

Журнал неорганической химии, 2022, T. 67, № 3, стр. 286-293
Пергалогенированные металлопорфирины: синтез, структура и спектральные свойства
Н. В. Чижова a, О. А. Дмитриева a, Н. Ж. Мамардашвили a, *
a Институт химии растворов им. Г.А. Крестова
1153040 Иваново, ул. Академическая, Россия
* E-mail: ngm@isc-ras.ru
Поступила в редакцию 10.09.2021
После доработки 26.10.2021
Принята к публикации 27.10.2021
- EDN: YIORZT
- DOI: 10.31857/S0044457X22030059
Аннотация
С целью получения пергалогенированных металлопорфиринов проведено исчерпывающее бромирование и хлорирование β-положений Ni(II)-, Zn(II)-5,10,15,20-тетра(2,6-дихлорфенил)- и 5,10,15,20-тетра(2,6-дифторфенил)порфиринов с помощью N-бромсукцинимида и N-хлорсукцинимида в диметилформамиде. При обработке Zn(II)-2,3,7,8,12,13,17,18-октабром-5,10,15,20-тетра(2,6-дифторфенил)порфирина и Zn(II)-2,3,7,8,12,13,17,18-октахлор-5,10,15,20-тетра(2,6-дифторфенил)порфирина трифторуксусной кислотой получены соответствующие порфирины-лиганды. Синтезированные соединения идентифицированы методами электронной абсорбционной, 1Н ЯМР-спектроскопии и масс-спектрометрии. Методом DFT рассчитаны структуры галогензамещенных порфиринов и их комплексов с цинком. Введение атомов брома и хлора в пиррольные кольца орто-замещенных порфиринов и их металлокомплексов приводит к искажению плоской структуры молекул до седлообразной. Оценено влияние β-галогенирования на квантовые выходы флуоресценции синтезированных соединений. Квантовый выход β-хлорзамещенного порфирина уменьшается в ~7 раз по сравнению с исходным тетра(2,6-дифторфенил)порфирином, а его порфирината цинка по сравнению с исходным комплексом – в ~15 раз. Полное тушение флуоресценции, вызванное влиянием тяжелых атомов брома в β-положениях макроцикла, наблюдается как для порфиринов, так и для их металлокомплексов. Пергалогенированные металлопорфирины обладают увеличенной n-проводимостью и могут быть использованы при создании новых материалов, проявляющих нелинейно-оптические и каталитические свойства.
ВВЕДЕНИЕ
Порфирины и их комплексы с металлами вызывают большой интерес в связи с их применением в качестве полупроводников, электронных переключателей, термохромных материалов, катализаторов, фотосенсибилизаторов для фотодинамической терапии [1–5].
Повышенное внимание к синтезу и исследованию комплексов никеля порфиринами обусловлено возможностью их применения в качестве молекулярных термометров, сенсоров и оптических материалов [6–8]. Галогензамещенные металлопорфирины используются при производстве координационных полимеров, обладающих каталитическими и нелинейно-оптическими свойствами [9, 10].
Исчерпывающее бромирование β-положений Cu(II)-тетрафенилпорфина Br2 описано в работе [11]. Бромирование Co(II)-тетрафенилпорфина c помощью N-бромсукцинимида (NBS) в смеси хлороформа и диметилформамида (ДМФА) приводило к образованию β-октабромзамещенного порфирината кобальта(II) [12]. При взаимодействии Ni(II)-тетрафенилпорфирина с NBS в кипящем о-дихлорбензоле синтезирован октабромзамещенный порфиринат никеля(II) [13]. В работе [14] показано, что при обработке Ni(II)-тетрафенилпорфирина NBS в зависимости от соотношения реагентов в смеси хлороформ–диметилформамид легко образуются моно-, тетра- и окта-β-бромзамещенные комплексы никеля. При взаимодействии Ni(II)-тетрафенилпорфирина с N-хлорсукцинимидом (NCS) в кипящем орто-дихлорбензоле получены β-октазамещенные порфиринаты никеля [15, 16]. Авторы [17] для синтеза β-октахлорзамещенных металлопорфиринов использовали тионилхлорид.
Хлорирование орто-дигалогензамещенных Zn(II)-порфиринов с помощью NCS в кипящем хлороформе выполнено в работе [18]. Авторами [19] показано, что при взаимодействии орто-дихлорзамещенного Zn(II)-порфирина с NBS в метаноле в течение 1 ч образуется β-октабромзамещенный комплекс с выходом 45%. При кипячении дихлорзамещенного комплекса цинка с NBS в смеси хлороформ–метанол время образования Zn(II)-октабромпорфирина сокращается до 30 мин, выход увеличивается до 73% [20]. При кипячении комплекса цинка с NCS в метаноле в течение 11 ч получен β-октахлорзамещенный Zn-порфирин [19]. При замене метанола на смесь хлороформа и метанола время реакции сокращается до 5 ч [20].
С целью получения пергалогенированных металлопорфиринов в настоящей работе исследованы реакции бромирования и хлорирования β-положений комплексов никеля и цинка с тетра(2,6-дихлорфенил)порфирином, тетра(2,6-дифторфенил)порфирином с помощью NBS и NCS в ДМФА. Найдены оптимальные условия синтеза замещенных по пиррольным и фенильным кольцам Ni(II)- и Zn(II)-порфиринов (схема 1 ).
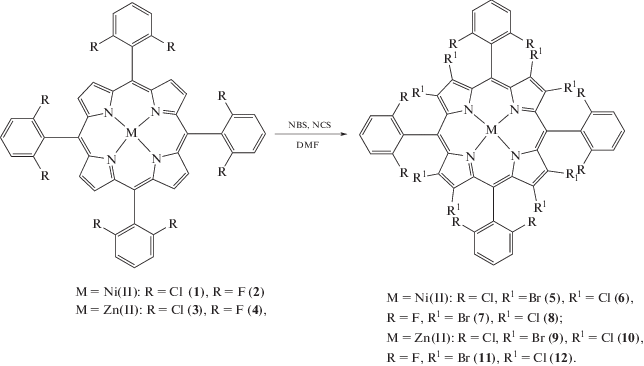
Схема 1 .
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
5,10,15,20-Тетра(2,6-дихлорфенил)порфирин и 5,10,15,20-тетра(2,6-дифторфенил)порфирин (PorphyСhem), NBS, NCS, ацетататы кадмия(II), цинка(II), никеля(II), хлориды цинка(II) и никеля(II), трифторуксусную кислоту (Acros), оксид алюминия, (Merck), С6D6, CDCl3 (Aldrich), растворители (х. ч.) использовали без дополнительной обработки. Cпектры 1Н ЯМР (500 МГц) получали на приборе Bruker AV III-500 (внутренний стандарт – ТМС). Масс-спектры регистрировали на масс-спектрометре MaldiToF Shimadzu Biotech Axima Confidence (матрица – дигидроксибензойная кислота). Электронные спектры поглощения и спектры флуоресценции записывали на спектрофотометрах Cary-100 (Varian) и Shimadzu RF-5301 соответственно.
Структуры порфиринов и их комплексов с цинком были рассчитаны с помощью DFT-метода [21] с использованием комбинации B3LYP функционала и def2-TZVP [22–25] для атомов Zn и 6-31G + (d, p) [26, 27] базисного набора для остальных атомов [28, 29]. Для расчетов Zn-комплексов использовали симметрию D4h, для порфиринов – D2h. Расчеты проводили с использованием пакета программ GAMESS (v.12) [30].
Спектры флуоресценции порфиринов и их Zn-комплексов были записаны в толуоле при комнатной температуре на длине волны возбуждения λmax = 415 нм (с < 10–7 моль/л). В качестве стандарта для комплексов цинка 11, 12 был выбран Zn(II)-тетра(2,6-дифторфенил)порфирин с квантовым выходом в толуоле 0.03 [31], для порфиринов 13, 14 – тетрафенилпорфин с квантовым выходом в толуоле 0.1 [32]. Квантовый выход флуоресценции рассчитывали по формуле [33]:
Cd(II)-5,10,15,20-тетра(2,6-дихлорфенил)порфирин синтезировали по методике, приведенной в [34]. ЭСП в ДМФА, λ, нм (lgε): 418 (4.77), 438 (5.53), 578 (4.34), 620 (3.98). Масс-спектр, m/z (Iотн, %): 1000.7 (82) [M]+. Для С44H20N4Cl8Cd вычислено 1000.7. Спектр 1H ЯМР (C6D6), δ, м.д.: 8.73 с (8Н, пиррол), 7.36 д (8Н, Phm, J = 7.6 Гц), 6.97 т (4Н, Phn, J = 7.65 Гц).
Ni(II)-5,10,15,20-тетра(2,6-дихлорфенил)порфирин (1). Смесь 0.02 г (0.020 ммоль) Cd(II)-тетра(2,6-дихлорфенил)порфирина и 0.052 г (0.400 ммоль) NiCl2 в 15 мл ДМФА нагревали в колбе с обратным холодильником до температуры кипения и кипятили в течение 2 мин. Смесь охлаждали, выливали в воду, добавляли NaClтв, осадок отфильтровывали, промывали водой, высушивали, хроматографировали на оксиде алюминия дихлорметаном, затем хлороформом. Выход составил 0.018 г (0.180 ммоль, 92%). Масс-спектр, m/z (Iотн, %): 947.5(98) [M + H]+. Для С44H20N4Cl8Ni вычислено 947. Спектр 1H ЯМР, CDCl3, δ, м.д.: 8.60 с (8Н, пиррол), 7.74 д (8Н, Phm, J = 7.6 Гц), 7.66 т (4Н, Phn, J = 7.65 Гц).
Ni(II)-5,10,15,20-тетра(2,6-дифторфенил)порфирин (2). Cмесь 0.02 г (0.026 ммоль) тетра(2,6-дифторфенил)порфирина и 0.093 г (0.528 ммоль) Ni(OAc)2 в 10 мл ДМФА нагревали до температуры кипения, кипятили в течение 12 мин. Обрабатывали аналогично 1. Выход 0.018 г (0.022 ммоль, 85%). Масс-спектр, m/z (Iотн, %): 814.9(98) [M]+. Для С44H20N4F8Ni вычислено 815.4. Спектр 1H ЯМР (CDCl3), δ, м.д.: 8.78 с (8Н, пиррол), 7.75 т (4Н, Phn, J = 7.65 Гц), 7.33 д (8Н, Phm, J = 7.6 Гц).
Zn(II)-5,10,15,20-тетра(2,6-дихлорфенил)порфирин (3) синтезировали по методике, описанной в [20]. Масс-спектр, m/z (Iотн, %): 952.9 (97) [M]+. Для С44H20Cl8N4Zn вычислено 953.7. Спектр 1Н ЯМР (CDCl3), δ, м.д.: 8.99 с (8Н, пиррол), 7.82 т (4Н, Phn, J = 7.65 Гц), 7.41 д (8Н, Phm, J = 7.6 Гц).
Zn(II)-5,10,15,20-тетра(2,6-дифторфенил)порфирин (4). Cмесь 0.02 г (0.026 ммоль) тетра(2,6-дифторфенил)порфирина и 0.049 г (0.264 ммоль) Zn(OAc)2 в 7 мл ДМФА нагревали до температуры кипения и кипятили в течение 2 мин. Обрабатывали аналогично 1. Выход 0.018 г (0.0219 ммоль, 83%). Масс-спектр, m/z (Iотн, %): 821.4 (96) [M]+. Для С44H20F8N4Zn вычислено 822.1. Спектр 1Н ЯМР (CDCl3), δ, м.д.: 8.99 с (8Н, пиррол), 7.82 т (4Н, Phn, J = 7.65 Гц), 7.41 д (8Н, Phm, J = 7.6 Гц).
Ni(II)-2,3,7,8,12,13,17,18-октабром-5,10,15,20-тетра(2,6-дихлорфенил)порфирин (5). К 0.02 г (0.021 ммоль) комплекса 1 в 5 мл ДМФА добавляли 0.15 г (0.844 ммоль) NBS, выдерживали в течение 4 ч при 30°С, 8 ч при 23°С. Обрабатывали аналогично 1. Выход 0.028 г (0.0177 ммоль, 82%). Масс-спектр, m/z (Iотн, %): 1578.6 (97) [M + Н]+. Для С44H12Br8Cl8N4Ni вычислено 1578.2. Спектр 1Н ЯМР (CDCl3), δ, м.д.: 7.74–7.64 м (8Н, Phm, 4Н, Phn).
Ni(II)-2,3,7,8,12,13,17,18-октахлор-5,10,15,20-тетра(2,6-дихлорфенил)порфирин (6). К раствору 0.02 г (0.021 ммоль) комплекса 1 в 5 мл ДМФА прибавляли 0.169 г (1.266 ммоль) NCS, выдерживали при комнатной температуре в течение 14 ч. Обрабатывали аналогично 1. Выход 0.021 г (0.0172 ммоль, 79%). Масс-спектр, m/z (Iотн, %): 1223.4 (96) [M + Н]+. Для С44H12Cl16N4Ni вычислено 1222.6. Спектр 1Н ЯМР (CDCl3), δ, м.д.: 7.61–7.54 м (8Н, Phm, 4Н, Phn).
Ni(II)-2,3,7,8,12,13,17,18-октабром-5,10,15,20-тетра(2,6-дифторфенил)порфирин (7). К 0.02 г (0.0245 ммоль) комплекса 2 в 5 мл ДМФА прибавляли 0.174 г (0.980 ммоль) NBS, выдерживали в течение 1.5 ч (30°С), 2.5–3 ч (25–22°С). Обрабатывали аналогично 1. Выход 0.03 г (0.0207 ммоль, 84%). Масс-спектр, m/z (Iотн, %): 1446.5 (98) [M + H]+. Для С44H12Br8F8N4Ni вычислено 1446.6. Спектр 1Н ЯМР (CDCl3), δ, м.д.: 7.78–7.72 м (8Н, Phm, 4Н, Phn).
Ni(II)-2,3,7,8,12,13,17,18-октахлор-5,10,15,20-тетра(2,6-дифторфенил)порфирин (8). К 0.02 г (0.024 ммоль) комплекса 2 в 5 мл ДМФА добавляли 0.196 г (1.47 ммоль) NCS, выдерживали при 22–25°С в течение 8 ч. Обрабатывали аналогично 1. Выход 0.022 г (0.020 ммоль, 82%). Масс-спектр, m/z (Iотн, %): 1090.7 (98) [M + H]+. Для С44H12Cl8F8N4Ni вычислено 1089.9. Спектр 1Н ЯМР (CDCl3), δ, м.д.: 7.09, 7.02 м (8Н, Phm, 4Н, Phn).
Zn(II)-2,3,7,8,12,13,17,18-октабром-5,10,15,20-тетра(2,6-дихлорфенил)порфирин (9). К 0.02 г (0.021 ммоль) комплекса 3 в 5 мл ДМФА добавляли 0.15 г (0.840 ммоль) NBS, выдерживали при 23°C в течение 16 ч. Реакционную смесь выливали в насыщенный раствор NaHCO3, выпавший осадок отфильтровывали, промывали водой, высушивали, хроматографировали на оксиде алюминия дихлорметаном, затем хлороформом. Выход 0.023 г (0.016 ммоль, 71%). Масс-спектр, m/z (Iотн, %): 1584.7 (97) [M – H]+. Для С44H12Br8 Cl8N4Zn вычислено 1584.9. Спектр 1Н ЯМР (CDCl3), δ, м.д.: 7.71–7.62 м (8Н, Phm, 4Н, Phn).
Zn(II)-2,3,7,8,12,13,17,18-октахлор-5,10,15,20-тетра(2,6-дихлорфенил)порфирин (10). К 0.02 г (0.021 ммоль) 3 в 5 мл ДМФА добавляли 0.168 г (1.260 ммоль) NСS, выдерживали при 23°C течение 36 ч. Обрабатывали аналогично 9. Выход 0.02 г (0.0163 ммоль, 78%). Масс-спектр, m/z (Iотн, %): 1231.04 (97) [M + 2H]+. Для С44H12Cl16N4Zn вычислено 1229.3. Спектр 1Н ЯМР (CDCl3), δ, м.д.: 7.73–7.65 м (8Н, Phm, 4Н, Phn).
Zn(II)-2,3,7,8,12,13,17,18-октабром-5,10,15,20-тетра(2,6-дифторфенил)порфирин (11). К 0.02 г (0.0243 ммоль) комплекса 4 в 5 мл ДМФА добавляли 0.174 г (0.972 ммоль) NBS, выдерживали в течение 3 ч при 30°С, 7 ч при 22°С. Обрабатывали аналогично 9. Выход 0.024 г (0.016 ммоль, 70%). Масс-спектр, m/z (Iотн, %): 1455.7 (55) [M + 2H]+. Для С44H12Br8F8N4Zn вычислено 1453.3. Спектр 1Н ЯМР (CDCl3), δ, м.д.: 7.80 т (4Н, Phn), 7.32 д (8Н, Phm).
Zn(II)-2,3,7,8,12,13,17,18-октахлор-5,10,15,20-тетра(2,6-дифторфенил)порфирин (12). К раствору 0.02 г (0.024 ммоль) комплекса 2 в 5 мл ДМФА добавляли 0.195 г (1.460 ммоль) NСS, выдерживали при комнатной температуре (23°С) в течение 24 ч. Обрабатывали аналогично 9. Выход 0.02 г (0.018 ммоль, 72%). Масс-спектр, m/z (Iотн, %): 1099.2 (98) [M + 2H]+. Для С44H12Cl8F8N4Zn вычислено 1097.6. Спектр 1Н ЯМР (CDCl3), δ, м.д.: 6.94–6.89 м (8Н, Phm, 4Н, Phn).
2,3,7,8,12,13,17,18-Октабром-5,10,15,20-тетра(2,6-дифторфенил)порфирин (13). К раствору 0.02 г (0.014 ммоль) комплекса 11 в 4 мл CHCl3 добавляли 4 мл трифторуксусной кислоты, перемешивали при комнатной температуре в течение 1 ч, добавляли воду, органический слой отделяли, промывали водой, раствором бикарбоната натрия, снова водой, высушивали Na2SO4, хроматографировали на оксиде алюминия дихлорметаном. Выход 0.014 г (0.010 ммоль, 75%). ЭСП в CHCl3, λ, нм, (lgε): 372 (4.48), 459 (5.22), 557 (4.23), 602 (3.94), 642 (3.84), 715 (3.58). Масс-спектр, m/z (Iотн, %): 1392.1 (96) [M + 2H]+. Для С44H14Br8F8N4 вычислено 1389.9. Спектр 1Н ЯМР (CDCl3), δ, м.д.: 8.06 т (4Н, Phn), 7.54 д (8Н, Phm), 0.31 с (NH-протоны).
2,3,7,8,12,13,17,18-Октахлор-5,10,15,20-тетра(2,6-дифторфенил)порфирин (14). К раствору 0.02 г (0.018 ммоль) комплекса 12 в 4 мл CHCl3 добавляли 4 мл трифторуксусной кислоты, перемешивали при комнатной температуре в течение 1.5 ч. Выход составил 0.014 г (0.0135 ммоль, 73%). ЭСП в CHCl3, λ, нм (lgε): 442 (5.18), 539 (4.30), 587 (4.11), 629 (4.02). Масс-спектр, m/z (Iотн, %): 1035.5 (97) [M + H]+. Для С44H14Cl8F8N4 вычислено 1034.3. Спектр 1Н ЯМР (CDCl3), δ, м.д.: 7.57 т (4Н, Phn), 7.09 д (8Н, Phm), –2.24 с (NH-протоны).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Исследования показали, что реакция комплексообразования орто-дихлорзамещенного порфирина 1 с ацетатом никеля (мольное соотношение 1 : 20) в кипящем ДМФА протекает медленно. Степень превращения исходного порфирина в комплекс никеля в течение 1 ч составляет ~15%. Увеличение времени реакции до 3 ч не приводит к образованию Ni(II)-тетра-2,6-(дихлорфенил)порфирина (1). Для получения соединения 1 использовали метод переметаллирования лабильных комплексов порфиринов [35]. Реакция Cd(II)-тетра(2,6-дихлорфенил)порфирина с NiCl2 в кипящем ДМФА приводит к образованию комплекса 1 уже в течение 2 мин. В спектре 1Н ЯМР соединения 1 в CDCl3 присутствуют сигналы пиррольных колец при 8.60 м.д., сигналы мета- и пара-протонов при 7.74 и 7.66 м.д. (рис. 1). В масс-спектре комплекса 1 зафиксирован сигнал с m/z = 947.5 (для С44H20N4Cl8Ni вычислено 947, экспериментальная часть). Напротив, при взаимодействии тетра(2,6-дифторфенил)порфирина с ацетатом Ni(II) в кипящем ДМФА в течение 12 мин образуется Ni(II)-тетра(2,6-дифторфенил)порфирин (2).
Показано, что бромирование орто-дихлорзамещенного комплекса никеля 1 NBS (мольное соотношение 1 : 40) в ДМФА в течение 4 ч при 30°С, в течение 7–8 ч при 23°С приводит к образованию Ni(II)-β-октабром-тетра(2,6-дихлорфенил)порфирина (5). В масс-спектре комплекса 5 зафиксирован сигнал c m/z = 1578.6, соответствующий молекулярному иону полученного соединения. В спектре 1Н ЯМР соединения 5 присутствует мультиплет сигналов мета- и пара-протонов при 7.74–7.64 м.д. Для получения Ni(II)-β-октахлор-тетра(2,6-дихлорфенил)порфирина (6) в ДМФА требуются более жесткие условия: 60-кратный избыток NCS, время реакции увеличивается до 14 ч.
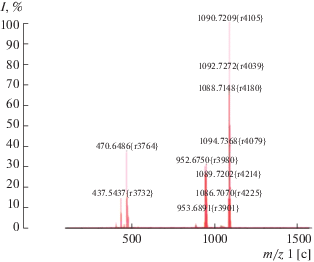
В сравнимых с 1 условиях бромирование комплекса никеля 2 в ДМФА уже в течение 1.5 ч при 30°С приводит к образованию Ni(II)-β-октабром-тетра(2,6-дифторфенил)порфирина (7). При мольном соотношении 2 : NCS = 1 : 60 в ДМФА в течение 4 ч (30°С) получен Ni(II)-β-октахлор-тетра(2,6-дифторфенил)порфирин (8). В масс-спектрах комплексов никеля 7, 8 зафиксированы сигналы с m/z = 1446.5 и 1090.7, соответствующие молекулярным ионам полученных соединений (экспериментальная часть, рис. 2).
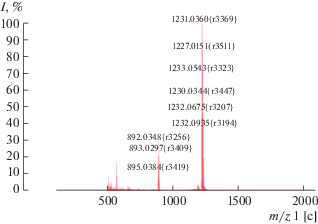
Бромирование комплексов цинка 3, 4 по сравнению с порфиринатами никеля в аналогичных условиях протекает в 2 раза медленнее. Так, время образования Zn(II)-β-октабром-тетра(2,6-дифторфенил)порфирина (11) составляет от 3 до 7 ч в зависимости от температуры (30–22°С). Этот факт можно объяснить сильным π-дативным взаимодействием между ионом никеля и порфириновым макроциклом dπ–eg (π*)-типа. Координация иона цинка с внутрициклическими атомами азота осуществляется только за счет ϭ-связи Zn ← N, способствуя уменьшению электронной плотности и на реакционных центрах макроцикла. Еще большее влияние металл оказывает на протекание реакции хлорирования. В сравнимых с 6 условиях Zn(II)-β-октахлор-тетра(2,6-дихлорфенил)порфирин (10) получен при 23°C в течение 36 ч, Zn(II)-β-октахлор-тетра(2,6-дифторфенил)порфирин (12) – в течение 48 ч (мольное соотношение реагентов 1 : 90). В масс-спектрах полученных соединений присутствуют сигналы с m/z = 1231 и 1099.2, соответствующие молекулярным ионам комплексов 10 и 12 (рис. 3, экспериментальная часть).
Характеристики ЭСП галогензамещенных комплексов никеля и цинка в ДМФА приведены в табл. 1. На рис. 4 представлены ЭСП порфирината никеля 2 и его β-бромзамещенного комплекса 7 в хлороформе.
Таблица 1.
Характеристики электронных спектров поглощения галогензамещенных Ni(II)- и Zn(II)-тетрафенилпорфиринов в ДМФА, λ, нм (lg ε)
| Соединение | Полоса Соре | Q-полосы |
|---|---|---|
| 1 | 410 (5.65) | 526 (4.55), 560 (4.33) |
| 2 | 404 (5.57) | 522 (4.55), 555 (4.30) |
| 3 | 405 (4.66), 426 (5.50) | 559 (4.35), 594пл |
| 4 | 399 (4.76), 419 (5.64) | 552 (4.44) |
| 5 | 446 (5.47) | 564 (4.41), 603 (4.37) |
| 6 | 449 (5.41) | 556 (4.33), 596 (4.28) |
| 7 | 374 (4.54), 445 (5.30), 457пл | 562 (4.24), 603 (4.25) |
| 8 | 364 (4.47), 446 (5.24) | 555 (3.94), 594 (4.04) |
| 9 | 368 (4.52), 474 (5.31) | 612 (4.16), 660 (4.06) |
| 10 | 364 (4.66), 455 (5.40) | 592 (4.34), 643пл |
| 11 | 366 (4.75), 473 (5.50) | 612 (4.42), 663 (4.32) |
| 12 | 456 (5.42) | 592 (4.45), 644пл (4.20) |
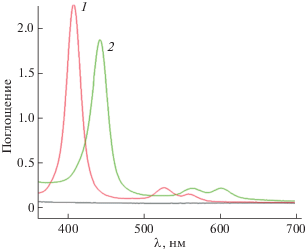
Рис. 4.
ЭСП в хлороформе: 1 – Ni(II)-тетра(2,6-дифторфенил)порфирина, 2 – Ni(II)-β-октабром-тетра(2,6-дифторфенил)порфирина.
При обработке комплексов цинка 11, 12 трифторуксусной кислотой получены октабром-5,10,15,20-тетра(2,6-дифторфенил)порфирин (13) и октахлор-5,10,15,20-тетра(2,6-дифторфенил)порфирин (14).
На рис. 5 приведены оптимизированные структуры исследуемых порфиринов и их Zn-комплексов, а выбранные длины связей, валентные и двугранные углы представлены в табл. 2.
Рис. 5.
Молекулярные конфигурации порфиринов и их Zn(II)-комплексов, вид сверху (вверх) и сбоку (вниз).
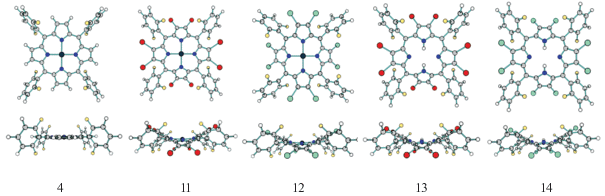
Таблица 2.
Структурные параметры порфиринов и их комплексов с цинком: длины связей, Å, валентные углы, град
| Соединение | 4 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|
| Расстояния между связями | |||||
| Cβ–${{{\text{C}}}_{{\beta '}}}$ | 1.365 | 1.370 | 1.366 | 1.376 | 1.377 |
| Cα–Cβ | 1.450 | 1.453 | 1.452 | 1.440 | 1.440 |
| Cα–N | 1.388 | 1.389 | 1.390 | 1.374 | 1.374 |
| Cα–Cm | 1.404 | 1.411 | 1.410 | 1.408 | 1.409 |
| Cm–C1 | 1.496 | 1.490 | 1.492 | 1.489 | 1.488 |
| N1–N3 | 4.105 | 4.060 | 4.074 | 4.180 | 4.188 |
| N–Zn/H | 2.053 | 2.035 | 2.041 | 1.012 | 1.012 |
| Углы связей | |||||
| ${{{\text{C}}}_{{\beta '}}}$CβCα | 107.4 | 107.8 | 107.9 | 108.4 | 108.4 |
| CβCαN | 109.2 | 107.5 | 107.4 | 105.2 | 105.1 |
| CαN${{{\text{C}}}_{{\alpha '}}}$ | 106.9 | 108.8 | 108.8 | 112.4 | 112.7 |
| NCαCm | 125.7 | 123.5 | 124.3 | 124.6 | 124.5 |
| CβCαCm | 125.1 | 128.8 | 128.1 | 129.9 | 130.2 |
| CαCmC1 | 117.3 | 118.0 | 118.0 | 117.4 | 117.9 |
| CαCmCα | 125.4 | 123.9 | 123.9 | 123.9 | 123.2 |
| NZnN | 179.7 | 172.1 | 172.7 | ||
| Двугранный угол | |||||
| Cβ${{{\text{C}}}_{{\beta '}}}$${{{\text{C}}}_{{\alpha '}}}$N | 0.3 | –4.7 | –4.1 | –3.2 | –3.1 |
| ${{{\text{C}}}_{{\beta '}}}$CβCαCm | 179.9 | –169.5 | –170.3 | –171.0 | –171.1 |
| NCαCm${{{\text{C}}}_{{\alpha '}}}$ | 1.3 | 17.1 | 15.4 | 17.6 | 18.7 |
| NCαCmCα | –1.3 | –17.1 | –15.4 | –17.6 | –18.7 |
| NCαCmC1 | 178.7 | –162.9 | –164.6 | –162.6 | –161.7 |
| CαCmC1Сf | 72.2 | 69.2 | 70.9 | 69.3 | 64.3 |
Введение атомов брома и хлора в пиррольные кольца макроцикла (соединения 11–14) приводит к искажению плоской структуры молекул до седлообразной, что отражается в увеличении угла CβCαCm при уменьшении углов NCαCm и MNCα [36, 37]. Непланарность порфиринового ядра вызвана стерическим отталкиванием периферийными заместителями, что усиливает снятие напряжения за счет длин связей и углов [38]. Наблюдаемое искажение в этих соединениях больше, чем в Zn(II)-2,3,12,13-тетрабромтетрафенилпорфирине [39]. Кроме того, углы NZnN отклонены от 180°, это указывает на то, что атом цинка лежит вне плоскости ядра макроцикла. Каждое пиррольное кольцо поочередно смещается вверх и вниз от средней плоскости в противоположных направлениях. В таком случае фенильные кольца наклонены к плоскости макроцикла, чтобы минимизировать стерическое скопление между заместителями. Это проявляется в уменьшении угла CαCmC1Сf β-замещенных соединений по сравнению с их незамещенным аналогом 4. Zn-комплексы меньше подвержены седлообразному искажению плоскости макроцикла по сравнению с его свободными основаниями, что вызвано координацией металла с внутрициклическими атомами азота пиррольных колец макроцикла. Стерическое отталкивание зависит от природы β-заместителей (Br или Cl), при котором угол наклона фенильного кольца (CαCmC1Сf) увеличивается с уменьшением искажения макроцикла (табл. 2, рис. 5). Природа галогена в β-положениях орто-замещенных комплексов цинка незначительно влияет на рассчитанные значения расстояний N–Zn и N1–N3. Расстояние N–Zn составляет 2.04 Å, что незначительно выше 2.03 Å для Zn(II)-β-октабромтетрафенилпорфирина [40].
При β-замещении в порфиринах 13, 14 происходит удлинение ядра макроцикла N1–N3 вдоль направления β-замещения в результате стеричес-кого напряжения, которое усиливается атомами брома и хлора, вызывая искажение макроцикла. По сравнению с комплексами цинка в порфиринах наблюдается уменьшение значений углов CβCαN и CαN${{{\text{C}}}_{{\alpha '}}}$ на ~4°–6° и большее сопряжение фенильных колец с макроциклом, что выражено в уменьшении длины связи мезо-углеродного атома с фенильным кольцом (Cm–C1) и его угла наклона к плоскости макроцикла (CαCmC1Сf).
Нами изучено влияние природы заместителей на спектрально-флуоресцентные свойства синтезированных порфиринов и их Zn-комплексов. Установлено, что квантовый выход β-хлорзамещенного порфирина 14 уменьшается в 7 раз по сравнению с исходным тетра(2,6-дифторфенил)порфирином, а порфирината цинка 12 – в 15 раз по сравнению с исходным комплексом 4. Значительное влияние β-галогенирования на тушение флуоресценции наблюдается в случае цинковых комплексов. Это вызвано увеличением плотности заряда на порфириновом кольце за счет включения иона металла, приводящим к усилению безызлучательного распада одновременно с уменьшением квантового выхода флуоресценции [41]. Полное тушение флуоресценции, вызванное влиянием тяжелых атомов брома в β-положениях макроцикла, наблюдается для соединений 11 и 13 (табл. 3).
ЗАКЛЮЧЕНИЕ
При взаимодействии орто-ди(галогенфенил)замещенных порфиринатов никеля и цинка с NBS и NCS в диметилформамиде синтезированы и идентифицированы методами электронной абсорбционной, 1Н ЯМР-спектроскопии и масс-спектрометрии комплексы Ni(II) и Zn(II) c октабром-тетра(2,6-дихлорфенил)порфирином, октахлор-тетра(2,6-дихлорфенил)порфирином, октабром-тетра(2,6-дифторфенил)порфирином, октахлор-тетра(2,6-дифторфенил)порфирином. При обработке β-замещенных тетра(2,6-дифторфенил)порфиринатов Zn(II) трифторуксусной кислотой получены соответствующие порфирины. Методом DFT рассчитаны структуры галогензамещенных порфиринов и их комплексов с цинком. Определено влияние β-галогенирования на квантовый выход флуоресценции синтезированных соединений.
Список литературы
Koifman O.I., Ageeva T.A., Beletskaya I.P. et al. // Macroheterocycles. 2020. V. 13. P. 311. https://doi.org/10.6060/mhc200814k
Tsivadze A.Y., Chernyad’ev A.Y. // Russ. J. Inorg. Chem. 2020. V. 65. № 11. P. 1662. https://doi.org/10.31857/S0044457X20110197
Chizhova N.V., Kumeev R.S., Mamardashvili N.Zh. // Russ. J. Inorg. Chem. 2013. V. 58. № 6. P. 740. https://doi.org/10.1134/S0036023613060089
Zdanovich S.A., Mamardashvili N.Zh., Golubchikov O.A. // Russ. J. Org. Chem. 1996. V. 32. № 5. P. 756.
Simonova O.R., Zdanovich S.A., Zaitseva S.V. et al. // Russ. J. Inorg. Chem. 2020. V. 65. № 7. P. 1006. https://doi.org/10.31857/S0044457X2007020X
Engeser M., Fabbrizzi L., Licchelli M. et al. // Chem. Commun. 1999. P. 1191. https://doi.org/10.1039/A901931F
Lupton J.M. // Appl. Phys. Lett. 2002. V. 81. № 13. P. 2478. https://doi.org/10.1063/1.1509115
Finikova O.S., Cheprakov A.V., Beletskaya I.P. et al. // J. Org. Chem. 2004. V. 69. № 2. P. 522. https://doi.org/10.1021/jo0350054
Stuzhin P.A., Goryachev M.Yu., Ivanova S.S. et al. // J. Porphyr. Phthalocyanines. 2013. V. 17. P. 905. https://doi.org/10.1142/S1088424613500892
Chumakov D.E., Khoroshutin A.V., Anisimov A.V. et al. // Chem. Heterocycl. Compd. 2009. V. 45. № 3. P. 259. https://doi.org/10.1007/s10593-009-0277-8
Bhyrappa P., Krishnan V. // Inorg. Chem. 1991. V. 30. № 2. P. 239. https://doi.org/10.1021/ic00002a018
Чижова Н.В., Иванова Ю.Б., Мамардашвили Н.Ж. // Макрогетероциклы. 2018. Т. 11. № 1. С. 85. https://doi.org/10.6060/mhc171265c
Vicente M. da G.H., Smith K.M. // Curr. Org. Synth. 2014. V. 11. № 1. P. 3. https://doi.org/10.2174/15701794113106660083
Chizhova N.V., Konakova A.V., Mal’tseva O.V. et al. // Russ. J. Org. Chem. 2017. V. 53. № 7. P. 1094. https://doi.org/10.1134/S1070363217070222
Spyroulias G.A.,Despotopoulos A.P., Raptopoulou C.P. et al. // Chem. Commun. 1997. № 8. P. 783. https://doi.org/10.1039/a608013h
Spyroulias G.A., Despotopoulos A.P., Raptopoulou C.P. et al. // Inorg. Chem. 2002. V. 41. № 10. P. 2648. https://doi.org/10.1021/ic000738h
Rumyantseva V.D., Aksenova E.A., Ponamoreva N.O. et al. // Russ. J. Bioorg. Chem. 2000. V. 26. № 6. P. 423. https://doi.org/10.1007/BF02758672
Gonsalves A.M., Johnstone R.A.W., Pereira M.M. et al. // Tetrahedron Lett. 1991. V. 32. № 10. P. 1355. https://doi.org/10.1016/S0040-4039(00)79666-3
Chorghade M.S., Dolphin D., Dupré D. et al. // Syhthesis. 1996. V. 11. P. 1320. https://doi.org/10.1055/s-1996-4401
Ivanova Yu.B., Chizhova N.V., Shumilova I.A. et al. // Russ. J. Org. Chem. 2020. V. 56. № 6. P. 1054. https://doi.org/10.1134/S1070428020060147
Lee N., Petrenko T., Bergmann U. et al. // J. Am. Chem. Soc. 2010. V. 132. № 28. P. 9715. https://doi.org/10.1021/ja101281e
Becke A.D. // Phys. Rev. A. 1988. V. 38. № 6. P. 3098. https://doi.org/10.1103/PhysRevA.38.3098
Stephens P.J., Devlin F.J., Chabalowski C.F. et al. // J. Phys. Chem. 1994. V. 98. № 45. P. 11623. https://doi.org/10.1021/j100096a001
Lee C., Yang W., Parr R.G. // Phys. Rev. B. 1988. V. 37. № 2. P. 785. https://doi.org/10.1103/PhysRevB.37.785
Vosko S.H., Wilk L., Nusair M. // Can. J. Phys. 1980. V. 58. № 8. P. 1200. https://doi.org/10.1139/p80-159
Weigend F. // Phys. Chem. Chem. Phys. 2006. V. 8. P. 1057. https://doi.org/10.1039/B515623H
Weigend F., Ahlrichs R. // Phys. Chem. Chem. Phys. 2005. V. 7. P. 3297. https://doi.org/10.1039/B508541A
Petersson G.A., Bennett A., Tensfeldt T.G. et al. // J. Chem. Phys. 1988. V. 89. P. 2193. https://doi.org/10.1063/1.455064
Petersson G.A., Al-Laham M.A. // J. Chem. Phys. 1991. V. 94. P. 6081. https://doi.org/10.1063/1.460447
Schmidt M.W., Baldridge K.K., Boatz J.A. et al. // J. Comput. Chem. 1993. V. 14. P. 1347. https://doi.org/10.1002/jcc.540141112
Yang S.I., Seth J., Strachan J.P. et al. // J. Porphyrins Phthalocyanines. 1999. V. 3. № 2. P. 117. https://doi.org/10.1002/(SICI)1099-1409(199902)3:2%3C117::AID-JPP110%3E3.0.CO;2-X
Seybold P.G., Gouterman M. // J. Mol. Spectrosc. 1969. V. 31. № 1–13. P. 1. https://doi.org/10.1016/0022-2852(69)90335-X
Principles of fluorescence spectroscopy / Ed. Lakowicz J.R. New York: Springer, 2006. V. 26. 954 p.
Чижова Н.В., Мамардашвили Г.М., Дмитриева О.А. и др. // Макрогетероциклы. 2019. Т. 12. № 4. С. 364. https://doi.org/10.6060/mhc190556m
Хембрайт P. // Успехи химии. 1977. Т. 46. С. 1207.
Senge M.O. // Chem. Commun. 2006. V. 3. P. 243. https://doi.org/10.1039/B511389J
Valicsek Z., Horváth O. // Microchem. J. 2013. V. 107. P. 47. https://doi.org/10.1016/j.microc.2012.07.002
Bhyrappa P., Sankar M., Varghese B. // Inorg. Chem. 2006. V. 45. № 10. P. 4136. https://doi.org/10.1021/ic052035b
Kumar P.K., Bhyrappa P., Varghese B. // Tetrahedron Lett. 2003. V. 44. P. 4849. https://doi.org/10.1016/S004039(03)01143-2
Pomarico G., Sabuzi F., Conte V. et al. // New J. Chem. 2019. V. 43. № 45. P. 17774. https://doi.org/10.1039/c9nj02503k
De Souza T.G.B., Vivas M.G., Mendonca C.R. et al. // J. Porphyrins Phthalocyanines. 2016. V. 20. P. 282. https://doi.org/10.1142/S1088424616500048
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии