 1. C. 55. https://doi.org/10.6060/mhc140486z
Simonova O.R., Zaitseva S.V., Tyurin D.V. et al. // Russ. J. Inorg. Chem. 2018. V. 63. № 9. С. 1164. [Симонова О.Р., Зайцева С.В., Тюрин Д.В. и др. // Журн. неорган. химии. 2018. Т. 63. № 9. С. 1139.]https://doi.org/10.1134/S003602361809019X
Tatsumi K., Hoffmann R., Whangbo M.-H. // J. Chem. Soc., Chem. Commun. 1980. № 11. P. 509. https://doi.org/10.1039/C39800000509
Mansuy D., Lecomte J.P., Chottard J.C. et al. // Inorg. Chem. 1981. V. 20. № 9. P. 3119. https://doi.org/10.1021/ic50223a076
Rossi G., Goedken V.L., Ercolani C. // J. Chem. Soc., Chem. Commun. 1988. V. 550. № 1. P. 46. https://doi.org/10.1039/C39880000046
Bakshi E.N., Delfs C.D., Murray K.S. et al. // Inorg. Chem. 1988. V. 27. № 23. P. 4318. https://doi.org/10.1021/ic00296a048
Paoletti A.M., Penned G., Rossi G. et al. // Inorg. Chem. 1995. V. 34. № 19. P. 4780. https://doi.org/10.1021/ic00123a010
Zaitseva S.V., Tyurin D.V., Zdanovich S.A. et al. // Russ. J. Inorg. Chem. 2019. V. 64. № 6. P. 815. [Зайцева С.В., Тюрин Д.В., Зданович С.А. и др. // Журн. неорган. химии. 2019. Т. 64. № 6. С. 660.]https://doi.org/10.1134/S0036023619060184
Rodrı’guez-Morgade M.S., Planells M., Torres T., et al. // J. Mater. Chem. 2008. V. 18. № 2. P. 176. https://doi.org/10.1039/b712004d
Kroitor A.P., Martynov A.G., Gorbunova Y.G. et al. // Eur. J. Inorg. Chem. 2019. V. 2019. № 14. P. 1923. https://doi.org/10.1002/ejic.201900029
Zanotti G., Angelini N., Notarantonio S. et al. // Corpor. Int. J. Photoenergy 2010. V. 2010. P. 136807. https://doi.org/10.1155/2010/136807
Zaitseva S.V., Tyulyaeva E.Y., Zdanovich S.A. et al. // J. Organomet. Chem. 2020. V. 912. P. 121164. https://doi.org/10.1016/j.jorganchem.2020.121164
Colomban C., Kudrik E.V., Briois V. et al. // Inorg. Chem. 2014. V. 53. № 21. P. 11517. https://doi.org/10.1021/ic501463q
Cailler L.P., Clemancey M., Barilone J. et al. // Inorg. Chem. 2020. V. 59. № 2. P. 1104. https://doi.org/10.1021/acs.inorgchem.9b02718
Kudrik E.V., Afanasiev P., Alvarez L.X. et al. // Nature Chem. 2012. V. 4. № 12. P. 1024. https://doi.org/10.1038/nchem.1471
Tatsumi K., Hoffmann R. // J. Am. Chem. Soc. 1981. V. 103. № 12. P. 3328. https://doi.org/10.1021/ja00401a057
Sorokin A.B., Kudrik E.V., Bouchu D. // Chem. Commun. 2008. № 22. P. 2562. https://doi.org/10.1039/b804405h
Kleinwächter J., Hanack M. // J. Am. Chem. Soc. 1997. V. 119. № 44. P. 10684. https://doi.org/10.1021/ja962663f
Lançon D., Kadish K.M. // Inorg. Chem. 1984. V. 23. № 24. P. 3942. https://doi.org/10.1021/ic00192a021
Ehudin M.A., Gee L.B., Sabuncu S. et al. // J. Am. Chem. Soc. 2019. V. 141. № 14. P. 5942. https://doi.org/10.1021/jacs.9b00795
Zaitseva S.V., Zdanovich S.A., Tyurin D.V. et al. // Russ. J. Gen. Chem. 2018. V. 88. № 6. P. 1142. [Зайцева С.В., Зданович С.А., Тюрин Д.В. и др. // Журн. общей химии. 2018. Т. 88. № 6. С. 962.]https://doi.org/10.1134/S1070363218060166
Zanotti G., Angelini N., Notarantonio S. et al. // Int. J. Photoenergy. 2015. V. 2010. Article ID 136807. https://doi.org/10.1155/2010/136807
Saka E.T., Cakir D., Biyikliogli Z. et al. // Dyes and Pigments. 2013. V. 98. № 2. P. 255. https://doi.org/10.1016/j.dyepig.2013.02.021
Zaitseva S.V., Zdanovich S.A., Golubchikov O.A. // Russ. J. Coord. Chem. 2002. V. 28. № 12. P. 843. [Зайцева С.В., Зданович С.А., Голубчиков О.А. // Коорд. химия. 2002. Т. 28. № 12. С. 900.]https://doi.org/10.1023/A:1021630228084
Praneeth V.K.K., Nather C., Peters G. et al. // Inorg. Chem. 2006. V. 45. № 7. P. 2795. https://doi.org/10.1021/ic050865j
Mamardashvili G.М., Mamardashvili N.Zh., Koifman O.I. // Macroheterocycles. 2013. V. 6. № 1. P. 67. [Мамардашвили Г.М., Мамардашвили Н.Ж., Койфмана О.И. // Макрогетероциклы. 2013. Т. 6. № 1. С. 67.]https://doi.org/10.6060/mhc130226m
Zaitseva S.V., Zdanovich S.A., Koifman O.I. // Macroheterocycles. 2012. V. 5. № 1. P. 81. [Зайцева С.В., Зданович С.А., Койфман О.И. // Макрогетероциклы. 2012. Т. 5. № 1. С. 81.]https://doi.org/10.6060/mhc2012.111149z
Afanasiev P., Bouchu D., Kudrik E.V. et al. // Dalton Trans. 2009. 44. P. 9828. https://doi.org/10.1039/B916047G
Stuzhin P.A., Migalova I.S., Berezin V.D. // Russ. J. Inorg Chem. 1998. V. 43. № 10. P. 1536. [Стужин П.А., Мигалова И.С., Березин Б.Д. // Журн. неорган. химии. 1998. Т. 43. № 10. С. 1655.]
Bélanger S., Keefe M.H., Welch J.L. et al. // Coord. Chem. Rev. 1999. № 190–192. P. 29. https://doi.org/10.1016/S0010-8545(99)00062-4
Krest C.M., Onderko E.L., Yosca T.H. et al. // J. Biol. Chem. 2013. V. 288. № 24. P. 17074. https://doi.org/10.1074/jbc.R113.473108
Gumiero A., Metcalfe C.L., Pearson A.R. et al. // J. Biol. Chem. 2011. V. 286. № 20. P. 1260. https://doi.org/10.1074/jbc.A110.183483
Hohenberger J., Ray K., Meyer K. // Nat. Commun. 2012. V. 3. P. 720. https://doi.org/10.1038/ncomms1718
Oszajca M., Franke A., Brindell M. et al. // Coord. Chem. Rev. 2016. V. 306. Part 2. P. 483. https://doi.org/10.1016/j.ccr.2015.01.013
Quesne M.G., Senthilnathan D., Singh D., et al. // ACS Catal. 2016. V. 6. № 4. P. 2230. https://doi.org/10.1021/acscatal.5b02720
Geng C., Ye S., Neese F. // Dalton Trans. 2014. V. 43. № 16. P. 6079. https://doi.org/10.1039/C3DT53051E
Tiago de Oliveira F., Chanda A., Banerjee D. et al. // Science. 2007. V. 315. № 5813. P. 835. https://doi.org/10.1126/science.1133417
Kundu S., Thompson J. Van K., Ryabov A.D. et al. // J. Am. Chem. Soc. 2011. V. 133. № 46. P. 18546. https://doi.org/10.1021/ja208007w
Bauer I., Knölker H.J. // Chem. Rev. 2015. V. 115. № 9. P. 3170. https://doi.org/10.1021/cr500425u
Simonova O.R., Zaitseva S.V., Tyulyaeva E.Y. et al. // Russ. J. Phys. Chem. A. 2018. V. 92. № 11. P. 2128. [Симонова О.Р., Зайцева С.В., Тюляева Е.Ю. и др. // Журн. физ. химии. 2018. Т. 92. № 11. С. 1692.]https://doi.org/10.1134/S0036024418110390
Накамото К. ИК-спектры и КР-спектры неорганических и координационных соединений / Под ред. Пентина Ю.А. М.: Мир, 1991. 535 с.
Grishina E.S., Makarova A.S., Kudrik E.V. et al. // J. Phys. Chem. 2016. V. 90. № 3. P. 477. https://doi.org/10.1134/S0036024416030134
Pandit Y.A., Shah S.J., Rath S.P. // Z. Anorg. Allg. Chem. 2018. V. 644. № 15 P. 856. https://doi.org/10.1002/zaac.201800247
Hu S., Spiro T.G. // J. Am. Chem. Soc. 1993. V. 115. № 25. P. 12029. https://doi.org/10.1021/ja00078a047
Dey S., Sil D., Pandit Y.A., Rath S.P. // Inorg. Chem. 2016. V. 55. № 7. P. 3229. https://doi.org/10.1021/acs.inorgchem.5b02065
Girolami G.S., Gorlin P.A., Milam S.N. et al. // J. Coord. Chem. 1994. V. 32. № 1–3. P. 173. https://doi.org/10.1080/00958979408024247
Sorokin A.B. // Bioinorg. React. Mech. 2012. V. 8. № 1–2. P. 59. https://doi.org/10.1515/irm-2012-0002
Franke A., Fertinger C., van Eldik R. // Chem. Eur. J. 2012. V. 18. № 22. P. 6935. https://doi.org/10.1002/chem.201103036
Tyurin D.V., Zaitseva S.V., Kudrik E.V. // Russ. J. Phys. Chem. A. 2018. V. 92. № 5. P. 870. [Тюрин Д.В., Зайцева С.В., Кудрик Е.В. // Журн. физ. химии. 2018. Т. 92. № 5. С. 723. https://doi.org/10.7868/S0044453718050084]https://doi.org/10.1134/S0036024418050321
Malakhova M.V., Orlova V.F., Karpov V.A. et al. // Bull. Exp. Biol. Med. Biophys. Biochem. 1998. V. 126. № 9. P. 321. https://doi.org/10.1007/BF02447377
Kennedy T.A., Liebler D. // Chem. Res. Toxicol. 1991. V. 4. № 3. P. 290. https://doi.org/10.1021/tx00021a005
Yamauchi R., Miyake N., Inoue H. et al. // Agric. Food Chem. 1993. V. 41. № 5. P. 708. https://doi.org/10.1021/jf00029a005
Benevides C.M.J., Veloso M.C.C., de Paula Pereira P.A. et al. // Food Chem. 2011. V. 126. № 3. P. 927. https://doi.org/10.1016/j.foodchem.2010.11.082
French R.R., Holzer P., Leuenberger M.G. et al. // Angew. Chem. Int. Ed. 2000. V. 39. № 7. P. 1267. https://doi.org/10.1002/(SICI)1521-3773(20000403)-39:7%3C1267::AID-ANIE1267%3E3.0.CO;2-7
Shing K.-P., Cao B., Liu Y. et al. // J. Am. Chem. Soc. 2018. V. 140. № 22. P. 7032. https://doi.org/10.1021/jacs.8b04470
Caris-Veyrat C., Amiot M.-J., Ramasseul R. et al. // New J. Chem. 2001. V. 25. № 2. P. 203. https://doi.org/10.1039/B006975M
Zhang R., Vanover E., Luo W. et al. // Dalton Trans. 2014. V. 43. № 23. P. 8749. https://doi.org/10.1039/C4DT00649F
Wang C., Shalyaev K.V., Bonchio M. et al. // Inorg. Chem. 2006. V. 45. № 12. P. 4769. https://doi.org/10.1021/ic0520566
Дополнительные материалы
- скачать ESM.docx
- Таблица S1. - Таблица S5.
Рис. S1. - Рис. S6.
Инструменты
Журнал неорганической химии