

Журнал неорганической химии, 2022, T. 67, № 3, стр. 360-372
Генерация и спектральные свойства окисленных форм порфириновых комплексов иридия и рения
Е. Ю. Тюляева a, *, Н. Г. Бичан a, Т. Н. Ломова a
a Институт химии растворов им. Г.А. Крестова РАН
153045 Иваново, ул. Академическая, 1, Россия
* E-mail: teu@isc-ras.ru
Поступила в редакцию 07.09.2021
После доработки 14.09.2021
Принята к публикации 20.09.2021
- EDN: XWNEPU
- DOI: 10.31857/S0044457X2203014X
Аннотация
Представлен краткий обзор результатов исследований в области реакционной активности комплексов иридия и рения в различных степенях окисления с замещенными, расширенными, N-fused, N-confused порфиринами, корролами, а также гетероатомными макроциклами в условиях химического и электрохимического окисления. Проанализированы характерные спектральные особенности форм, а также факторы, определяющие стабилизацию того или иного заряда иона-комплексообразователя и место локализации заряда при окислении соединений: ароматическая часть молекулы, центральный атом металла или аксиальный лиганд. Иридий и рений в степенях окисления от +1 до +7 образуют с порфиринами и их аналогами устойчивые комплексы, которые представляют особый интерес благодаря необычным свойствам и потенциалу применения в различных областях науки и техники, включая современные материалы и катализаторы. Показано, что высокая реакционная способность в окислительно-восстановительных процессах с реакционным центром на макроцикле или центральном ионе представляет главную особенность порфириновых комплексов иридия и рения. Высокая устойчивость не только молекулярных, но и заряженных радикальных форм комплексов вызывает огромный интерес для дальнейшего прогресса в изучении механизмов их химических и фотофизических превращений, необходимого для развития прикладной химии порфириновых комплексов иридия, рения и их аналогов.
ВВЕДЕНИЕ
Иридий и рений относятся к одним из самых редких элементов из-за их ничтожно малого содержания в природе. Однако разнообразие формальных степеней окисления (от +1 до +5 у Ir, от +1 до +7 у Re) в сочетании со способностью ароматических тетрапиррольных лигандов стабилизировать необычные степени окисления металлов обеспечивает порфириновым комплексам иридия (IrP) и рения (ReP) уникальные свойства, перспективные в радиофармакологии при диагностике и лечении заболеваний и в разработке и использовании люминесцентных материалов и катализаторов [1–10]. Благодаря сочетанию богатых окислительно-восстановительных свойств и координационной ненасыщенности, тетрапиррольные комплексы иридия и рения рассматриваются в качестве подходящих синтетических моделей для изучения одно- и двухэлектронных окислительных процессов при переносе кислорода в каталитических циклах природных ферментов наряду с такими комплексами, как порфирины железа и марганца [11–14]. Обсуждается также перспектива применения IrP и ReP в качестве катализаторов реакций органических соединений [15–30], окисления воды [31, 32] и восстановления кислорода [33]. Опубликованы данные по использованию таких комплексов в качестве доноров электронов в системах с фотоиндуцированным электронным транспортом, где реализуются их короткоживущие катион-радикальные формы [34–37], и в качестве биосовместимых и биоразлагаемых кислородных зондов [38].
ТЕОРЕТИЧЕСКАЯ ЧАСТЬ
Предметом настоящей работы является обобщение и анализ имеющихся в литературе и собственных данных по генерации и свойствам окисленных, в том числе радикальных, форм комплексов иридия и рения в различных степенях окисления с замещенными, расширенными, N-fused, N-confused порфиринами, корролами, а также гетероатомными макроциклами. Специфика свойств окисленных и радикальных форм комплексов IrP и ReP диктует применимость тех или иных физических и физико-химических методов в их описании. Электронные спектры поглощения (ЭСП) окисленных форм IrP и ReP однотипны по причинам, описанным в следующем разделе, и весьма характерны (рис. 1).
Рис. 1.
Электронные спектры поглощения окисленных форм металлопорфиринов: 1 – ClMnTPP•+ в 2.52 M H2O2; 2 – (HSO4)RhTPP•+ в 17.41 М H2SO4; 3 – O=Re(HSO4)MPOEP•+ в 17.77 М H2SO4; 4 – (CH3COO)(CH3COOH)IrTPP•+в 100%-ной AcOH (TPP и OEP – дианионы 5,10,15,20-тетрафенил21H,23H-порфирина и 2,3,7,8,12,13,17,18-октаэтил-21H,23H-порфирина).
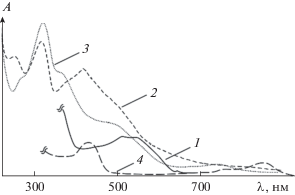
Метод ЭПР информативен лишь при определенном распределении спина неспаренного электрона по атомам макроцикла и неприменим при слабой связи между радикальным и металлическим центрами [39]. Поэтому метод УФ-видимой спектроскопии был признан лучшим на ранних стадиях исследований для характеристики радикальных форм металлопорфиринов [40, 41]. Несмотря на то, что электронный спектр поглощения до сих пор остается наиболее простым методом идентификации π-катион-радикалов, в настоящее время совместное использование методов РСА, ЭПР, ЯМР-спектроскопии и квантово-химических расчетов позволяет охарактеризовать распределение спиновой плотности неспаренного электрона в более сложных системах. Так, в работах [42–44] на основании результатов исследования методом ЭПР и DFT (B3LYP/6-311G(d,p)) расчетов определено распределение спина неспаренного электрона по всем атомам диазапорфиринового кольца в комплексах с 3d-металлами. Кроме того, отмечается наличие ферромагнитного перекрывания между спинами парамагнитного центрального иона (CuII) и π-радикала [45]. Доступность количественной энергетической характеристики граничных МО π-катион-радикалов CoIIP•+ и MnIIIP•+ в составе фотоиндуцированных короткоживущих радикальных солей с фуллеренсодержащим анион-радикалом продемонстрирована с помощью квантово-химических методов DFT и TD-DFT (B3LYP* + D3BJ/6-31G уровень) и фемтосекундной переходной спектроскопии поглощения [46].
Образование окисленных форм MP отражается на их ИК-спектрах. Для одноэлектронно-окисленных по макроциклу форм фиксируются полосы, относящиеся к колебаниям связей Cα–Cβ и Cα–N в пиррольных кольцах в области 1300–1600 см–1. Самыми заметными из них, подтверждающими тип порфирина, а не состояние симметрии, являются полосы при ~1280 см–1, отвечающие π-катион-радикальным комплексам H2TPP, и при ~1550 см–1, характерные для MOEP•+ [47]. Однако присутствие и природа аксиальных и периферийных заместителей (в частности, в мезо-фенилах) значительно влияют на величину частотного сдвига указанных типов колебаний в окисленных формах комплексов [48]. Переход к двухэлектронно-окисленным дикатион-радикальным формам димерных комплексов характеризуется увеличением интенсивности сигналов с небольшим их смещением [49, 50].
Для комплексов низкозарядных катионов металлов (Zn, Cu, Ni, Co, Mg, Fe, Ru) с порфиринами выполнено исследование структуры π-катион-радикалов (деформация, длина связей) методом рентгеноструктурного анализа и расчетным DFT-методом [51–55]. Согласно расчетам, при образовании окисленных по ароматической части тетрапиррольных молекул (порфиринов и фталоцианинов) имеет место изменение длин связей кольца по сравнению с нейтральной молекулой. При образовании катион-радикальных форм увеличивается разница в длине связей, чередующихся в порфириновом кольце, и величине углов между ними, в отличие от делокализованных связей в нейтральной молекуле. В табл. 1 на примере ZnTPP и его замещенных по фенильным кольцам аналогов показано изменение величины двугранных углов (ψ, град) между плоскостями порфиринового кольца и мезо-фенильных заместителей в зависимости от состояния окисления молекулы. Структурные изменения способствуют переносу электрона между нейтральной, катион-радикальной и дикатионной формами.
Таблица 1.
Полученные расчетным методом DFT (B3LYP 6-31G(d,p)) значения двугранных углов (ψ, град) между плоскостями порфиринового кольца и мезо-фенильных заместителей в различных состояниях окисления [51]
| Комплекс | Ψ* | Ψ** |
|---|---|---|
| ZnTPP | 66.01 | 65.43 |
| ZnT(2′-thienyl)P | 65.26 | 63.57 |
| ZnT(3′-furyl)P | 58.37 | 57.13 |
| ZnT(3′-thienyl)P | 61.86 | 61.18 |
| ZnTPP•+ | 55.86 | 56.00 |
| ZnT(2′-thienyl)P•+ | 44.78 | 44.74 |
| ZnT(3′-furyl)P•+ | 44.52 | 44.20 |
| ZnT(3′-thienyl)P•+ | 49.12 | 49.04 |
| ZnTPP2+ | 45.36 | 55.09 |
| ZnT(2′-thienyl)P2+ | 34.46 | 34.46 |
| ZnT(3′-furyl)P2+ | 35.53 | 34.61 |
| ZnT(3′-thienyl)P2+ | 39.24 | 39.32 |
* Оптимизировано для изолированных молекул и радикалов. ** Оптимизировано в дихлорметане. ZnTPP – (5,10,15,20-тетрафенил-21Н,23Н-порфинато)цинк(II), ZnT(2′-thienyl)P – (5,10,15,20-(2'-тиенил)-21Н,23Н-порфинато)цинк(II), ZnT(3′-furyl)P – (5,10,15,20-(3'-фурил)-21Н,23Н-порфинато)цинк(II), ZnT(3′-thienyl)P – (5,10,15,20-(3'-тиенил)-21Н,23Н-порфинато)цинк(II).
Однако следует учитывать, что существует различие между данными теоретических расчетов и рентгеноструктурного анализа, отражающее проблему эффекта Яна–Теллера, согласно которому взаимосвязь вырожденных электронных состояний с искажениями приводит к снятию вырождения и понижению симметрии. Экспериментальные данные демонстрируют важность эффектов окружения, которые не учитываются и не обнаруживаются в некоторых расчетах [52].
Методом циклической вольтамперометрии определены редокс-потенциалы CoIIPc•+/CoIIPc и MnIIIPc•+/MnIIIPc (Pc – дианион фталоцианина) в 0.1 M (н-Bu)4NClO4 в дихлорметане, равные соответственно 0.94 и 1.25 В, когда комплексы входили в состав донорно-акцепторной пары с аксиально координированным 1′-N-метил-2′-(1H-имидазол-1-ил)-фенилпирролидино[3′,4′:1,2] [60]фуллереном (ImC60) [46]. Здесь же продемонстрирован эффект тушения флуоресценции MnIIIPc (λexs = 365 нм) в составе радикальной соли MnIIIPc•+/${\text{ImC}}_{{{\text{60}}}}^{{\centerdot - }}$.
Детальное изучение влияния локализации заряда, степени окисления и спинового состояния центрального металла в высокоокисленных формах на спектры 1H ЯМР и ЭПР представлено в литературе для порфириновых комплексов железа [55].
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Для прогнозирования каталитических свойств соединений и установления механизма катализа на металлопорфиринах (MP) важно знать характеристики свойств окисленных и восстановленных форм катализатора – интермедиата редокс-процессов. Устойчивость MP к окислению определяется в основном электронной природой катиона-комплексообразователя, макроцикла и его функциональных заместителей и лигандов в аксиальном положении. Многие окисленные катионные и катион-радикальные формы IrP и ReP наблюдаются в химических, электрохимических или фотофизических превращениях как сравнительно устойчивые или короткоживущие интермедиаты. Удаление электрона при электрохимическом и химическом воздействии на МР может проходить по центральному атому или по ароматической части молекулы и сопровождаться либо повышением степени окисления катиона-комплексообразователя в составе комплекса, либо образованием π-катион-радикальной формы молекул MP•+ с локализацией положительного заряда на макроцикле. В зависимости от распределения спина неспаренного электрона различают два типа π-катион-радикалов металлопорфиринов: 2A1u радикал с плотностью спина, сосредоточеной на атомах Смезо метиновых мостиков и внутрициклических атомах N, и 2A2u радикал, характеризующийся небольшой плотностью спина в мезо-положениях [40, 41]. Поскольку электронные свойства этих форм близки, симметрию состояния можно изменить, заменив аксиальный лиганд в составе комплекса. Из-за близости энергий ВЗМО электронные спектры поглощения π-катион-радикалов обоих типов аналогичны, что и является причиной характеристичности ЭСП π-катион радикалов металлопорфиринов. По результатам ZINDO-расчета молекул, катионов и анионов ZnPc и ZnTPP с использованием 16-орбитальной модели и данным магнитного кругового дихроизма, опубликованным в основополагающей работе [56], как в случае одноэлектронного восстановления ZnTPP, так и в случае одноэлектронного окисления ZnPc имеет место снятие вырождения двух граничных орбиталей (ВЗМО), характерное для исходных молекул. Это катастрофически изменяет ЭСП упомянутых соединений. В спектре ZnTPP появляются полосы поглощения с λmax = 538 и 910 нм π* → π*-переходов в пределах вакантных МО. Одновременно Q-полосы (0,0) и (0,1), расположенные при 605 и 560 нм в спектре исходного ZnTPP, смещаются батохромно на 145 и 168 нм. При удалении одного электрона (переход от ZnPc к ZnPc+) аналогичные новые полосы принадлежат π → π-переходам внутри заполненных оболочек. Наряду с ними проявляются смещенные батохромно Q-полосы при 958 и 925 нм, полоса второго π → π*-перехода при 300–450 нм, полосы B1, B2 и π → π*. Общим результатом становится резкое увеличение поглощения на границе УФ и видимой части спектра, появление новых полос в ближней ИК-области и сравнительное падение адсорбции на месте исходных Q-полос, что и наблюдается в приведенных на рис. 1 спектрах других одноэлектронно-окисленных металлопорфиринов. Наличие или отсутствие полос поглощения MP в области 550−700 нм является основой интерпретации имеющихся экспериментальных данных для идентификации окисленных по макроциклу соединений.
Генерирование окисленных форм тетрапиррольных соединений иридия и рения в условиях электрохимических экспериментов представлено в литературе, однако идентификация процессов окисления с указанием места локализации положительного заряда выполнена не в каждой работе. Для порфириновых комплексов Ir3+ с различными аксиальными лигандами и мезо-фенильными и/или β-алкильными заместителями макроцикла величины первого потенциала окисления, отнесенного к процессу образования π-катион-радикальной формы, определены в интервале 0.51−1.31 В [2, 57]. Образование дважды окисленных форм происходит при 1.15−1.45 В, причем для комплексов с H2TPP наличие этого процесса зависит от природы аксиальных лигандов. Исследования комплексов иридия(III) с близкими порфириновыми аналогами три(пентафтор- и три(пара-X-фенил)корролами (Х = CF3, H, Me, OMe (рис. 2, структура 1)) свидетельствуют об окислении по центральному катиону до Ir4+ при Е = 0.20−1.69 В и возможности второго окисления как по макроциклу, так и по катиону иридия при Е = 0.89−1.18 В [3, 58].
Рис. 2.
Химическое строение трифенилкорролов. L = = пиридин (py), триметиламин (tma), изохинолин (isoq), 4-диметиламинопиридин (dmap), 4-пиколиновая кислота (4pa).
Аксиально координированные алкильными группами комплексы иридия Ir(C8H13)OEP и Ir(C8H13)(СО)OEP являются первыми примерами σ-связанных по аксиальной оси металлопорфиринов, которые могут быть обратимо окислены по аксиальному лиганду в среде тетрагидрофурана при 0.68 и 0.80 В соответственно [59]. Окисление аксиальной части молекулы также было обнаружено в реакции (CH2COAr)IrTTP (TTP – дианион 5,10,15,20-тетратолил21H,23H-порфина) с 1-окси-2,2,6,6-тетраметилпиперидинилом, когда предполагаемый интермедиат с локализацией радикала на атоме углерода IrIII(CH2CHR•) был стабилизирован центральным катионом, а результатом реакции являлся комплекс IrII(CH2=CHR) с неспаренным электроном на металле [60].
Биядерный комплекс иридия(I) (TPP)[IrI(CO)3]2 (рис. 3, структура 2) характеризуется двумя окислительными потенциалами: 0.92 и 1.5 В в бензонитриле (PhCN) или CH2Cl2, содержащих 0.2 M перхлорат тетрабутиламмония [61]. При первом (быстром и необратимом) электроокислении по металлу IrI → IrIII с отрывом СО, фиксируемом по исчезновению полос колебаний связей Ir−CO при 2054 и 1979 см−1 в ИК-спектре исходного соединения, образуется [(TPP)IrIII]+${\text{ClO}}_{4}^{ - }$. Дальнейшее удаление электрона происходит с макроцикла при 1.5 В аналогично другим порфириновым комплексам трехзарядного иридия.
Рис. 3.
Химическое строение биядерных комплексов иридия(I) и рения (I) с H2TPP (структура 2) и моноядерного комплекса рения(I) c замещенным H2Pc (структура 3).
В изоструктурных комплексах рения(I) (TPP)[ReI(CO)3]2 (рис. 3, структура 2) и (tBuPc)[ReI(CO)3]2, в отличие от (TPP)[IrI(CO)3]2, при электрохимическом воздействии изменения степени окисления металла не происходит. Наблюдаемые спектральные изменения и изменения величины Еox характерны для процессов образования форм, окисленных по макроциклу: 1.01 (CH2Cl2), 1.16 В (PhCN) для порфиринового [62] и 0.52 В для фталоцианинового [63] комплексов. В то же время моноядерный комплекс (tBuPc)ReI(CO)3 (рис. 3, структура 3) подвергается окислению дважды: при 0.32 и 0.76 В, однако отнесение первого процесса удаления электрона в цитируемой работе не проведено [63].
Окислительные потенциалы замещенных N-confused-порфириновых (NCP) комплексов рения(I) и рения(V), представленных на схеме 1 , изменяются в пределах 0.1−0.6 В в зависимости от степени окисления центрального атома и протяженности π-электронной системы. Значительные батохромные сдвиги в ЭСП химически окисленных форм и меньшие значения потенциалов окисления по сравнению с соответствующими ReNCP свидетельствуют о протекании процесса окисления по макроциклу и включении периферийных заместителей в π-сопряжение, что подтверждается также теоретическими расчетами [64].
Значительно более высокие потенциалы, характеризующие окисление макроцикла, демонстрируют уникальные моно- и дигетерокомплексы Re(CO)3 с тиапорфирином, селенпорфирином и оксапорфирином, в которых одно или два из пиррольных колец порфирина заменены на тиофен, фуран и селенофен соответственно. Для них определены потенциалы: 1.10 и 1.43 В (STPPReI(CO)3) (рис. 4, структура 4); 1.65 В (S2TTPReI(CO)3), 1.15 и 1.74 В (Se2TTPReI(CO)3) (рис. 4, структура 5); 0.88 и 1.33 В (ОTPPReI(CO)3) (рис. 4, структура 6) [65, 66]. Несмотря на сильное искажение молекулы при комплексообразовании с ионом Re+ по сравнению с соответствующим свободным макроциклическим основанием, такие комплексы высокоустойчивы к окислению.
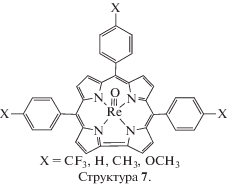
Электрохимическое поведение тетрапиррольных комплексов рения(V) представлено в литературе данными по оксорений(V)триарилкорролам (рис. 5, структура 7) [67]. Результаты исследования редокс-свойств и величины Еох (0.93−1.10 В) свидетельствуют о возможности окисления соединений по макроциклу.
Приведенные данные демонстрируют, что ни одно из этих соединений рения не может быть окислено по центральному атому металла при электрохимическом воздействии в изученных условиях в области потенциалов 0−2 В. Переход Re(I) → Re(V) химическим путем (схема 1 ) возможен для соединения ReI(NCTPP), упомянутого выше [64]. Химическое окисление Re(I) → Re(VII) оказалось успешным в единственном случае. На примере получения триоксокомплекса рения(VII) NFPReO3 (NFP – N-fused порфирин) окислением соответствующего комплекса рения(I) NFPRe(CO)3 с использованием Me3NO ⋅ 2H2O в 1,2-Cl2C6H4 при 140°C в течение 30 мин была представлена впечатляющая способность N-fused порфиринов стабилизировать комплексы металлов в высокой степени окисления за счет координации с тремя атомами азота и сравнительно небольшой координирующей полости (схема 2 ) [68].
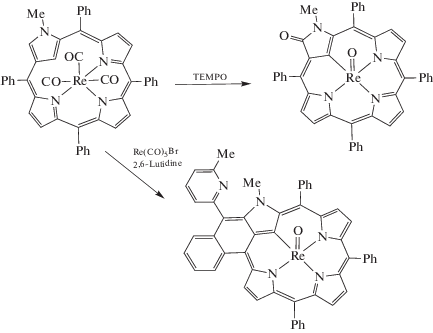
Схема 1 . Химическое окисление ReI(NCTPP).
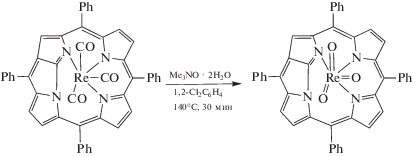
Схема 2 . Химическое окисление ReI(NFTPP).
Попытка получить комплекс рения(VII) из исходного рений(V)коррола с использованием окислителя не увенчалась успехом, подтвердив предварительно проведенные теоретические расчеты [67].
В общем случае реакционная способность порфириновых комплексов рения(V) в растворах в основном представлена процессами обмена аксиальных лигандов [34, 69]. Однако соединения рения(V) с H2TPP, H2OEP и его мезо-фенилзамещенными аналогами оказались в группе соединений, для которых была обнаружена и изучена авторами данной статьи химическая генерация окисленных форм в среде аэрированных кислот HOAc, H2SO4, CF3COOH и в смесях HOAc–H2SO4 (табл. 2, [70–73]). Одноэлектронное окисление протекает вследствие взаимодействия координационного центра с молекулярным O2 в условиях избытка протонов:
Таблица 2.
Окисленные формы некоторых порфириновых комплексов иридия и рения в протонсодержащих растворителях
| Комплекс | Растворитель (C, моль/л) | Форма |
|---|---|---|
| [O=ReTPP]2O | 5–10 H2SO4/HOAc | О=Re(HSO4)TPP•+ |
| O=Re(Cl)OEP | 16.78–18.1 H2SO4 | O=Re(Cl)(O2)OEPа |
| O=Re(OPh)OEP | 16.8–18.1 H2SO4 | O=Re(OPh)(O2)OEPа |
| O=Re(OPh)MPOEP | 16.8–18.2 H2SO4 | O=Re(OPh)(O2)MPOEPа |
| O=Re(Cl)MPOEP | 16.8–18.2 H2SO4 | O=Re(HSO4)MPOEP•+ |
| O=Re(Cl)5,15DPOEP | 16.8–18.1 H2SO4 | O=Re(HSO4)5,15DPOEP•+ |
| O=Re(X)Pб | 16.8–18.2 H2SO4 | O=Re(X)(O2)Pа |
| (Cl)(H2O)IrTPP | 0.043 H2O/HOAc | (OAc)(HOAc)IrTPP•+ |
| 99в CF3COOH | (CF3COO)2IrIVTPP | |
| 16.78–18.09 H2SO4 | (HSO4)2IrIVTPP•+ |
Отнесение интермедиатов и контроль за скоростью реакций окисления MP проводили на основании характеристичности УФ-видимых спектров π-катион-радикальных форм MP. Химическое строение промежуточных и конечных продуктов реакции подтверждали, когда это было возможно, дополнительными исследованиями методами ИК- и одно- и двумерной ЯМР-спектроскопии.
На примере O=ReV(X)P с различным составом координационной сферы по экваториальным и аксиальным направлениям установлено влияние природы лигандов на процессы, протекающие в сернокислых растворах комплексов по общей схеме 3 [70–73].
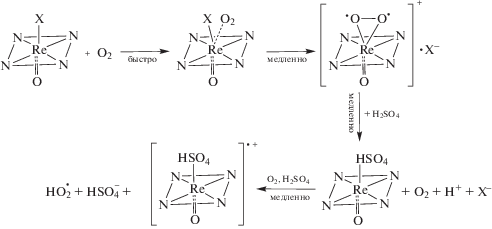
Схема 3 . Химическое окисление O=ReV(X)P в концентрированной H2SO4.
В комплексах, содержащих мезо-фенильные заместители и аксиальные хлорид-ионы в цис-положении к оксолиганду, замещение Cl– на гидросульфат-ионы благоприятствует удалению электрона с π-системы макроциклического лиганда. В случае O=Re(Cl)OEP, O=Re(OPh)OEP, O=Re(OPh)MPOEP, а также комплексов O=Re(Cl), O=Re(OPh) и O=Re(OH) с 5,15-бис(4'-метоксифенил)-3,7,13,17-тетраметил-2,8,12,18-тетраэтилпорфирином реакция останавливается на стадии образования катионного комплекса с аксиально координированным кислородом и внешнесферным ионом (схема 3 , верхняя строка).
Попытка химической генерации π-катион-радикала μ-оксодимерного [O=ReTPP]2O под действием трет-бутилгидропероксида (С = 0.19 моль/л в бензоле), контролируемая ЭСП, приводила к реакции, включающей разрыв µ-оксомостика при координации молекулы гидропероксида с сохранением координационного центра [74]. При этом спектр ни одной из конечных форм, находящихся в равновесии в растворе, не соответствовал спектру окисленной по макроциклу формы.
В условиях избытка CF3COOH в растворе CH2Cl2 гетеропорфирины STTPReI(CO)3, S2TTPReI(CO)3 и Se2TTPReI(CO)3 (структуры 4, 5) не образуют π-катион-радикалов, а лишь подвергаются протонированию по внутрициклическим атомам азота. 1Н ЯМР-титрование соединений с использованием CF3COOH в CDCl3, демонстрирующее низкопольный сдвиг сигналов протонов пирролов, подтверждает протекание процесса [65, 66]. Оксапорфирин рения(V) (структура 6) в аналогичных условиях подвергается диссоциации с образованием протонированной формы макроциклического лиганда [66]. Устойчивость к окислению этих соединений объясняется их высокими окислительными потенциалами, упомянутыми выше.
Реакции аксиального замещения на ионы и молекулы среды протекания реакций, характерные для комплексов рения, предшествуют также генерации высокоокисленных форм порфиринов иридия(III) в средах (табл. 2) с высоким содержанием протонов [75, 76]. Место локализации неспаренного электрона при химическом окислении (Cl)(H2O)IrTPP в кислотах определяется кислотностью среды и природой аксиальных лигандов. В 100%-ной AcOH комплекс медленно образует окисленную по ароматической части форму, в CF3COOH имеет место окисление по центральному катиону металла с образованием комплекса иридия(IV), а в концентрированной H2SO4 последовательно проходят оба эти процесса (схема 4 ).
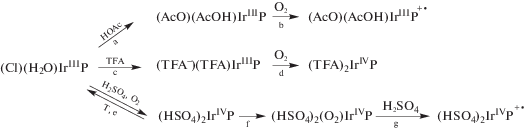
Схема 4 . Трансформации (Cl)(H2O)IrTPP в протонодонорных растворителях.
Координированные молекулы и анионы трифторуксусной кислоты (схема 4 , реакция c), обладая электроноакцепторными свойствами, оказывают цис-влияние на координированный макроцикл и уменьшают π-электронную плотность на его атомах, поэтому окисление комплекса иридия(III) проходит не по макроциклу (схема 4 , реакция d). Смешанная электронодонорно-акцепторная природа гидросульфат-ионов в (HSO4)2IrIVTPP, имеющих как неподеленные электронные пары, так и доступные по энергии вакантные d-орбитали, в сочетании с высокой кислотностью серной кислоты способствует прохождению двух последовательных реакций окисления: по центральному атому и по макроциклу (схема 4 , реакции e, g). Возможность перехода IrIII → IrIV подтверждается электрохимическими исследованиями: окислительно-восстановительная пара [(TPP)Ir]+/[(TPP)Ir]2+ имеет относительно низкое значение E1/2 ~ 1.4 В [2].
Окисление по центральному атому металла отмечено и в ходе весьма редкой для порфириновых комплексов реакции окислительного присоединения AcOH к комплексу иридия(I) с молекулярным порфирином [IrCl(H2О)2]2Н2ТРР (SAT-комплекс) (схема 5 ) [77].
Электронодонорно-акцепторная природа молекул окислителей, вероятно, обусловливает также различие продуктов взаимодействия комплекса (Cl)(CO)IrIIITTP с некоторыми окислителями [78]. Так, реакция с пиридин-N-оксидом и триметиламин-N-оксидом ведет к координации молекул окислителя в аксиальное положение комплексов с замещением хлорид-иона без образования продуктов окисления. Пероксид водорода Н2О2 и мета-хлорнадбензойная кислота не вступают в реакцию с исследуемым соединением, о чем свидетельствуют данные используемых в работе методов 1Н ЯМР и масс-спетрометрии. Лишь в ходе реакции с иодозилбензолом (PhIO)n наблюдалось образование окисленной парамагнитной формы комплекса, предположительно, иридия(V) с потерей аксиального СО. Однако убедительных доказательств образования и структуры высоковалентной формы или интермедиата в исследовании не представлено.
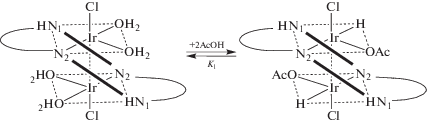
Схема 5 . Реакция окислительного присоединения AcOH к [IrCl(H2О)2]2Н2ТРР.
Для аксиального σ-комплекса Ir(C8H13)TPP продемонстрирована [79] возможность как химического окисления одноэлектронным окислителем [(4-BrC6H4)3N](SbCl6)) по алкильному заместителю, так и стабилизации центрального катиона в степени окисления >3+ благодаря прохождению реакций аксиального присоединения и замещения лигандов PPh3, Cl−, (LOEt)Ru(N)Cl2 (LOEt = (η5-C5H5)Co{P(O)${{({\text{OEt}})}_{2}}\} _{3}^{ - }$). Выводы в работе были подтверждены результатами электрохимических измерений и метода РСА.
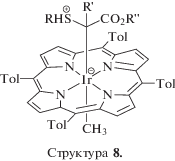
Рассмотренные выше свойства окисленных форм IrP и ReP определяют перспективы практического применения порфириновых комплексов этих металлов. Высокая стабильность комплексов и в окисленном, и в восстановленном состоянии открывает возможность их применения не только в катализе, но и при создании переключателей окислительно-восстановительного потенциала. Богатая редокс-химия приведет в перспективе к разработке индикаторов оптических сенсоров на кислород. Уже описан пример ячеек для расщепления воды с фотоанодом, основанным на порфириновом комплексе IrO2 · nH2O [80]. “Работа” устойчивых редокс-интермедиатов IrP и ReP в катализируемых ими процессах отмечается во многих исследованиях. Так, при введении карбенов, полученных из этил-, метил-, метилфенил- и метил-(п-толил)диазоацетата, в S–H-связи ароматических и алифатических тиолов, катализируемом (5,10,15,20-тетратолилпорфиринато)метилиридием(III), при температуре окружающей среды таким интермедиатом служит нейтральная форма катализатора с избыточным электроном на иридии (рис. 6, структура 8) [21].
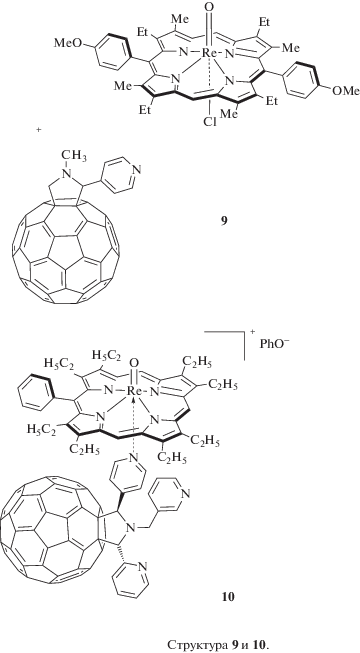
С учетом высокой реакционной способности по аксиальной оси можно предполагать успешное использование электронодонорных свойств IrP и ReP в координационных донорно-акцепторных парах со свойством фотоиндуцированного электронного транспорта. Две такие системы (рис. 7, структуры 9, 10) уже получены и охарактеризованы в основном состоянии физико-химическими методами в наших работах [34, 81].
ЗАКЛЮЧЕНИЕ
Краткий обзор работ последних десятилетий по комплексам иридия и рения с порфиринами и их аналогами показал, что основная проблема на пути продвижения этой темы – недостаточность описания конкретных промежуточных форм комплексов в химических и фотофизических превращениях. Координационные соединения этих редких металлов с замещенными, расширенными, N-fused, N-confused и модифицированными порфиринами, корролами, а также гетероатомными макроциклами являются отличными системами для изучения свойств и реакционной способности по различным связям. Тот факт, что эти комплексы формируют особую группу с замечательными свойствами, очевиден из приведенных данных. Они одновременно проявляют высокую устойчивость по связям металл–азот и высокую реакционную способность в реакциях замещения аксиальных лигандов, что наряду с богатой редокс-химией делает их многообещающими кандидатами для применения в катализе. Комплексы с корролами структурно охарактеризованы и исследованы в качестве фотосенсибилизаторов в фотодинамической терапии рака. Несмотря на то, что IrP и ReP могут выступать в качестве отличных электронных доноров, практически совсем не изученной остается область фотоиндуцированного формирования радикальных солей в составе донорно-акцепторных пар для органических солнечных ячеек.
Для изучения окисленных форм IrP и ReP необходимо их получать в условиях, при которых они являются долгоживущими. Поэтому способы генерации этих форм должны получить дальнейшее развитие. Данные о существовании и поведении окисленных форм в различных средах и в различных реакциях позволят моделировать превращения с каталитическим участием IrP и ReP, расшифровать механизмы таких превращений, а также расширить перечень изучаемых технических процессов и биохимических реакций. Надеемся, что результаты настоящей работы помогут лучше понимать основы химии окислительно-восстановительных процессов с участием макроциклических комплексов иридия и рения и поспособствуют разработке новых комплексов для потенциального применения в междисциплинарных областях и каталитических исследованиях в будущем.
Список литературы
Koren K., Dmitriev R.I., Borisov S.M. et al. // ChemBioChem. 2012. V. 13. P. 1184. https://doi.org/10.1002/cbic.201200083
Lam T.L., Ka Chung T., Yang C. et al. // Chem. Sci. 2019. V. 10. P. 293. https://doi.org/10.1039/c8sc02920b
Thomassen I.K., McCormick-McPherson L.J., Borisov S.M. et al. // Scientific Reports. 2020. V. 10. Art. 7551.https://doi.org/10.1038/s41598-020-64389-3
Naoda K., Osuka A. // J. Porphyrins and Phthalocyanines. 2014. V. 18. P. 652. https://doi.org/10.1142/s1088424614500382
Alemayehu A.B., Vazquez-Lima H., Teat S.J. et al. // ChemistryOpen. 2019. V. 8. P. 1298. https://doi.org/10.1002/open.201900271
Borisov S.M., Einrem R.F., Alemayehu A.B. et al. // Photochem. Photobiol. Sci. 2019. https://doi.org/10.1039/c8pp00473k
Majumder S., Borah B.P., Bhuyan J. // Dalton Transactions. 2020. https://doi.org/10.1039/d0dt00813c
Xie J., Liang C., Luo S. et al. // ACS Appl. Mater. Interfaces. 2021. V. 13. P. 27934. https://doi.org/10.1021/acsami.1c06381
Zhang L.P., Geng Y., Li L.J. et al. // Chem. Sci. 2021. V. 12. P. 5918. https://doi.org/10.1039/d1sc00126d
Majumder S., Borah B.P., Bhuyan J. // Dalton Trans. 2020. V. 49. P. 8419. https://doi.org/10.1039/d0dt00813c
Huang X., Groves J.T. // Chem. Rev. 2018. V. 118. P. 2491. https://doi.org/10.1021/acs.chemrev.7b00373
Machan C.W. // ACS Catal. 2020. V. 10. P. 2640. https://doi.org/10.1021/acscatal.9b04477
Passard G., Dogutan D.K., Qiu M. et al. // ACS Catal. 2018 V. 8. P. 8671. https://doi.org/10.1021/acscatal.8b01944
Lomova T., Tsaplev Y., Klyueva M., Ovchenkova E. // J. Organomet. Chem. 2021. V. 945. P. 121880. https://doi.org/10.1016/j.jorganchem.2021.121880
Hartwig J.F., Key H.M., Dydio P. et al. // U.S. Patent, WO 2017/066562 A2, 2017.
Wang J.-C., Xu Z.-J., Guo Z. et al. // Chem. Commun. 2012. V. 48. P. 4299. https://doi.org/10.1039/C2CC30441D
Anding B.J., Ellern A., Woo L.K. // Organometallics. 2012. V. 31. P. 3628. https://doi.org/10.1021/om300135f
Anding B.J., Woo L.K. // Organometallics. 2013. V. 32. P. 2599. https://doi.org/10.1021/om400098v
Dydio P., Key H.M., Hayashi H. et al. // J. Am. Chem. Soc. 2017. V. 139. P. 1750. https://doi.org/10.1021/jacs.6b11410
Wang Y., Cui H., Wei Z.-W. et al. // Chem. Sci. 2017. V. 8. P. 775. https://doi.org/10.1039/c6sc03288e
Dairo T.O., Woo L.K. // Organometallics. 2017. V. 36. P. 927. https://doi.org/10.1021/acs.organomet.6b00947
Yang W., Zhang H., Li L. et al. // Organometallics. 2016. V. 35. P. 3295. https://doi.org/10.1021/acs.organomet.6b00490
Shum W.P., Kesling H.S. // U.S. Patent. 5103027. 1992.
Buchler J., Schmidt M., Prascher G. // U.S. Patent. 4973718.1990.
Buchler J., Kruppa S., Schmidt M., Prascher G. // U.S. Patent. 4987226. 1991.
Zaragoza J.P.T., Siegler M.A., Goldberg D.P. // Chem. Commun. 2016. V. 52. P. 167. https://doi.org/10.1039/C5CC07956J
Toganoh M., Fujino K., Ikeda S., Furuta H. // Tetrahedron Lett. 2008 V. 49. P. 1488. https://doi.org/10.1016/j.tetlet.2007.12.117
Yamamoto T., Toganoh M., Furuta H. // Dalton Trans. 2012. V. 41. P. 9154. https://doi.org/10.1039/c2dt30885a
Wang H.X., Wu K., Che C.M. // Synlett. 2021. V. 32. P. 249. https://doi.org/10.1055/s-0040-1707221
Bian Y.J., Qu X.Y., Chan K.S. // Organometallics. 2020. V. 39. P. 1376. https://doi.org/10.1021/acs.organomet.0c00100
Manas S., Armando J.L.P., José Armando L. da Silva // Coord. Chem. Rev. 2014. V. 439. P. 213911. https://doi.org/10.1016/j.ccr.2021.213911
Sorokin A.B. // Coord. Chem. Rev. 2019. V. 389. P. 141. https://doi.org/10.1016/j.ccr.2019.03.016
Xiao M.L., Zhu J.B., Li G.R. et al. // Angew. Chem. Int. Ed. 2019. V. 58. P. 9640. https://doi.org/10.1002/anie.201905241
Бичан Н.Г., Тюляева Е.Ю., Ломова Т.Н., Семейкин А.С. // Журн. орг. химии. 2014. Т. 50. № 9. С. 1376. [Bichan N.G., Tyulyaeva E.Yu., Lomova T.N., Semeikin A.S. // Russ. J. Org. Chem. 2014. V. 50. P. 1361. https://doi.org/10.1134/S1070428014090218]
Tritton D.N., Bodedla G.B., Tang G.L. et al. // J. Mater. Chem. A. 2020. V. 8. P. 3005. https://doi.org/10.1039/c9ta12492f
Lomova T.N. // Appl. Organomet. Chem. 2021. Art. e6254. https://doi.org/10.1002/aoc.6254
Tański T., Jarka P., Szindler M. et al. // Appl. Surf. Sci. 2019. V. 491. P. 807. https://doi.org/10.1016/j.apsusc.2019.04.274
Castro M.C.R., Ben Sedrine N., Monteiro T., Machado A.V. // Spectrochim. Acta, Part A: Molecular and Biomolecular Spectroscopy. 2020. V. 235. P. 118309. https://doi.org/10.1016/j.saa.2020.118309
Morishima I., Takamuki Y., Shiro Y. // J. Am. Chem. Soc. 1984. V. 106. P. 7666. https://doi.org/10.1021/ja00337a002
Dolphin D., Forman A., Borg D.C. et al. // Proc. Natl. Acad. Sci. USA. 1971. V. 68. P. 614.
Carnieri N., Harriman A. // Inorg. Chim. Acta. 1982. V. 62. P. 103. https://doi.org/10.1016/S0020-1693(00)88485-6
Satoh T., Minoura M., Nakano H. et al. // Angew. Chem. Int. Ed. 2016. V. 55. P. 2235. https://doi.org/10.1002/anie.201510734
Sudoh K., Satoh T., Amaya T., et al. // Chem. Eur. J. 2017. V. 23. P. 16364. https://doi.org/10.1002/chem.201703664
Mutoh M., Sudoh K., Furukawa K. et al. // Asian J. Org. Chem. 2019. V. 8. P. 352. https://doi.org/10.1002/ajoc.201900085
Matano Y. // 11th International Conference on Porphyrins and Phthalocyanines. Buffalo. USA. 2021. Book of Abstacts. Society of Porphyrins & Phthalocyanines. P. 145.
Ovchenkova E.N., Bichan N.G., Tsaturyan A.A. et al. // J. Phys. Chem. C. 2020. V. 124. P. 4010. https://doi.org/10.1021/acs.jpcc.9b11661
Shimomura E.T., Phillippi M.A., Goff H.M. et al. // J. Am. Chem. Soc. 1981. V. 103. P. 6778. https://doi.org/10.1021/ja00412a055
El-Attar M.A., Xu N., Awasabisah D. et al. // Polyhedron. 2012. V. 40. P. 105. https://doi.org/10.1016/j.poly.2012.03.034
Dey S., Sil D., Pandit Y.A., Rath S.P. // Inorg. Chem. 2016. V. 55. P. 3229. https://doi.org/10.1021/acs.inorgchem.5b02065
Pandit Y.A., Shah S.J., Rath S.P. // Z. Anorg. Allg. Chem. 2018. V. 644. P. 856. https://doi.org/10.1002/zaac.201800247
Tran T.T.H., Chang Y.-R., Hoang T.K.A. et al. // J. Phys. Chem. A. 2016. V. 120. P. 5504. https://doi.org/10.1021/acs.jpca.6b03538
Scheidt W.R., Cheng B., Reddy K.V., Brancato K.E. // J. Porphyrins Phthalocyanines. 2017. V. 21. P. 273. https://doi.org/10.1142/S1088424617500080
Nemykin V.N., Dudkin S.V., Fathi-Rasekh M. et al. // Inorg. Chem. 2015. V. 54. P. 10711. https://doi.org/10.1021/acs.inorgchem.5b01614
Nevonen D.E., Ferch L.S., Chernii V.Y. et al. // J. Porphyrins Phthalocyanines. 2020. V. 24. P. 894. https://doi.org/10.1142/s1088424619502043
Walker F.A. // Inorg. Chem. 2003. V. 42. P. 4526. https://doi.org/10.1021/ic026245p
Mack J., Stillman M.J. // J. Porphyrins Phthalocyanines. 2001. V. 5. P. 67. https://doi.org/10.1002/1099-1409(200101)5:1<67::AID-JPP300>3.0.CO;2-3
Swistak C., Cornillon J.L., Anderson J.E., Kadish K.M. // Organometallics. 1987. V. 6. P. 2146. https://doi.org/10.1021/om00153a020
Palmer J.H., Brock-Nannestad T., Mahammed A. et al. // Angew. Chem. Int. Ed. 2011. V. 50. P. 9433. https://doi.org/10.1002/anie.201102913
Cornillon J.-L., Anderson J.E., Swistak C., Kadish K.M. // J. Am. Chem. Soc. 1986. V. 108. P. 7633. https://doi.org/10.1021/ja00284a030
Yeung S.K., Chan K.S. // Organometallics. 2005. V. 24. P. 6426. https://doi.org/10.1021/om050661a
Kadish K.M., Deng Y.J., Yao C.-L., Anderson J.E. // Organometallics. 1988. V. 7. P. 1979. https://doi.org/10.1021/om00099a012
Kadish K.M., Schaeper D., Bottomley L.A. et al. // J. Inorg. Nucl. Chem. 1980. V. 42. P. 469. https://doi.org/10.1016/0022-1902(80)80030-3
Murata K., Koike Y., Ishii K. // Chem. Commun. 2020. V. 56. P. 13760. https://doi.org/10.1039/d0cc04625f
Yamamoto T., Toganoh M., Mori S. et al. // Chemical Science. 2012. V. 3. P. 3241. https://doi.org/10.1039/c2sc20708g
Kaur T., Lee W.-Z., Ravikanth M. // Inorg. Chem. 2016. V. 55. P. 5305. https://doi.org/10.1021/acs.inorgchem.6b00214
Ghosh A., Ravikanth M. // Inorg. Chem. 2012. V. 51. P. 6700. https://doi.org/10.1021/ic300344g
Einrem R.F., Gagnon K.J., Alemayehu A.B., Ghosh A. // Chem. Eur. J. 2016. V. 22. P. 517. https://doi.org/10.1002/chem.201504307
Toganoh M., Ikeda S., Furuta H. // Chem. Commun. 2005. P. 4589. https://doi.org/10.1039/b508208k
Buchler J.W., Kruppa S.B. // Z. Naturforsch. 1990. V. 45. P. 518.
Тюляева Е.Ю., Бичан Н.Г., Ломова Т.Н. // Журн. неорган. химии. 2013. Т. 58. № 11. С. 11522. [Tyulyaeva E.Yu., Bichan N.G., Lomova T.N. // Russ. J. Inorg. Chem. 2013. V. 58. С. 1366. https://doi.org/10.1134/S0036023613110223]
Бичан Н.Г., Тюляева Е.Ю., Ломова Т.Н. // Журн. неорган. химии. 2014. Т. 59. С. 1692. [Bichan N.G., Tyulyaeva E.Yu., Lomova T.N. // Russ. J. Inorg. Chem. 2014. V. 59. P. 1445. https://doi.org/10.1134/S0036023614120079]https://doi.org/10.7868/S0044457X14120071
Бичан Н.Г., Тюляева Е.Ю., Ломова Т.Н. // Журн. физ. химии. 2014. Т. 88. С. 1528. [Bichan N.G., Tyulyaeva E.Yu., Lomova T.N. // Russ. J. Phys. Chem. 2014. V. 88. P. 1719. https://doi.org/10.1134/S0036024414100045]https://doi.org/10.7868/S0044453714100057
Бичан Н.Г., Тюляева Е.Ю., Ломова Т.Н., Семейкин А.С. // Журн. неорган. химии. 2017. Т. 62. № 12. С. 1585. [Bichan N.G., Tyulyaeva E.Yu., Lomova T.N., Semeikin A.S. // Russ. J. Inorg. Chem. 2017. V. 62. P. 1576. https://doi.org/10.1134/S0036023617120208]https://doi.org/10.7868/S0044457X17120054
Симонова О.Р., Зайцева С.В., Бичан Н.Г., Тюляева Е.Ю. // Тез докл. XII Всерос. конф. с междунар. уч. “Проблемы сольватации и комплексообразования в растворах. От эффектов в растворах к новым материалам”. Иваново, 2015. С. 201.
Тюляева Е.Ю., Можжухина Е.Г., Бичан Н.Г., Ломова Т.Н. // Журн. неорган. химии. 2015. Т. 60. № 2. С. 194. [Tyulyaeva E.Yu., Mozhzhukchina E.G., Bichan N.G., Lomova T.N. // Russ. J. Inorg. Chem. 2015. V. 60. P. 157. https://doi.org/10.1134/S0036023615020199]https://doi.org/10.7868/S0044457X15020208
Тюляева Е.Ю., Бичан Н.Г., Можжухина Е.Г., Ломова Т.Н. // Журн. физ. химии. 2016. Т. 90. № 1. С. 28. [Tyulyaeva E.Yu., Bichan N.G., Mozhzhukchina E.G., Lomova T.N. // Russ. J. Phys. Chem. 2016. V. 90. P. 37. https://doi.org/10.1134/S0036024416010325]https://doi.org/10.7868/S0044453716010325
Ломова Т.Н. Аксиально координированные металлопорфирины в науке и практике. М.: Красанд, 2018. 704 с. https://www.rfbr.ru/rffi/ru/books/o_208717
Park-Gehrke L.S. // A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy; University of Washington. 2010. 171 p.
So S.-C., Cheung W.-M., Chiu W.-H. et al. // Dalton Trans. 2019. V. 48. P. 8340. https://doi.org/10.1039/c9dt00244h
Sherman B.D., Pillai S., Kodis G. et al. // Can. J. Chem. 2011. V. 89. P. 152. https://doi.org/10.1139/V10-118
Бичан Н.Г., Овченкова Е.Н., Ломова Т.Н. // Журн. физ. химии. 2019. Т. 93. № 4. С. 558. [Bichan N.G., Ovchenkova E.N., Lomova T.N. // Russ. J. Phys. Chem. A. 2019. V. 93. № 4. P. 703. https://doi.org/10.1134/S003602441904006X]
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии