

Журнал неорганической химии, 2021, T. 66, № 10, стр. 1387-1395
Синтез и ионоселективные свойства композита MoO2/C
Г. С. Захарова a, *, З. А. Фаттахова a
a Институт химии твердого тела УрО РАН
620990 Екатеринбург, ул. Первомайская, 91, Россия
* E-mail: volkov@ihim.uran.ru
Поступила в редакцию 11.03.2021
После доработки 05.04.2021
Принята к публикации 07.04.2021
Аннотация
Изучены условия получения композита MoO2/C при гидротермальной обработке пероксомолибденовой кислоты в присутствии глюкозы, выполняющей роль источника углерода и восстановителя, с последующим отжигом в инертной атмосфере. С помощью рентгенофазового и термогравиметрического анализа, а также КР-спектроскопии и низкотемпературной адсорбции азота определены особенности формирования композитов. Показана возможность использования синтезированных композитов в качестве активного материала твердофазных ионоселективных электродов для определения концентрации ионов калия в растворе. Электроды на основе MoO2/C проявляют калийную функцию в интервале 1 ≤ ${\text{p}}{{C}_{{{{{\text{K}}}^{ + }}}}}$ ≤ 5 при кислотности рабочих растворов 5 ≤ рH ≤ 6 с угловым коэффициентом 57 мВ/${\text{p}}{{C}_{{{{{\text{K}}}^{ + }}}}}.$ Определены коэффициенты селективности электродов в ряду одно- и двухзарядных катионов.
ВВЕДЕНИЕ
Композит на основе диоксида молибдена MoO2/C представляет интерес для материаловедения благодаря уникальным электрохимическим [1–4], фотоэлектрическим [5], сенсорным [6, 7] и фотокаталитическим [8, 9] свойствам. Его отдельные составляющие, образующие единую систему при совместном формировании, обеспечивают синергический эффект за счет взаимодействия компонентов. Аморфный углерод как химически инертное, высоко износостойкое и теплопроводное соединение является одним из важнейших и широко используемых материалов [10]. Известно, что введение углерода в металл-оксидную матрицу позволяет улучшить каталитическую активность [11] и увеличить электронную составляющую проводимости, повысив электрохимические характеристики электродных материалов литиевых источников тока [12, 13].
Известно, что физико-химические свойства, морфология, текстурные и размерные характеристики в значительной степени определяются условиями получения соединений. Все известные методы синтеза композита MoO2/C условно можно разделить на две группы. К первой группе относятся способы, в которых источник углерода остается неизменным на протяжении всего процесса получения. Такими источниками углерода, механически диспергируемыми в процессе синтеза композита MoO2/C, являются углеродные нанотрубки [14, 15], оксид графена [16–18]. Использование углеродсодержащих соединений, которые легко карбонизируются при синтезе композита, характерно для второй группы методов. Такой подход к получению MoO2/C является наиболее эффективным и обеспечивает равномерное распределение углерода в композите. Несомненным его преимуществом является возможность вводить углерод в состав композита in situ, т.е. в процессе формирования композита при разложении органической компоненты. В качестве источника углерода могут быть использованы полиэтиленгликоль [19], поливиниловый спирт [20], анилин [21], аскорбиновая [22], винная [4] и олеиновая [23] кислоты. Наиболее привлекательным источником углерода, выполняющим одновременно и роль сильного восстановителя в процессе получения композита MoO2/C, является глюкоза [24–27]. В отличие от карбоновых кислот и других используемых углеродсодержащих органических соединений, глюкоза, имеющая циклическое строение, легко подвергается карбонизации в гидротермальных условиях при температуре ~200°С с образованием сферических частиц углерода [28]. Кроме того, глюкоза относится к экологически чистым и дешевым источникам углерода.
Цель настоящей работы – исследование условий образования композита MoO2/C при гидротермальной обработке пероксомолибденовой кислоты в присутствии глюкозы с последующим отжигом в инертной атмосфере, изучение его ионоселективных свойств.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исходных веществ использовали порошок металлического молибдена (99.9 мас. % Мо), 30%-ный раствор пероксида водорода марки “ос. ч.”, глюкозу C6H12O6 марки “х. ч.”. Получение композита MoO2/C проводили в две стадии. На первой стадии порошок молибдена растворяли при охлаждении (5–10°С) в Н2О2 с образованием желтого раствора пероксомолибденовой кислоты H2Mo(O2)x (x = 2–4). К полученному раствору при перемешивании добавляли C6H12O6 в молярном соотношении Мо : C6H12O6 = = 1 : (0.25–2). Реакционную смесь помещали в автоклав, нагревали до 160°С, выдерживали в течение 24 ч, а затем охлаждали до комнатной температуры. Полученные осадки отфильтровывали, промывали водой и сушили на воздухе. Продукты гидротермальной реакции, используемые в дальнейшем в качестве прекурсоров, обозначали как МоОn-Х, где Х – мольное содержание глюкозы в реакционной массе, n = 2 или 3. Следует отметить, что при гидротермальной обработке реакционной смеси состава Мо : C6H12O6 = 1 : 0.5 осадок не формируется. Поэтому реакционный раствор после завершения гидротермальной реакции выпаривали до получения твердого остатка. На второй стадии прекурсоры MoOn-Х подвергали прокаливанию в токе азота со скоростью 7 град/мин при температуре 500°С в течение 1 ч. Полученные образцы обозначали как МоОn/C-Х. Рентгенофазовый анализ (РФА) образцов выполняли на дифрактометре Shimadzu XRD 7000 (CuKα‑излучение, λ = 1.5418 Å). Морфологию порошков изучали на сканирующем электронном микроскопе (СЭМ) JSM 6390 LA (JEOL). Спектры комбинационного рассеяния регистрировали на конфокальном рамановском дисперсионном спектрометре in Via Reflex (Renishaw) с использованием твердотельного лазера RL532-08 с длиной волны 532 нм и мощностью 100 мВт. Для понижения поглощения лазерного излучения, приводящего к нагреву образца и изменению его структуры, мощность лазера была уменьшена до 1%. Термический анализ выполняли на анализаторе STA 449 F3 Jupiter (Netzsch), совмещенном с масс-спектрометром QMS 403 при скорости нагрева 10 град/мин в атмосфере воздуха (ТГ–ДСК–МС). Текстурные характеристики (удельную поверхность, пористость) композитов определяли методом низкотемпературной адсорбции азота на анализаторе Gemini VII (Micromeritics). Пробоподготовку образцов проводили вакуумированием при 150°С в течение 2 ч. На основании полученных изотерм сорбции азота рассчитывали удельную поверхность по методу Брунауэра–Эммета–Теллера (БЭТ). Анализ пористости материалов выполняли с использованием данных изотерм сорбции по методу Баррета–Джойнера–Халенда.
Электроды на основе композита MoO2/C были изготовлены по методике [29]. Измерение потенциала проводили иономером И-130.2М в режиме вольтметра с точностью ±1 мВ. Для определения коэффициентов селективности использовали метод непрерывных растворов [30].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Согласно данным РФА, прекурсоры общей формулы МоОn-Х являются хорошо окристаллизованными порошками (рис. 1а). Гидротермальная обработка реакционной смеси с молярным соотношением реагентов Мо : C6H12O6 = 1 : 0.25 приводит к образованию прекурсора, изоструктурного орторомбической фазе α-МоО3. Дальнейшее повышение содержания глюкозы в реакционной массе сопровождается появлением наряду с основной фазой (MoO3) примеси MoO2. Образование однофазного MoO2 моноклинной модификации наблюдается при Х ≥ 0.75. Таким образом, условием, определяющим фазовый состав продуктов гидротермальной обработки пероксомолибденовой кислоты в присутствии глюкозы, является молярное соотношение указанных компонентов. Термолиз синтезированных МоОn-Х прекурсоров в атмосфере азота приводит к формированию композитов, фазовый состав которых также определяется исходным содержанием глюкозы в реакционной массе (рис. 1б). При X ≤ 0.25 образуется композит на основе орторомбической фазы α-MoO3 (пр. гр. Pbnm), а при X ≥ 0.5 – композиты на основе моноклинной фазы MoO2 (пр. гр. Р21/с) с параметрами элементарной ячейки, представленными в табл. 1.
Рис. 1.
Дифрактограммы порошков прекурсоров (а) и композитов (б) при молярном соотношении Mo : C6H12O6 = 1 : : 0.25 (1), 1 : 0.5 (2), 1 : 0.75 (3), 1 : 1 (4), 1 : 2 (5) и позиции брегговских пиков α-МоО3 (JCPDS 5-508) и MoO2 (JCPDS 72-4534).
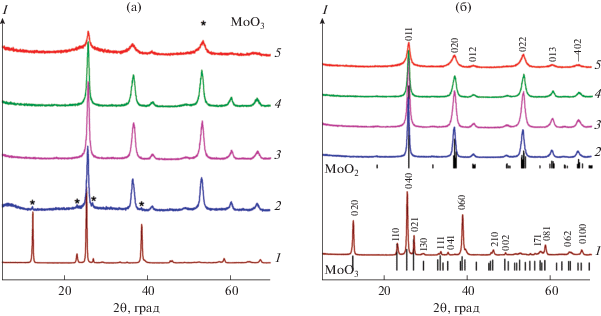
Таблица 1.
Параметры элементарной ячейки, средний размер кристаллитов и удельная поверхность композитов МоО3/С-0.25 и МоО2/C-X
| Композит | a, Å | b, Å | c, Å | β, град | V, Å3 | Dср, нм | SБЭТ, м2/г |
|---|---|---|---|---|---|---|---|
| МоО3/С-0.25 | 3.996(2) | 13.861(3) | 3.696(2) | – | 204.7(5) | 24.5 | 3.2 |
| МоО2/С-0.5 | 5.544(8) | 4.844(2) | 5.625(8) | 119.53(4) | 131.4(8) | 13.3 | 66 |
| МоО2/С-1 МоО2/С-2 α-МоО3 (JCPDS 5-508) МоО2 (JCPDS 72-4534) |
5.603(9) 5.610(2) 3.962 5.6109 |
4.839(5) 4.813(5) 13.858 4.8562 |
5.628(0) 5.613(7) 3.697 5.6285 |
121.08(3) 120.48(2) – 120.95 |
130.7(2) 130.6(5) 202.99 131.53 |
10.6 7.2 – – |
126 116 – – |
С использованием уравнения Шеррера был рассчитан средний размер кристаллитов МоО3 и МоО2 соответствующих композитов:
где Dср – размеры кристаллитов, k – безразмерный коэффициент формы частиц (постоянная Шеррера), Δ(2θ) – ширина дифракционного пика на половине высоты, θ – брегговский угол, λ – длина волны рентгеновского излучения. Расчет вели по интенсивным и хорошо разрешенным дифракционным пикам. Средний размер кристаллитов МоО3 и МоО2 композитов, уменьшающийся с увеличением содержания глюкозы в реакционной массе, представлен в табл. 1.Методом СЭМ изучена морфология синтезированных композитов в зависимости от содержания С6Н12О6 в реакционной массе (рис. 2). Композит МоО3/С-0.25 состоит из частиц удлиненно-призматического габитуса. Линейные размеры частиц МоО3/С-0.25 составляют до 11 мкм в длину и 0.1–0.9 мкм в ширину. Композит МоО2/С-X образован сильно агломерированными хлопьевидными частицами. Их размерные характеристики МоО2/С-X изменяются в широком диапазоне (30–260 нм), при этом наблюдается незначительное увеличение размеров с ростом С6Н12О6 в реакционной массе (X).

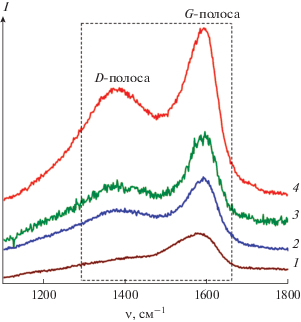
В спектрах КР композитов МоОn/С-Х в интервале 1100–1800 см–1 наблюдаются полосы, типичные для углеродных материалов (рис. 3). Пик при 1385 см–1 описывает D-линию, которая соответствует колебаниям атомов углерода с sp3-типом гибридизации, и свидетельствует о наличии разупорядочения [31]. Пик при 1594 см–1 описывает G-линию, отвечающую колебаниям атомов углерода в sp2-гибридизации, и указывает на наличие в образце мелкокристаллического графита. В выбранных условиях съемки в интервале частот 100–1100 см–1 пики, характерные для MoO2, не фиксируются (рис. S1 ). В спектрах КР в низкочастотной области проявляются только вибрационные моды MoO3 композита МоО3/С-0.25 [32]. Таким образом, исследования, выполненные с использованием КР-спектроскопии показали, что формирование композита МоOn/C происходит с образованием разупорядоченной мелкокристаллической графитовой составляющей. Для оценки степени упорядоченности углерода использовали показатель, определяемый соотношением интенсивностей D- и G-линий (ID/IG). Для композитов МоO3/C-0.25, МоO2/C-0.5, МоO2/C-1, МоO2/C-2 величина ID/IG равна 0.65, 0.67, 0.73, 0.76 соответственно. Увеличение показателя ID/IG с ростом содержания глюкозы в реакционной массе свидетельствует об увеличении степени разупорядочения углеродной составляющей композитов МоOn/C-Х. Полученные данные хорошо согласуются с результатами РФА, свидетельствующими об уменьшении степени кристалличности композитов МоОn/С-Х с увеличением содержания глюкозы в реакционной массе.
Рис. 3.
КР-спектры композитов МоО3/С-0.25 (1), МоО2/С-0.5 (2), МоО2/С-1 (3), МоО2/С-2 (4) в диапазоне частот 1100–1800 см–1.
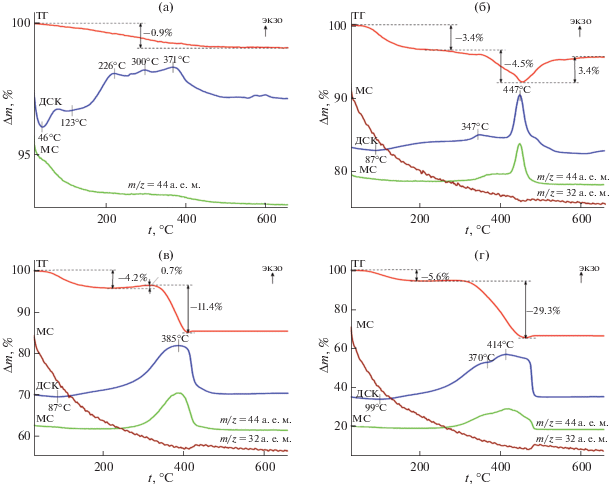
Для количественного определения содержания углерода в композитах MoOn/C-Х использовали ТГ–ДСК–МС-анализ (рис. 4). При термическом разложении композита МоO3/C-0.25 наблюдается убыль массы (0.9 вес. %). Процесс описывается сложным эндоэффектом, сопровождающим дегидратацию образца, и широким экзоэффектом с максимумами при 226, 300 и 371°С. Указанный экзоэффект обусловлен окислением углеродной составляющей композита, что подтверждается появлением слабого пика на МС-кривой (зависимость ионного тока от температуры), соответствующего молекулярному иону ${\text{CO}}_{2}^{ + }$ с m/z = 44 а. е. м. Следует отметить, что количественно определить содержание углерода в композите МоO3/C-0.25 не представляется возможным, так как на плавно изменяющейся ТГ-кривой сложно выделить участки, отвечающие удалению воды и углерода. Термолиз композитов МоO2/C-Х (Х ≥ 0.5) проходит в несколько стадий. В интервале температур 20–220°С наблюдается убыль массы, которую можно отнести к удалению адсорбированной воды. Ее количество увеличивается с ростом содержания углерода в композите. Относительно сложный характер изменения ТГ-кривой композитов МоO2/C-Х, обусловленный последовательным протеканием процессов окисления углеродной составляющей композита до CO2 и МоО2 до МоО3, наблюдается в интервале температур 250–600°С. Выделение диоксида углерода подтверждается интенсивным пиком на МС-кривой, характерным для молекулярного иона ${\text{CO}}_{2}^{ + }$ (m/z = 44 а. е. м.). Процесс окисления MoO2, сопровождающийся поглощением кислорода, описывается изменением ТГ-кривой, а также появлением пика на МС-кривой, отвечающего молекулярному иону ${\text{O}}_{2}^{ - }$ (m/z = 32 а. е. м.). Ход ТГ-кривой композита МоO2/C-1 в интервале температур 220–400°С отличается от других составов и свидетельствует об особенностях его формирования. В процессе нагревания образца убыли массы (10.7 вес. %), описывающей окисление органической компоненты композита, предшествует незначительная прибыль веса (0.7 вес. %), соответствующая окислению MoO2. Такое термическое поведение МоO2/C-1 можно объяснить химической неоднородностью композита. Мы полагаем, что на поверхности частиц MoO2 образуется углеродный слой, содержащий в своем составе также и MoO2. Неоднородность структуры композита обусловлена процессом созревания Оствальда (Ostwald-ripening process), включающим растворение термодинамически нестабильных мелких частиц MoO2 с последующей их рекристаллизацией на углеродной поверхности композита с образованием более крупных частиц [33]. Термогравиметрический анализ МоO2/C-X позволил оценить содержание углерода в образцах, которое составило 4.5, 10.7 и 29.3 вес. % для МоO2/C-0.5, МоO2/C-1 и МоO2/C-2 соответственно.
Рис. 4.
Кривые ТГ–ДСК–МС композитов MoO3/C-0.25 (а), MoO2/C-0.5 (б), MoO2/C-1 (в) и MoO2/C-2 (г) в атмосфере воздуха.
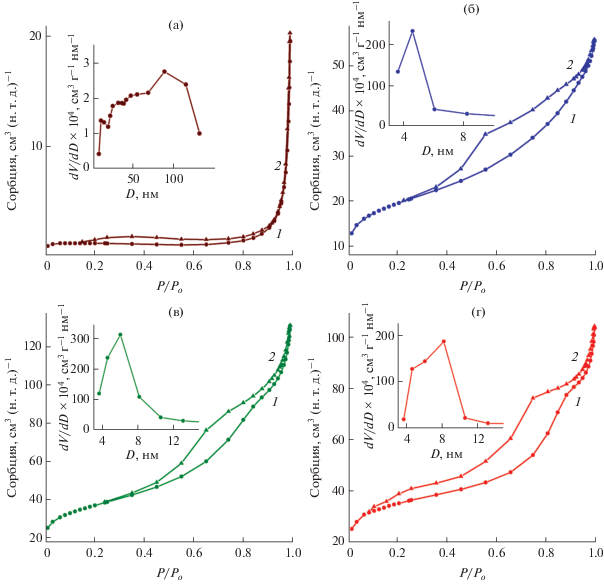
На рис. 5 представлены изотермы сорбции азота и соответствующие кривые распределения пор по размерам композитов MoO3/C-0.25 и MoO2/C-Х. Согласно классификации ИЮПАК [34], представленные изотермы сорбции относятся к IV типу. Изотермы сорбции композитов MoO3/C-0.25 и MoO2/C-Х имеют петлю гистерезиса H4 и Н3 соответственно. Петля Н3 характерна для агрегированных пластинчатых образцов с щелевидными порами. Гистерезис типа H4 описывает процессы сорбции в порах клиновидной формы. Полученные величины удельной поверхности композитов (SБЭТ) приведены в табл. 1. Наибольшие значения удельной поверхности наблюдаются для композитов с высоким содержанием углерода. Широкое распределение пор по размерам для композита MoO3/C-0.25 свидетельствует о наличии в его структуре мезо- и макропор. Мономодальное распределение пор наблюдается для композитов на основе MoO2 с преобладанием мезопор размером 5–8 нм.
Рис. 5.
Изотермы сорбции (1 − адсорбция, 2 − десорбция) и кривые распределения пор по размерам (вставки) композитов MoO3/C-0.25 (а), MoO2/C-0.25 (б), MoO2/C-1 (в) и MoO2/C-2 (г).
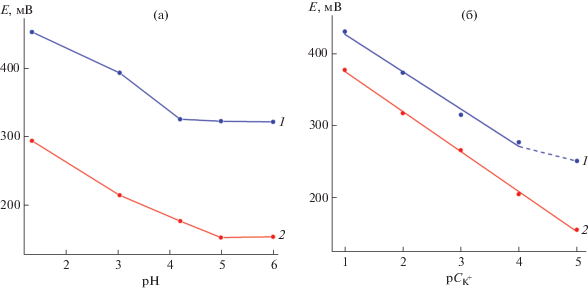
Композит МоО2/С-1, отличающийся высоким значением удельной поверхности, исследовали в качестве электродно-активного вещества для определения концентрации ионов калия в растворе. Для сравнения также была изучена электродная функция композита МоО3/С-0.25. На рис. 6а приведены концентрационные зависимости потенциала электродов на основе композитов МоО3/С-0.25 и МоО2/С-1, позволяющие определить рабочий интервал рН раствора, не зависящий от потенциала системы. Установлено, что в интервале 5 ≤ рН ≤ 6 электроды практически не реагируют на изменение концентрации ионов водорода. Поэтому электродное поведение композитов изучали при рН 6 (рис. 6б). Исследование K+-функции позволило установить, что наилучшими характеристиками обладает электрод на основе композита МоО2/С-1, реагирующий на изменение концентрации ионов калия в интервале 1 ≤ ${\text{p}}{{C}_{{{{{\text{K}}}^{{\text{ + }}}}}}}$ ≤ 5 с угловым коэффициентом, равным 57 ${{{\text{мВ}}} \mathord{\left/ {\vphantom {{{\text{мВ}}} {{\text{р}}{{С}_{{{{{\text{K}}}^{ + }}}}}}}} \right. \kern-0em} {{\text{р}}{{С}_{{{{{\text{K}}}^{ + }}}}}}}.$ Полученное значение углового коэффициента калибровочной кривой близко к теоретическому значению для одновалентных ионов (59 ${{{\text{мВ}}} \mathord{\left/ {\vphantom {{{\text{мВ}}} {{\text{р}}{{С}_{{{{{\text{M}}}^{ + }}}}}}}} \right. \kern-0em} {{\text{р}}{{С}_{{{{{\text{M}}}^{ + }}}}}}}$). Ионоселективная мембрана на основе композита МоО3/С-0.25 проявляет K+-функцию в интервале 1 ≤ ${\text{р}}{{С}_{{{{{\text{K}}}^{ + }}}}}$≤ 4 с угловым коэффициентом, равным 52 ${{{\text{мВ}}} \mathord{\left/ {\vphantom {{{\text{мВ}}} {{\text{р}}{{С}_{{{{{\text{K}}}^{ + }}}}}}}} \right. \kern-0em} {{\text{р}}{{С}_{{{{{\text{K}}}^{ + }}}}}}}.$ Вероятно, угловой коэффициент калибровочных кривых, а также интервал чувствительности электродных материалов к ионам калия в значительной степени зависят от величины удельной поверхности и наличия мезопор, снимающих диффузионные затруднения K+ в процессе интеркаляции/деинтеркаляции. Следует отметить, что использование углеродсодержащих материалов позволяет на порядок увеличить нижний концентрационный предел обнаружения ионов калия по сравнению с электродом на основе MoO3 [35]. Положительный эффект присутствия углерода может быть обусловлен увеличением проводимости системы, а также созданием буферного слоя, предотвращающего структурные изменения молибден-оксидной матрицы при потенциометрических измерениях. Потенциал K+-функции при использовании МоО2/С-1 в качестве электродно-активного вещества определяется следующей потенциалобразующей реакцией:
(2)
${\text{Mo}}{{{\text{O}}}_{{\text{2}}}} + y{\text{M}}\left( {{{{\text{H}}}_{{\text{2}}}}{\text{O}}} \right)_{n}^{ + } + y\bar {e} \leftrightarrow {{{\text{M}}}_{y}}{\text{Mo}}{{{\text{O}}}_{{\text{2}}}} + n{{{\text{H}}}_{{\text{2}}}}{\text{O}}{\text{.}}$Рис. 6.
Зависимости потенциала электродов на основе МоО3/С-0.25 (1) и МоО2/С-1 (2) от рН (а) и концентрации ионов калия в растворе (б).
Образование твердого раствора замещения также лежит в основе потенциалобразующей реакции электродной функции композита МоО3/С-0.25:
(3)
${\text{Mo}}{{{\text{O}}}_{{\text{3}}}} + y{\text{M}}\left( {{{{\text{H}}}_{{\text{2}}}}{\text{O}}} \right)_{n}^{ + } + y\bar {e} \leftrightarrow {{{\text{M}}}_{y}}{\text{Mo}}{{{\text{O}}}_{{\text{3}}}} + n{{{\text{H}}}_{{\text{2}}}}{\text{O}}{\text{.}}$Для установления влияния посторонних ионов на K+-функцию электродов были определены значения потенциометрических коэффициентов селективности $\left( {{{{\text{K}}}_{{{{{\text{K}}}^{ + }}{\text{/}}{{{\text{M}}}^{{n + }}}}}}} \right)$ в присутствии мешающих катионов (табл. S1 ). Мешающие катионы по степени их влияния на работу электродов на основе МоО2/С-1 и МоО3/С-0.25 можно соответственно расположить в следующей последовательности:
Электроды на основе МоО2/С-1 селективны к ионам калия в присутствии Na+ и умеренно селективны к двухвалентным катионам. Следует отметить, что K+-селективность электродного материала на основе МоО3/С-0.25 в присутствии двухзарядных катионов значительно выше по сравнению с электродом, изготовленным из МоО2/С-1. Такое различие обусловлено структурными особенностями молибден-оксидной матрицы. Слоистая структура MoO3 является более предпочтительной для реализации процесса интеркаляции ионов, отличающихся большим размером.
ЗАКЛЮЧЕНИЕ
Установлено, что гидротермальной обработкой водного раствора пероксомолибденовой кислоты и глюкозы с последующим отжигом в инертной атмосфере могут быть получены композитные материалы на основе диоксида молибдена MoO2/C. Термическая стабильность и текстурные характеристики композитов зависят от содержания углерода, которое составляет 4.5–29.3 вес. %. Установлено, что MoO2/C может быть использован в качестве электродно-активного материала при изготовлении твердофазных ионоселективных электродов, позволяющих определять содержание ионов калия в растворе в интервале 1 ≤ ${\text{р}}{{С}_{{{{{\text{K}}}^{ + }}}}}$ ≤ 5 с угловым коэффициентом калибровочной кривой, равным 57 ${{{\text{мВ}}} \mathord{\left/ {\vphantom {{{\text{мВ}}} {{\text{р}}{{С}_{{{{{\text{K}}}^{ + }}}}}}}} \right. \kern-0em} {{\text{р}}{{С}_{{{{{\text{K}}}^{ + }}}}}}}.$
Список литературы
Zhang X., Wang J.-G., Hua W. et al. // J. Alloys Compd. 2019. V. 787. P. 301. https://doi.org/10.1016/j.jallcom.2019.02.042
Huo J., Xue Y., Liu Y. et al. // J. Electroanal. Chem. 2020. V. 857. P. 113751. https://doi.org/10.1016/j.jelechem.2019.113751
Herdt T., Bruns M., Schneider J.J. // Dalton Trans. 2018. V. 47. № 42. P. 14897. https://doi.org/10.1039/C8DT02076K
Zakharova G.S., Singer L., Fattakhova Z.A. et al. // J. Alloys Compd. 2021. V. 863. P. 158353. https://doi.org/10.1016/j.jallcom.2020.158353
Li L., Sui H., Zhao K. et al. // Electrochim. Acta. 2018. V. 259. P. 188. https://doi.org/10.1016/j.electacta.2017.10.171
Dong Y., Zhou M., Zhang L. // Electrochim. Acta. 2019. V. 302. P. 56. https://doi.org/10.1016/j.electacta.2019.02.006
Li B., Liu L.-H., Zhang X.-F. et al. // Anal. Chim. Acta. 2021. V. 1142. P. 73. https://doi.org/10.1016/j.aca.2020.11.038
Wang M., Peng Z., Li H. et al. // J. Mater. Res. 2018. V. 33. № 6. P. 685. https://doi.org/10.1557/jmr.2018.32
Ji H., Fei T., Zhang L. et al. // Appl. Surf. Sci. 2018. V. 57. P. 1142. https://doi.org/10.1016/j.apsusc.2018.06.134
Lee J.-H., Park S.-J. // Carbon. 2020. V. 163. P. 1. https://doi.org/10.1016/j.carbon.2020.02.073
Liu W., Cai J., Huang B. et al. // New J. Chem. 2021. V. 45. № 5. P. 2775. https://doi.org/10.1039/D0NJ05272H
Thauer E., Zakharova G.S., Wegener S.A. et al. // J. Alloys Compd. 2021. V. 853. P. 157364. https://doi.org/10.1016/j.jallcom.2020.157364
Zakharova G.S., Ottmann A., Möller L. et al. // J. Mater. Sci. 2018. V. 53. № 17. P. 12244. https://doi.org/10.1007/s10853-018-2488-9
Qiu S., Lu G., Liu J. et al. // RSC Adv. 2015. V. 5. № 106. P. 87286. https://doi.org/10.1039/C5RA17147D
Bhaskar A., Deepa M., Rao T.N. // ACS Appl. Mater. Interfaces. 2013. V. 5. № 7. P. 2555. https://doi.org/10.1021/am3031536
Hwang J., Yoon D., Kweon B. et al. // RSC Adv. 2016. V. 6. № 110. P. 108298. https://doi.org/10.1039/C6RA24632J
Zhang X., Huang X., Xia L. et al. // Ceram. Int. 2017. V. 43. № 6. P. 4753. https://doi.org/10.1016/j.ceramint.2016.11.117
Guo L., Wang Y. // J. Mater. Chem. A. 2015. V. 3. № 8. P. 4706. https://doi.org/10.1039/C4TA05520A
Li X., Xiao Q., Gao Y. et al. // J. Alloys Compd. 2017. V. 723. P. 1113. https://doi.org/10.1016/j.jallcom.2017.06.274
Chen Z., Yang T., Shi H. et al. // Adv. Mater. Interfaces. 2017. V. 4. № 3. P. 1600816. https://doi.org/10.1002/admi.201600816
Gao Q., Yang L., Lu X. et al. // J. Mater. Chem. 2010. V. 20. № 14. P. 2807. https://doi.org/10.1039/B921001F
Liu B., Zhao X., Tian Y. et al. // Phys. Chem. Chem. Phys. 2013. V. 15. № 22. P. 8831. https://doi.org/10.1039/C3CP44707C
Wang Y., Huang Z., Wang Y. // J. Mater. Chem. A. 2015. V. 3. № 42. P. 21314. https://doi.org/10.1039/C5TA05345E
Ni J., Zhao Y., Li L., Mai L. // Nano Energy. 2015. V. 11. P. 129. https://doi.org/10.1016/j.nanoen.2014.10.027
Jiang J., Yang W., Wang H. et al. // Electrochim. Acta. 2017. V. 240. P. 379. https://doi.org/10.1016/j.electacta.2017.04.103
Avendaño C., Briceño A., Méndez F.J. et al. // Dalton Trans. 2013. V. 42. № 8. P. 2822. https://doi.org/10.1039/C2DT31248D
Фаттахова З.А., Захарова Г.С. // Журн. неорган. химии. 2020. Т. 65. № 4. С. 458. [Fattakhova Z.A., Zakharova G.S. // Russ. J. Inorg. Chem. 2020. V. 65. № 4. P. 480. https://doi.org/10.1134/S0036023620040051]
Higgins L.J.R., Brown A.P., Harrington J.P. et al. // Carbon. 2020. V. 161. P. 423. https://doi.org/10.1016/j.carbon.2020.01.060
Подвальная Н.В., Захарова Г.С. // Журн. неорган. химии. 2020. Т. 65. № 7. С. 880. [Podval’naya N.V., Zakharova G.S. // Russ. J. Inorg. Chem. 2020. V. 65. № 7. P. 967. https://doi.org/10.1134/S0036023620070153]
Окунев М.С., Хитрова Н.В., Корниенко О.И. // Журн. аналит. химии. 1982. Т. 37. № 1. С. 5.
Ferrari A.C., Robertson J. // Phys. Rev. B. 2020. V. 61. № 20. P. 14095. https://doi.org/10.1103/PhysRevB.61.14095
Фаттахова З.А., Вовкотруб Э.Г., Захарова Г.С. // Журн. неорган. химии. 2021. Т. 66. № 1. С. 41. [Fattakhova Z.A., Vovkotrub E.G., Zakharova G.S. // Russ. J. Inorg. Chem. 2021. V. 66. P. 41. https://doi.org/10.1134/S0036023621010022]
Zhang L., Yao J., Xia F. et al. // Inorg. Chem. Front. 2018. V. 5. № 3. P. 550. https://doi.org/10.1039/C7QI00819H
Sing K.S.W., Everett D.H., Haul R.A.W. et al. // Pure Appl. Chem. 1985. V. 57. № 4. P. 603. https://doi.org/10.1351/pac198557040603
Zakharova G.S., Fattakhova Z.A., Zhu Q., Enyashin A.N. // J. Electroanal. Chem. 2019. V. 840. P. 187. https://doi.org/10.1016/j.jelechem.2019.03.072
Дополнительные материалы
- скачать ESM.docx
- Рис. S1. КР-спектры композитов МоО3/С-0.25 (1), МоО2/С-0.5 (2), МоО2/С-1 (3), МоО2/С-2 (4) в диапазоне частот 100‒1100 см-1.
Таблица S1. Коэффициенты селективности ионоселективного электрода на основе композитов МоО3/С-0.25 и МоО2/С-1.
Инструменты
Журнал неорганической химии