

Журнал неорганической химии, 2022, T. 67, № 3, стр. 334-341
Координационные и спектральные свойства оксазамещенных производных тетрафенилпорфирина
С. Г. Пуховская a, *, Ю. Б. Иванова b, Н. Н. Крук c, А. О. Плотникова a, А. С. Вашурин a, С. А. Сырбу b
a Ивановский государственный химико-технологический университет
153000 Иваново,
Шереметевский пр-т, 7, Россия
b Институт химии растворов им. Г.А. Крестова РАН
153045 Иваново,
ул. Академическая, 1, Россия
c Белорусский государственный технологический университет
220006 Минск,
ул. Свердлова, 13а, Беларусь
* E-mail: svetlana.puhovskaya@mail.ru
Поступила в редакцию 08.09.2021
После доработки 18.09.2021
Принята к публикации 22.09.2021
- EDN: NMEKXG
- DOI: 10.31857/S0044457X22030102
Аннотация
С использованием теоретических выводов из четырехорбитальной модели Гоутермана выполнен детальный анализ формирования спектров поглощения и флуоресценции свободных оснований и их комплексов с цинком для 5,10,15,20-тетрафенил-21-оксапорфирина и 5,10,15,20-тетрафенил-21,22-диоксапорфирина. Показано влияние симметрии молекулы на положение и форму полос в электронных спектрах поглощения и спектрах флуоресценции. Впервые определены кинетические параметры реакций образования комплексов оксазамещенных производных 5,10,15,20-тетрафенилпорфина с солями d-металлов (Cu(II), Zn(II), Co(II)) при 288–308 K. Проведено сравнение полученных кинетических данных для оксазамещенных производных с результатами исследований для их классического аналога – тетрафенилпорфина.
ВВЕДЕНИЕ
Простейший по структуре порфирин – порфин – представляет собой тетрапиррольный макрогетероцикл. В реакционном центре порфина расположены четыре атома азота, которые в большей части и определяют свойства молекулы [1, 2]. Модификация архитектуры молекулы, как правило, осуществляется двумя путями: во-первых, путем варьирования природы и числа периферических заместителей, во-вторых, путем изменения самого макрокольца (гидрирование, присоединение дополнительных циклов, введение других гетероатомов). Замещение пиррольного азота на атомы VIА группы приводит к образованию новых макрогетероциклических систем – порфириноидов или гетерозамещенных порфиринов с уникальными малоизученными свойствами, которые значительно отличаются от свойств классических порфиринов [3]. Преобразование реакционного центра неизбежно влияет на электронную структуру макроцикла, при этом изменяются как физические, так и химические свойства соединения при сохранении ароматического характера π-электронной системы, устойчивости и способности образовывать комплексы с различными катионами металлов. В последние годы химия гетеропорфиринов стремительно развивается: получен практически каждый аналог порфирина и их производные, такие как хлорины, корролы, тетрабензопорфирины, определены возможности их практического использования в качестве более эффективной замены обычных металлопорфиринов [4].
Образование комплексов – неотъемлемое свойство тетрапиррольных макроциклов, поэтому изучение закономерностей их получения является актуальной и своевременной задачей современной координационной химии.
Известно, что тетрапиррольные макрогетероциклы являются эффективными преобразователями первичного аналитического сигнала в оптический отклик сенсора. Для них характерны достаточно интенсивные спектрально-люминесцентные, в частности флуоресцентные, свойства, которые могут быть использованы для этих целей. Тетрапиррольные молекулы интенсивно изучаются и в ряде случаев используются в качестве источника аналитического сигнала, что способствует решению задач, связанных с детектированием и количественным определением содержания различных ионов в растворах. Это направление химии в последнее время активно развивается с целью создания ион-чувствительных материалов [5].
В настоящей работе представлены результаты изучения и анализ оптических и координационных свойств 5,10,15,20-тетрафенил-21-оксапорфирина и 5,10,15,20-тетрафенил-21,22-диоксапорфирина, а также их структурного аналога – 5,10,15,20-тетрафенилпорфина.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
5,10,15,20-Тетрафенилпорфин (Н2TФП, I), 5,10,15,20-тетрафенил-21-оксапорфирин (HOTФП, II) и 5,10,15,20-тетрафенил-21,22-диоксапорфирин (O2TФП, III) синтезировали и выделяли по известным методикам. Спектральные характеристики полученных соединений соответствуют литературным данным [6–8].
5,10,15,20-Tетрафенилпорфин (I). ЭСП (хлороформ), λ (lg ε): 413 (5.60), 513 (4.26), 546 (3.90), 590 (3.70), 650 (3.73); 1H ЯМР (500 MГц, CDCl3), δ, м.д.: 8.30 (m, 8H, фенил o-H), 7.80 (m, 12H, фенил m- и p-H), 8.75 (8H, β-C), –3.75 (s, 2H, NH).
FAB-спектр: найдено m/z = 613 (для C44H29N4 рассчитано m/z = 613.24).
5,10,15,20-Тетрафенил-21-оксапорфирин (II). ЭСП (хлороформ), λ (lgε): 418 (5.41), 514 (4.36), 548 (3.81), 617 (3.48), 678 (3.69); 1H ЯМР (500 MГц, CDCl3), δH, м.д.: 7.67 (m, m- и p-фенил), 8.09 (t, о-фенил), 8.46 (d, пиррол), 8.52 (d, пиррол), 8.80 (s, пиррол), 9.10 (s, фуран).
FAB-спектр: найдено m/z = 615 (для C44H29N3O рассчитано m/z = 615.73).
5,10,15,20-Тетрафенил-21,22-диоксапорфирин (III). ЭСП (ДМФА), λ (lgε): 417 (4.93), 512 (3.91), 547 (3.72), 587 (3.53), 646 (3.41). 1H ЯМР (CDCl3), δ, м.д.: 9.77 и 9.68 (d, 4H, фуран); 9.01 и 8.94 (d, пиррол); 8.16 (m, 8H, о-фенил); 7.71–7.76 (m, 12H, m- и p-фенил).
FAB-спектр: найдено m/z = 617.23 (для C44H28N2O2 рассчитано m/z = 616.71).
Синтез и очистку комплексов цинка с лигандами (I–III) проводили по известным методикам [6, 9].
Растворители (уксусная кислота, ДМФА) и ацетаты металлов марки “ч. д. а.” очищали стандартными методами [10, 11].
Использованный для измерения спектров флуоресценции толуол фирмы Aldrich (содержание воды не более 0.03%) применяли без дополнительной очистки.
1Н ЯМР-спектры растворов соединений I–III регистрировали на спектрометре Bruker-500 c рабочей частотой 500 МГц в CDCl3 (внутренний стандарт – тетраметилсилан).
Спектры флуоресценции тетрапиррольных соединений и их комплексов измеряли на флуориметре Cary Eclipse фирмы Varian при температуре 298 K.
ЭСП растворов порфиринов и скорость реакций образования комплексов порфиринов I–III определяли на спектрофотометрах Shimadzu UV-1800 и Hitachi U-2000.
Скорость реакции комплексообразования измеряли с применением термостатируемых кювет в интервале температур от 288 до 348 K. Колебание температуры не превышало 0.1 K.
Первый кинетический порядок реакции образования металлопорфиринов был определен на основании прямолинейной зависимости lg($c_{{{{{\text{H}}}_{{\text{2}}}}{\text{P}}}}^{0}$/${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{P}}}}}$) – τ, где $c_{{{{{\text{H}}}_{{\text{2}}}}{\text{P}}}}^{0}$ и ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{P}}}}}$ – начальная и текущая концентрации порфирина.
Концентрацию растворов в ходе эксперимента контролировали по изменению оптической плотности. Кинетический эксперимент выполняли при ~50–100-кратном избытке концентрации раствора соли по сравнению с раствором макрогетероцикла, что позволило рассчитать эффективные константы скорости (kэф) реакции комплексообразования по уравнению псевдопервого порядка:
(1)
${{k}_{{{\text{эф}}}}} = {\text{ }}\left( {1{\text{/}}\tau } \right){\text{ln}}\left[ {({{A}_{o}}--{{A}_{\infty }}){\text{/}}(A--{{A}_{\infty }})} \right],$Константы скорости (n + 1)-порядка рассчитывали по уравнению:
(2)
${{k}_{{n + 1}}} = {{k}_{{{\text{эф}}}}}{\text{/}}с_{{{\text{M}}{{{\left( {{\text{OAc}}} \right)}}_{{\text{2}}}}}}^{n},$Энергию активации (Еа) для изученного температурного диапазона рассчитывали по уравнению Аррениуса:
где k2, k1 – эффективные константы скорости реакции при Т2 и Т1 соответственно, а энтропию процесса образования переходного состояния (ΔS≠) – по уравнению:РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Замена одного или двух центральных атомов азота атомами халькогенов приводит к существенным изменениям в электронно-оптических свойствах полученных тетрапиррольных и макрогетероциклов по сравнению с классическими аналогами (табл. 1).
Таблица 1.
Электронные спектры поглощения свободных оснований и цинковых комплексов порфиринов I–III в толуоле
| Порфирин | Длина волны, нм | ||||
|---|---|---|---|---|---|
| Bxy | Qy(0.1) | Qy(0.0) | Qx(0.1) | Qx(0.0) | |
| Н2TФП | 419.0 | 514.0 | 547.5 | 593.5 | 650.5 |
| HOTФП | 420.5 | 507.5 | 541.5 | 614.0 | 674.0 |
| O2TФП | 419.0 | 513.5 | 548.5 | 590.0 | 649.0 |
| ZnTФП | 423.0 | – | – | 549.0 | 589.0 |
| ZnOTФП | 428.0/444.5 | 548.0 | 591.0 | 583.0 | 636.0 |
| ZnO2TФП | 423.0 | – | – | 549.5 | 589.0 |
Детальный анализ формирования спектров поглощения свободных оснований при 21- и 21,23-гетерозамещении, представленный нами в работе [12], позволил сделать заключение о значительных изменениях в конфигурационном составе электронных переходов.
В результате данных изменений полоса Qx(0,0) длинноволнового электронного перехода 21-ОНТФП в толуоле по сравнению с соответствующей полосой молекулы Н2ТФП батохромно сдвигается от 650 к 674 нм, а максимум полосы поглощения Qy(0,0) гипсохромно смещается к 542 нм. Противоположное направление спектральных сдвигов полос поглощения Qx(0,0) и Qy(0,0) объясняется с привлечением четырехорбитальной модели Гоутермана [13] (на рис. 1 показано распределение электронной плотности на молекулярных орбиталях макроцикла).
Рис. 1.
НВМО и ВЗМО порфиринового макроцикла: а – НВМО с1; б – НВМО с2; в – ВЗМО b1; г – ВЗМО b2. Размер кругов пропорционален атомным орбитальным коэффициентам. Положительный и отрицательный знаки коэффициентов показаны соответственно белыми и черными кругами. На панели в) показана ось х, направленная вдоль NH–NH (O–NH или O–O), и ось y, направленная вдоль N–N.
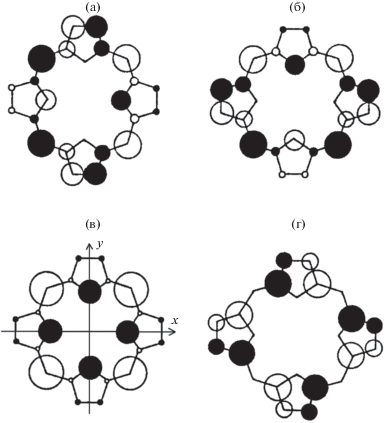
При введении фенильных заместителей в положения Сm макроцикла энергия орбитали b1 заметно повышается, и одноэлектронная конфигурация b1 → с1 вносит наибольший вклад в конфигурационный состав перехода. Монооксазамещение приводит к понижению энергии орбиталей b1 и с1 из-за меньшей электронной плотности на гетероатоме, а увеличение электронной плотности на атомах углерода Ca и Cb обусловливает слабое увеличение энергии орбиталей b2 и с2. Таким образом, полоса Qx(0,0) длинноволнового электронного перехода испытывает батохромный сдвиг, а полоса Qy(0,0) смещается гипсохромно. Аналогичный механизм был предложен для объяснения прогрессивных батохромных спектральных cдвигов при увеличении количества гетероатомов в макроцикле в 21,23-гетеропорфиринах [12].
Замещение соседних пиррольных колец фурановыми (21,22-замещение) приводит к несколько иному характеру спектральных сдвигов (табл. 1). Это обусловлено тем, что, во-первых, симметрия 21,22-замещенных производных ниже, чем таковая для 21,23-замещенных производных, и, во-вторых, иным характером сдвигов молекулярных орбиталей. Так, энергии орбиталей b1 и с1 уменьшаются примерно одинаково, это приводит к тому, что энергия доминирующей одноэлектронной конфигурации b1 → с1 практически не изменяется: максимум полосы поглощения Qx(0,0) наблюдается при 649 нм. Орбиталь с2 испытывает тенденцию к понижению энергии из-за меньшей электронной плотности на гетероатоме, что обусловлено совместным действием отрицательного индуктивного и более сильного мезомерного эффектов. Однако в то же время орбиталь с2 стремится к увеличению энергии из-за увеличения электронной плотности на атомах углерода Ca и Cb. В результате положение орбитали с2 практически не меняется. Аналогичная ситуация наблюдается для орбитали b2. В результате совокупности данных взаимодействий электронный спектр поглощения 21,22-О2ТФП практически не отличается от спектра поглощения исходного Н2ТФП (табл. 1).
Образование металлокомплексов 21- и 21,22-оксазамещенных молекул ТФП с ионом Zn2+ сопровождается значительными изменениями электронных спектров поглощения, однако они имеют более сложную структуру, чем спектры металлокомплексов “классических” порфиринов. Рассмотрим формирование электронного спектра поглощения молекулы ZnОНТФП (рис. 2).
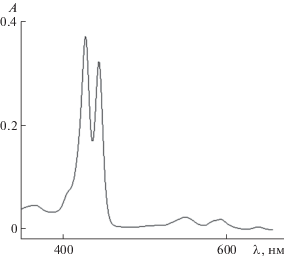
Спектр характеризуется двумя особенностями: во-первых, в видимой области спектра наблюдаются три Q-полосы, а не две, как у “классических” металлокомплексов; во-вторых, полоса Соре расщепляется на две полосы. Основная причина таких спектральных проявлений заключается, по нашему мнению, в более низкой симметрии молекулы Zn-ОНТФП по сравнению с классическими порфиринами. Если последние относятся к точечной группе симметрии D4h, то для металлокомплексов 21-гетеропорфиринов симметрия не может быть выше, чем С2v, равно как и для их свободных оснований, в то время как свободные основания классических порфиринов обладают симметрией D2h. Переход к металлокомплексу в классических порфиринах сопровождается двукратным вырождением орбиталей, величина расщепления между которыми у свободного основания порфирина составляет ~3000 см–1 [14]. Вследствие более низкой симметрии металлокомплексов 21-гетеропорфиринов молекулярные орбитали сближаются, однако вырождения не происходит. В результате полосы Qy(0,1) и Qy(0,0) вибронного спутника соответственно первого и второго электронных переходов перестраиваются. Действительно, вторая полоса поглощения имеет асимметричный контур с максимумом при 591 нм и плечом при 583 нм. Если принять плечо при 583 нм за полосу Qy(0,1), а максимум – за полосу Qy(0,0), то для каждого из электронных переходов частота вибронного повтора составит 1380 ± 50 см–1, а расщепление между электронными состояниями – 1200 см–1, что сопоставимо с расщеплением монопротонированной формы порфиринов, относящейся к той же точечной группе симметрии [14]. Соответственно, из-за низкой симметрии полоса Соре расщепляется на две компоненты: Вх с максимумом при 444.5 нм и Ву с максимумом при 428 нм. Заметим, что у свободного основания 21-ОНТФП расщепления не наблюдается, хотя полоса Соре заметно уширена по сравнению с молекулой Н2ТФП. По нашему мнению, отличия вызваны различной степенью вовлечения гетероатома в формирование сопряженной π-системы. У свободного основания гетероатом “включается” слабее, а у металлокомплексов гетероатом непосредственно вовлечен в формирование координационной связи с ионом металла и оказывает бóльшее влияние.
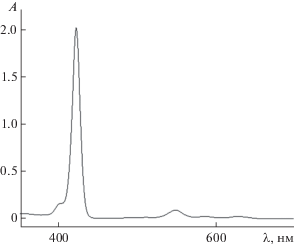
Симметрия молекулы ZnО2ТФП возрастает (точечная группа симметрии С2h), в результате чего полоса Соре уширена, но не расщеплена на компоненты, как у молекулы ZnОНТФП (рис. 3). В видимой области спектра наблюдаются две полосы поглощения (табл. 1), как и у “классических” порфиринов.
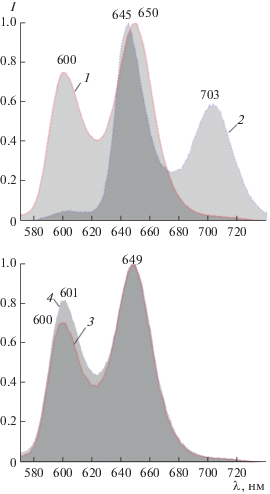
Исследуемые гетеропорфирины и в форме свободного основания, и в форме металлокомплекса флуоресцируют. Для спектров флуоресценции выполняется правило частот, т.е. положение максимумов в спектрах флуоресценции примерно зеркально симметрично положению максимумов в спектрах поглощения (рис. 4). Величина сдвига Стокса для молекулы ZnОНТФП составляет 220 см–1, для молекулы ZnО2ТФП она возрастает до 340 см–1. Рост величины при переходе от 21-окса- к 21,22-диоксазамещенному производному указывает на возрастание релаксационных процессов в нижнем возбужденном синглетном состоянии. Действительно, согласно рентгеноструктурным данным [15], молекула ZnОНТФП имеет неплоскую молекулярную конформацию макроцикла. С одной стороны, это обусловлено тем, что к иону цинка присоединяется аксиальный лиганд (противоион соли, ацетат), который индуцирует формирование куполообразной структуры. С другой стороны, несколько меньший радиус атома кислорода по сравнению с азотом обусловливает сжатие макроцикла и частичный выход из плоскости фуранового кольца с формированием макроцикла рифленого типа. Перераспределение электронной плотности в возбужденном состоянии создает предпосылки для структурной релаксации, величина которой растет с увеличением числа гетероатомов в ядре макроцикла.
Рис. 4.
Нормированные на максимум спектры флуоресценции цинковых комплексов: 1 – ZnТФП, 2 – ZnОНТФП, 3 – ZnТФП, 4 – ZnО2ТФП. Длина волны возбуждения 500 нм.
Введение атомов кислорода в макроциклическое кольцо приводит не только к изменению основности соединений [16, 17] и размера внутренней полости, но и к отсутствию одного или двух протонов ядра. Эти изменения существенно влияют на такие характерные свойства порфиринов, как ароматичность и способность связывать металлы.
К настоящему времени хорошо изученная реакция координации порфиринов двухзарядными катионами переходных металлов в неводных растворителях происходит в соответствии с уравнением (5), что убедительно показано в работах [1, 2]:
(5)
$\begin{gathered} {{{\text{Н}}}_{{\text{2}}}}{\text{P}} + \left[ {{\text{М}}{{{\text{Х}}}_{{\text{2}}}}{{{\left( {{\text{Solv}}} \right)}}_{n}}_{{--2}}} \right] \to \\ \to {\text{МР}} + {\text{ 2НХ}} + (n--2){\text{Solv}}. \\ \end{gathered} $Однако в литературе отсутствуют данные об особенностях протекания процессов координации для гетерозамещенных аналогов порфиринов. В настоящей работе впервые измерены константы скорости и определены энергетические параметры реакции образования комплексов цинка, меди и кобальта гетерозамещенных порфиринов II и III в сравнении с тетрафенилпорфирином в ДМФА (табл. 2).
Таблица 2.
Кинетические параметры реакции координации 5,10,15,20-тетрафенил-21-оксапорфирина в сравнении с 5,10,15,20-тетрафенилпорфином ацетатами цинка, кобальта и меди в ДМФА*
| Порфирин | ${{C}_{{{\text{М(OAc}}{{{\text{)}}}_{{\text{2}}}}}}}$ × 103, моль/л | $k_{{{\text{эф}}}}^{{298}}$ × 103 | $k_{v}^{{298}}$ , л моль–1 с–1 | Ea, кДж/моль | ΔS≠, Дж/(моль K) |
|---|---|---|---|---|---|
| Zn(OAc)2 | |||||
| O2TФП | 1.3 | Реакция не идет | |||
| HOTФП | 1.3 | 8.97 ± 0.10 | 6.9 ± 0.02 | 57 ± 4 | –45 ± 6 |
| Н2ТФП | 1.3 | 6.52 ± 0.10 | 5.02 ± 0.02 | 61 ± 4 | –35 ± 6 |
| Co(OAc)2 | |||||
| O2TФП | 1.5 | Реакция не идет | |||
| HOTФП | 1.5 | 2.25 ± 0.10 | 1.5 ± 0.02 | 62 ± 4 | –100 ± 8 |
| Н2ТФП | 1.7 | 0.15 ± 0.10 | 0.1 ± 0.02 | 91 ± 4 | –46 ± 6 |
| Cu(OAc)2 | |||||
| O2TФП | 0.11 | Реакция не идет | |||
| HOTФП | 0.11 | 5.99 ± 0.02** | 0.57 ± 0.02 | 8 ± 1 | –232 ± 10 |
| Н2ТФП | 0.11 | 0.02 ± 0.002** | 0.002 ± 0.0002 | 20 ± 1 | –200 ± 20 |
* Порядок реакции по соли в ДМФА определен авторами [18]. ** Размерность $k_{v}^{{298}}$, л0.5 моль–0.5 с–1.
Известно, что для классических порфиринов реакция (5) подчиняется кинетическому уравнению первого порядка по макроциклу [1, 2]:
(6)
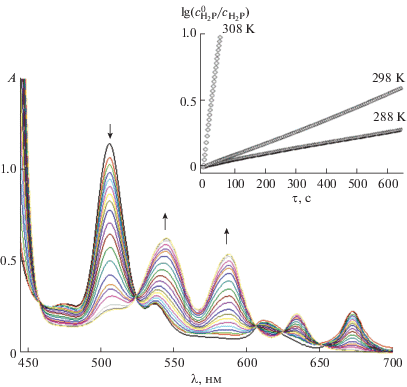
$--d{{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{P}}}}}{\text{/}}d\tau = k{{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{P}}}}}c_{{{\text{M}}{{{\text{X}}}_{{\text{2}}}}}}^{n},$Для всех изученных систем нами установлено, что реакция образования металлопорфиринов II, III также имеет первый кинетический порядок, что подтверждается прямолинейностью зависимостей в координатах lg($c_{{{{{\text{H}}}_{{\text{2}}}}{\text{P}}}}^{0}$/${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{P}}}}}$)–τ ($c_{{{{{\text{H}}}_{{\text{2}}}}{\text{P}}}}^{0}$ и ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{P}}}}}$ – начальная и текущая концентрации лиганда) и наличием четких изобестических точек. Характерные спектральные изменения в процессе комплексообразования показаны на рис. 5.
Рис. 5.
Изменение ЭСП в ходе реакции координации HOTФП ацетатом цинка в ДМФА при 298 K; на вставке представлена зависимость lg($с_{{{{{\text{H}}}_{{\text{2}}}}{\text{P}}}}^{0}$/${{с}_{{{{{\text{H}}}_{{\text{2}}}}{\text{P}}}}}$) от τ для реакции образования ZnOTФП при 288, 298 и 308 K (${{с}_{{{\text{Zn(OAC}}{{{\text{)}}}_{{\text{2}}}}}}}$ = 1.3 × 10–3 моль/л).
Как следует из данных табл. 2, не удалось измерить кинетику реакций координации 5,10,15,20-тетрафенил-21,22-диоксапорфирина ацетатами перечисленных металлов в ДМФА, однако при использовании в качестве растворителя уксусной кислоты наблюдается комплексообразование [17].
Замена одного из атомов азота пиррольного фрагмента макроциклического соединения на атом кислорода приводит к возрастанию основных свойств лиганда примерно на два порядка [17]. Рост частичного отрицательного заряда на атомах реакционного центра способствует упрочнению связей с третичными атомами азота N → M в переходном состоянии и тем самым обусловливает увеличение скорости реакции комплексообразования по сравнению с классическим аналогом. В координирующем слабоосновном растворителе ДМФА при переходе от HOTФП к Н2TФП константа скорости реакции комплексообразования (5) возрастает при одновременном снижении энергии активации. Наиболее ярко это наблюдается при образовании комплексов меди: константа скорости возрастает в ~280 раз при снижении энергии активации на ~12 кДж/моль.
ЗАКЛЮЧЕНИЕ
Проведен детальный анализ формирования ЭСП и спектров флуоресценции как свободных оснований, так и комплексов цинка оксазамещенных производных тетрафенилпорфина. Показано, что на положение и интенсивность полос в спектрах оказывает существенное влияние не только введение гетероатома, но и геометрия (симметрия) молекулы. Взаимно противоположное влияние этих факторов приводит к спектрам 5,10,15,20-тетрафенил-21,22-диоксапорфирина, близким к классическому Н2TФП, большие различия наблюдаются для 5,10,15,20-тетрафенил-21-оксапорфирина.
Впервые изучена кинетика реакций координации солей переходных металлов оксазамещенными производными в сравнении с классическим аналогом Н2TФП.
Показано, что модификация макроцикла путем замены одного из атомов азота пиррольного фрагмента на атом кислорода способствует увеличению скорости реакции при закономерном снижении энергии и энтропии активации.
Список литературы
Березин Б.Д. Координационная химия порфиринов и фталоцианина. М.: Наука, 1978. 280 с.
Березин Б.Д., Ениколопян Н.С. Металлопорфирины. М.: Наука, 1988. 158 с.
Syrbu S.A., Pukhovskaya S.G., Dao T.N. et al. // Macroheterocycles. 2019. V. 12. № 2. P. 135. [Сырбу С.А., Пуховская С.Г., Дао Тхе Нам и др. // Макрогетероциклы. 2019. Т. 12. № 2. С. 135.]https://doi.org/10.6060/mhc190557s
Ziolkowski P., Milach J., Symonowicz K. et al. // Tumori. 1995. V. 81. № 5. P. 364. https://doi.org/10.1177/030089169508100512
Kamaljit Singh, Amit Sharma, Shivali Sharma // Adv. Heterocycl. Chem. 2012. V. 106. P. 111. https://doi.org/10.1016/B978-0-12-396531-8.00002-X
You Y., Gibson S.L., Hilf R. et al. // J. Med. Chem. 2003. V. 46. № 17. P. 3734. https://doi.org/10.1021/jm030136i
Buchler J.W. Synthesis and properties of metalloporphyrins. Porphyrins. N.Y.: Academic Press, 1978. V. 1. 483 p. https://doi.org/10.1016/B978-0-12-220101-1.50017-2
Chmielewski P.J., Latos-Gra˙zy’nski L., Olmstead M.M. et al. // Chem. Eur. J. 1997. V. 3. № 2. P. 268. https://doi.org/10.1002/chem.19970030216
Won-Seob Cho, Chang-Hee Lee // Bull. Korean Chem. Soc. 1998. V. 19. № 3. P. 314.
Карякин Ю.В., Ангелов И.И. Чистые химические реактивы. М.: Химия, 1974. 407 с.
Вайсбергер А., Проскауэр Э., Риддик Дж. Органические растворители. Физические свойства и методы очистки. М.: Изд-во иностр. литер., 1958. 518 с.
Вершиловская И.В., Люлькович Е.С., Пуховская С.Г. и др. // Журн. прикл. спектроскопии. 2020. Т. 87. № 2. С. 181.
Gouterman M. The Porphyrins. V. 3. New York: Academic Press, 1978. 165 p.
Kruk M., Starukhin A., Maes W. // Macroheterocycles. 2011. V. 4. № 2. P. 69. [Крук Н., Старухин А., Маес В. // Макрогетероциклы. 2011. Т. 4. № 2. С. 69.]https://doi.org/10.6060/mhc2011.2.01
Gloe R., Suite S., Goetzke L. et al. // Inorg. Chem. 2013. V. 52. № 3. P. 1515. https://doi.org/10.1021/ic302268h
Syrbu S.A., Ivanova Y.B., Pukhovskaya S.G. et al. // Russ. J. Gen. Chem. 2019. V. 89. № 2. P. 255. [Сырбу С.А., Иванова Ю.Б., Пуховская С.Г. и др. // Журн. общей химии. 2019. Т. 89. № 2. С. 258.]https://doi.org/10.1134/S0044460X19020148
Pukhovskaya S.G., Ivanova Y.B., Plotnikova A.O. et al. // Russ. J. Gen. Chem. 2020. V. 90. № 7. P. 1292. [Пуховская С.Г., Иванова Ю.Б., Плотникова А.О. и др. // Журн. общей химии. 2020. Т. 90. № 7. С. 1110.]https://doi.org/10.31857/S0044460X2007015X
Кувшинова Е.М., Пуховская С.Г., Голубчиков О.А. и др. // Коорд. химия. 1993. Т. 19. № 8. С. 630.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии