

Российская археология, 2022, № 2, стр. 20-28
Геномика древних патогенов на примере чумной палочки (Yersinia pestis) в историческом контексте
И. Л. Кузнецова 1, 3, *, Т. В. Андреева 1, 2, 3, А. Б. Малярчук 2, С. С. Кунижева 1, 2, 3, Т. В. Тяжелова 1, Ф. Е. Гусев 1, 3, А. Д. Манахов 1, 3, М. В. Добровольская 4, Е. И. Рогаев 1, 2, 3, **
1 Научно-технологический университет “Сириус”, Научный центр генетики и наук о жизни
г. Сочи, Россия
2 Московский Государственный университет им. М.В. Ломоносова
Москва, Россия
3 Институт общей генетики им. Н.И. Вавилова РАН
Москва, Россия
4 Институт археологии РАН
Москва, Россия
* E-mail: irakuzn@gmail.com
** E-mail: rogaev.ei@talantiuspeh.ru
Поступила в редакцию 28.10.2021
После доработки 28.10.2021
Принята к публикации 16.11.2021
- EDN: SHHGPH
- DOI: 10.31857/S086960632202012X
Аннотация
Развитие методов высокопроизводительного секвенирования способствовало значительному прогрессу в области изучения древней ДНК. Хотя объектом большей части палеогенетических исследований являются геномы людей, новые технологии позволяют также исследовать отдельные микробные патогены из разных древних объектов. К настоящему времени больше всего данных получено по последовательностям древних геномов Yersinia pestis (возбудитель чумы). В статье приводится анализ данных о распространении и эволюции возбудителей чумы в периоды неолита, бронзы, средневековья и нового времени. Обсуждаются гипотезы, связывающие возникновение эпидемий и пандемий чумы в Евразии с историческими, социальными и демографическими процессами. Отмечается, что перспективы получения актуальных знаний об эволюции возбудителя чумы лежат в области расширения спектра изучаемых археологических материалов. Геномные данные этого патогена дополнили сведения, полученные ранее палеопатологами, и позволили не только идентифицировать штаммы возбудителей пандемий прошлого, но и выявить ныне не существующие линии патогена, уточнить хронологию появления патогена в популяциях человека, а также реконструировать эволюционную историю Y. pestis, которая актуальна для общественного здравоохранения и сегодня. В настоящем обзоре рассмотрены последние достижения в области генетических исследований Y. pestis и полученные с их помощью новые данные о наиболее известных эпидемиях чумы в истории человечества.
Методы палеогенетики дают исследователям возможность изучать ДНК вирусов и бактерий, обитавших в организмах древних людей. С приходом пандемии COVID-19 палеогеномные данные о патогенах в археологическом контексте приобретают новую актуальность и лишаются столь привычной отделенности от современности. Археологические источники позволяют проводить анализ возникновения, развития и завершения пандемии во времени, учитывая миграции, динамику социальных и культурных новаций, вызванных этой кризисной ситуацией. В ряде современных исследований пандемия рассматривается как один из типов глобального кризиса, сопоставимый с климатическими изменениями, но имеющий свою специфику (длительность, отложенные социальные и демографические последствия и пр.) (Gamble et al., 2021; Hull, 2009).
Ранее изучение перенесенных инфекционных заболеваний проводилось путем палеопатологической оценки костей древних скелетов из археологических раскопок. Однако этот подход имеет существенные ограничения, связанные с тем, что стремительно развивающиеся губительные инфекции не оставляют видимых следов на костях, а другой материал оказывается недоступен для исследования (Buikstra, 2012; Бужилова, 2005). Первые успехи в изучении ДНК древних бактерий и вирусов стали возможны с появлением метода полимеразной цепной реакции (ПЦР) (Spigelman, Lemma, 1993). Этот подход позволяет обнаружить присутствие инфекционных агентов, но дает ограниченную информацию об эволюционной истории патогена, поскольку генетический материал микроорганизмов в таком случае анализируется по одному или нескольким коротким фрагментам ДНК, выделенных из останков древних людей (Arriaza et al., 1995; Drancourt et al., 1998). Присутствие в древних образцах смеси бактериальных организмов может привести к неспецифическим или даже ложноположительным результатам ПЦР-анализа, как, например, в случае с ДНК почвенных бактерий, содержащих последовательности, сходные с ДНК M. Tuberculosis и Y. pestis (Gilbert et al., 2004; Müller, Roberts, Brown, 2016). Кроме того, древняя ДНК, извлеченная из археологического материала, обычно присутствует в нем в небольших количествах, сильно фрагментирована и содержит химические модификации (Briggs et al., 2007; Paabo, 1989).
За последнее десятилетие достижения в области геномики, в частности, разработка методов высокопроизводительного секвенирования, также называемого секвенированием следующего поколения (NGS), позволили существенно увеличить объем данных, получаемых из древних останков, что дало возможность исследовать также геномы древних патогенных микроорганизмов. Первым таким геномом стал геном Y. pestis из захоронения 1348–1349 гг., опубликованный в 2011 г., и с тех пор у исследователей появилась возможность более достоверно идентифицировать патоген и реконструировать его эволюционную историю.
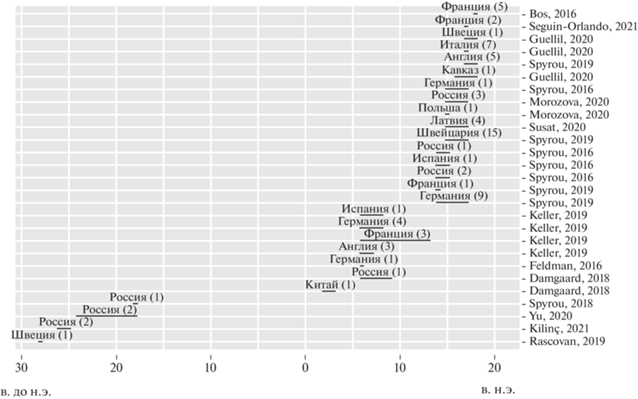
Рис. 1.
Опубликованные геномные последовательности Y. pestis. Примечание: на рисунке отмечены предполагаемый возраст образцов (горизонтальными линиями), место обнаружения (над линиями) и количество образцов (в скобках). Справа приведены ссылки на соответствующие исследования.
Fig. 1. Published Yersinia pestis genomic sequences
Особенности генома Yersinia pestis. Современный род Yersinia представлен более чем 20 видами, большинство из которых не патогенны для человека, за исключением чумной палочки Y. pestis и двух энтеропатогенов – Y. enterocolitica и Y. pseudotuberculosis. На основании полногеномного секвенирования коллекций современных видов бактерий были выявлены основные возможные причины высокой патогенности определенных представителей этого вида. Так, Y. pseudotuberculosis и Y. pestis, скорее всего, произошли от непатогенной формы Y. enterocolitica путем приобретения плазмиды pYV (Chain et al., 2004; Reuter et al., 2014). Плазмида pYV дала возможность эффективной доставки непосредственно в иммунные клетки хозяина белков Yops. Этот процесс получил название “смертельный поцелуй Yersinia” (Cornelis, Wolf-Watz, 1997) – проникнувшие в клетки белки Yops прежде всего препятствуют опознаванию клетками иммунной системы. Также были выявлены еще несколько локусов в геноме, оказавшихся ключевыми для появления высоко-патогенного вида Y. pestis: кодирующего токсин Yersinia murine toxin (ymt), за счет которого Y. pestis приобрела способность переноситься блохами (Hinnebusch et al., 2002; Hinnebusch, Jarrett, Bland, 2021), и кодирующего белок Pla, ускоряющая легочную инфекцию (Zimbler et al., 2015). Всего на сегодняшний день выявлено 80 хромосомных и 42 плазмидных гена Y. pestis, потенциально связанных с вирулентностью (Zhou et al., 2004; Zhou, Yang, 2009).
Пандемические находки. Y. pestis является возбудителем чумы – болезни, унесшей жизни около 60% населения Старого Света во время трех разрушительных пандемий: первой, или юстиниановой, чумы (6–8 вв. н.э.), второй (14–18 вв.), включая самый высоколетальный период Черной смерти (1347–1351 гг.), и третьей, или современной, пандемии (18–19 вв.), начавшейся в Китае (Frith, 2012; Scott, Dunkan, 2001; Stenseth et al., 2008). Сейчас чума считается проблемой прошлого, хотя до сих пор ежегодно регистрируются сотни случаев заболевания в африканских странах (Barbieri et al., 2020).
Чума – одно из самых изученных с помощью методов палеогенетики заболеваний (Schuenemann et al., 2011, Duchêne et al., 2020); на данный момент секвенировано более 70 древних геномов Y. pestis (рисунок). Каждая из трех пандемий была подтверждена геномными методами. Так, было реконструировано восемь геномов чумной палочки и доказано присутствие юстиниановой чумы на Британских островах, а также параллельное появление ее штаммов во Франции, Испании (Keller et al., 2019). В захоронениях на территории Баварии было выявлено наличие Y. pestis, филогенетически схожей со штаммами первой пандемии (Harbeck et al., 2013), что дало возможность предположить распространение чумы на юге Германии в первую пандемию, несмотря на отсутствие упоминания в исторических источниках о чуме в данном регионе (Feldman et al., 2016; Wagner et al., 2014).
Большинство древних геномов Y. pestis было получено для второй пандемии на территориях 10 европейских стран (рисунок). Были выдвинуты две гипотезы, объясняющие большую продолжительность второй пандемии чумы, основанные на наличии внутреннего и внешнего резервуаров чумы (Bramanti et al., 2021).
Согласно первой гипотезе, существовал внутренний резервуар чумной палочки в Западной Европе (Carmichael, 2014), которым, скорее всего, являлись черные крысы (McCormick, 2003). В случае множественного занесения чумной палочки в Западную Европу было бы ожидаемо обнаружить высокое генетическое разнообразие геномов Y. pestis из разных частей Европы. Тем не менее была выявлена генетическая преемственность между штаммами бактерии во время второй пандемии в Европе (Bos et al., 2016; Seguin-Orlando et al., 2021; Spyrou et al., 2019; Susat et al., 2020). Так, 34 генома Y. pestis из Англии, Франции, Германии, Швейцарии и России принадлежат к одной генетической линии и имеют общее происхождение (Spyrou et al., 2019). Однако генетически подтвержденное наличие резервуара в Восточной Европе и/или Азии и активные торговые связи с Западной Европой, в том числе Шелковый путь и торговля ценным мехом, не исключают постоянной “подпитки” новыми штаммами со стороны Центральной Азии, Персии и России (Guellil et al., 2020; Morozova et al., 2020; Spyrou et al., 2019). Этот аргумент свидетельствует в пользу второго сценария – Y. pestis могла быть реинтродуцирована из Азии или Восточной Европы в Западную Европу несколько раз в течение всего периода пандемии (Namouchi et al., 2018; Schmid et al., 2015).
Также остается открытым вопрос об основной причине окончания столь долго длившейся пандемии. Одной из классических версий является совершенствование гигиенических норм в Европе, а также формирование иммунитета и связанное с ним или независимое ослабление вирулентности штаммов Y. pestis. С помощью сравнительного анализа геномов Y. pestis из различных захоронений Западной Европы, датированных последовавшим за Черной Смертью периодом (15–17 вв.), было выявлено постепенное “истощение” гена pla, связанного с вирулентностью чумной палочки (Susat et al., 2020).
С накоплением данных секвенирования геномов второй пандемии чумы в последний год стало возможно делать первые предположения о влияниях штаммов Y. pestis на конкретные исторические события и наоборот. В частности, были проанализированы геномы Y. pestis из Италии и французских Альп 17 в. (Seguin-Orlando et al., 2021). Данный период исторически известен крупным социально-экономический спадом в Италии, предположительно являвшимся последствием высокопатогенного штамма чумы. Как показал сравнительный анализ, геномы Y. pestis, выявленные в Италии, не отличались от других европейских штаммов вариациями, связанными с повышением патогенности. На основании этих данных авторы сделали вывод, что события в экономике Италии были связаны не с мутированным штаммом чумной палочки, а с тяжелым социальным и демографическим статусом населения.
Геномы Y. pestis из допандемических образцов. Наиболее важными с исторической точки зрения являются результаты палеогеномных исследований допандемических штаммов Y. pestis. Ведь по палеоантропологическим материалам мы не можем получить сведения об этом заболевании, так как оно стремительно развивается и не оставляет следов на скелете. Самые ранние следы возбудителя были обнаружены в образцах, полученных из скелетов людей, похороненных в мегалитической галерейной гробнице Фральсегагардене, Фалбигден, Западная Швеция. Всего в этом сооружении похоронено до 78 человек за период 5100–4900 л.н. Авторы исследования рассматривают этот период как довольно короткий для такого большого числа индивидуумов (Rascovan et al., 2019). Как показали более ранние археологические исследования (Skoglund et al., 2012; Skoglund et al., 2014), люди, погребавшие своих соплеменников в этих сооружениях, жили небольшими (возможно, семейными) коллективами. Филогения чумы, построенная на основании этих находок и образцов, полученных из различных памятников эпохи бронзы Евразии в целом, позволяет авторам связать финал европейского неолита с неизвестной до сих пор пандемией чумы, которая привела к упадку поздненеолитических культур Европы. В свою очередь, возникновение вирулентных возбудителей авторы связывают с процессом неолитизации в Европе, который включал формирование крупных стационарных поселений с высокой плотностью. В качестве претендента на роль наиболее “благоприятных” условий рассматривается трипольская культура с ее крупными поселениями на территории Молдовы, Румынии, Украины. Далее чума по торговым путям распространилась в другие регионы, и несколько генетических изменений привели к появлению в Европе и Азии бронзового века более вирулентных штаммов (Rascovan et al., 2019). Другие палеогеномные находки чумного вибриона были сделаны при изучении палеоантропологических материалов из погребений степных культур раннего бронзового века, или культур изначально степного происхождения (ямной, афанасьевской, культуры шнуровой керамики), наиболее ранние даты которых – около 4800 г. до н.э. (Rasmussen et al., 2015, Valtueña et al., 2017). В целом авторы гипотетически связывали приход чумы в Европу с расселением пастушестких племен ямной общности. Филогенетическая картина неолитической чумы еще не создана, поэтому здесь мы остаемся в поле догадок. Вызывает некоторое удивление обнаружение отдельных инфицированных индивидов в мегалитических сооружениях. Ведь мы знаем, как выглядят кладбища периода чумных вспышек. Здесь же мы имеем дело с отдельными заболевшими на протяжении двух сотен лет. Значит ли это, что древний штамм был менее вирулентен? Тогда можно ли связывать “закат” неолитической Европы с эпидемией и даже пандемией? Пока эти вопросы остаются без ответа. Важно подчеркнуть, что рассмотрение вопросов филогении чумного возбудителя невозможно без комплексного исследования, включающего палеогеномные и археологические исследования. Изучение останков неолитических земледельцев, у которых был выявлен самый древний на настоящий момент штамм Y. pestis, позволяет предположить, что он вызывал легочную форму болезни (Rascovan et al., 2019).
Один из поворотных моментов в эволюции Y. pestis произошел в среднем или позднем бронзовом веке, когда в геном бактерии встроился локус, содержащий токсин ymt, вследствие чего чума начала распространяться через блох (Rasmussen et al., 2015). В период 3000–1000 лет до н.э. выявляются и ymt-содержащие, и ymt-несодержащие геномы Y. pestis, что указывает на постепенное вытеснение менее эффективно распространяющегося штамма. Очевидно, что до появления возможности передачи возбудителя эктопаразитом скорость распространения заболевания была гораздо ниже. Она была такова, что за несколько сот лет охватила всю Северную Евразию (Yu et al., 2020). К эпохе бронзы, началу III тыс. до н.э., относятся свидетельства чумы в Забайкалье. В настоящее время возбудитель чумы обнаружен в 4400-летнем образце из Прибайкалья (Аносово-1) и в образце возраста ~3800 л.н. из Якутии (Каменка-2) (Kılınç et al., 2021). Важно отметить, что в последнем случае в захоронении было три молодых человека, все родственники, в том числе пара родитель–ребенок.
Филогения древних штаммов возбудителя чумы свидетельствует о том, что большинство из них исчезли, за исключением одного, имеющего сходство со штаммом, выделенным из скелетных останков людей – жертв Юстиниановой чумы (Damgaard et al., 2018). Авторы исследований эволюции чумных возбудителей предполагают, что “обрывы” большинства линий, имевших место в эпоху ранней и средней бронзы, связаны с крупными вспышками заболевания, которые приводили к вымиранию неолитического населения (Rascovan et al., 2019).
Одним из ключевых регионов для изучения распространения чумной палочки является Восточная Европа (контактная зона между Европой и Азией). К настоящему времени получено несколько древних геномов Y. pestis из этого региона (из Польши и России) и показано, что данные восточноевропейские линии являются филогенетически родственными ранее описанным вариантам из западной и юго-восточных частей Европы (Morozova et al., 2020; Spyrou et al., 2016; Spyrou et al., 2019). Разнообразие европейских штаммов средневековой чумы позволяет предполагать, что Черная смерть распространялась по Европе не только благодаря внутренним контактам, но и пополнению из Восточной Европы.
Таким образом, на сегодняшний день собрана достаточно обширная коллекция древних геномов Y. pestis разных исторических времен с территории Западной Европы, однако для Восточной Европы и Азии количество таких данных намного меньше, хотя именно они могут уточнить особенности филогении и эволюции Y. pestis.
Ближайшие перспективы. Y. pestis на сегодняшний день является наиболее изученным древним патогеном человека. Как показывает накопленный опыт, при анализе образцов из задокументированных чумных захоронений следы ДНК Y. pestis удается найти не более, чем в 25% случаев, что диктует необходимость расширения объема исследуемого археологического материала для получения достоверных результатов.
Прогресс в области NGS привел к постепенному увеличению скорости и уменьшению стоимости получения геномных данных, в том числе для образцов древней ДНК. Благодаря этому с каждым годом становится более вероятным, что помимо анализа геномов, выделенных из единичных или десятков исторических образцов, будет проведен анализ многих массовых древних захоронений. Такие данные позволят намного шире взглянуть, в частности, на влияние пандемий на человечество в историческом и генетическом аспекте.
Необходимо отметить основные технические сложности в исследованиях, связанных с палеогенетикой, которые важно решить в ближайшем будущем. Как отмечалось выше, существенной проблемой для правильной интерпретации результатов секвенирования является сходство патогенных форм и непатогенных форм бактерий, представленных в почве в большом количестве. Один из путей решения – параллельный анализ почвы и седиментов из коллективных захоронений. Работы, опубликованные за последние два года в области генетических исследований древних седиментов, демонстрируют большой потенциал этого подхода. Во-первых, ДНК в пробах из седиментов может случайно совпадать с таковой из останков скелетов (Vernot et al., 2021), а также патогенов, населявших внутренние органы (Ramirez, Saka, Nores, 2021). Во-вторых, появляется возможность анализировать ДНК окружающей среды исследуемого периода (Zavala et al., 2021).
Заключение. Чума сыграла огромную роль в истории человечества. Не удивительно, что специалисты в области палеогенетики проявляют значительный интерес к этому объекту исследований. Уже накоплено существенное количество геномных данных. Благодаря имеющимся результатам, новые работы могут дополнять, подтверждать или опровергать исторические данные. В результате геномных исследований был выявлен эволюционный путь возникновения патогенной формы чумной палочки из непатогенной, а также вариации, усиливающие и ослабляющие ее патогенность, включая пути передачи. Основываясь на этих данных, были сделаны выводы об очагах чумы в конце неолита и бронзовом веке, а также связь штаммов и путей трансмиссии в периоды известных пандемий. Успехи в области геномного изучения пандемий в историческом контексте особенно актуальны в современности. Они способствуют лучшему пониманию, в том числе, эволюционных процессов формирования патогенных микроорганизмов, взаимодействия “хозяин-патоген” и адаптации человечества к этим процессам.
Работа выполнена при финансовой поддержке проекта Минобрнауки России, системный номер № 075-10-2020-116 (номер гранта 13.1902.21.0023)
Список литературы
Бужилова А.П. Homo Sapiens: История болезни. Москва: Языки славянской культуры, 2005. 321 с.
Arriaza B.T. et al. Pre-Columbian tuberculosis in Northern Chile: Molecular and skeletal evidence // American Journal of Physical Anthropology. 1995. V. 98. № 1. P. 37–45.
Barbieri R. et al. Yersinia pestis: the Natural History of Plague // Clinical Microbiology Reviews. 2020. V. 34. № 1. 00044-19.
Bos K.I. et al. Eighteenth century Yersinia pestis genomes reveal the long-term persistence of an historical plague focus // Elife. 2016. V. 5. 12994.
Bramanti B. et al. Plague: A Disease Which Changed the Path of Human Civilization // Yersinia pestis: Retrospective and Perspective / Eds. R. Yang, A. Anisimov. Dordrecht: Springer, 2016 (Advances in Experimental Medicine and Biology; 918). P. 1–26.
Bramanti B. et al. Assessing the origins of the European Plagues following the Black Death: A synthesis of genomic, historical, and ecological information // Proceedings of the National Academy of Sciences. 2021. V. 118. № 36. e2101940118.
Briggs A.W. et al. Patterns of damage in genomic DNA sequences from a Neandertal // Proceedings of the National Academy of Sciences. 2007. V. 104. № 37. P. 14616–14621.
Buikstra J.E., Roberts C.A. The Global History of Paleopathology: Pioneers and Prospects. Oxford; New York: Oxford University Press, 2012. 800 p.
Carmichael A.G. Plague Persistence in Western Europe: A Hypothesis // Medieval Globe. 2014. V. 1. № 1. P. 157–191.
Chain P.S.G. et al. Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis // Proceedings of the National Academy of Sciences. 2004. V. 101. № 38. P. 13826–13831.
Cornelis G.R., Wolf-Watz H. The Yersinia Yop virulon: a bacterial system for subverting eukaryotic cells // Molecular Microbiology. 1997. V. 23. № 5. P. 861–867.
Damgaard P. de B. et al. 137 ancient human genomes from across the Eurasian steppes // Nature. 2018. V. 557. № 7705. P. 369–374.
Drancourt M. et al. Detection of 400-year-old Yersinia pestis DNA in human dental pulp: An approach to the diagnosis of ancient septicemia // Proceedings of the N-ational Academy of Sciences. 1998. V. 95. № 21. P. 12637–12640.
Duchêne S. et al. The Recovery, Interpretation and Use of Ancient Pathogen Genomes // Current Biology. 2020. V. 30. № 19. P. R1215–R1231.
Feldman M. et al. A High-Coverage Yersinia pestis Genome from a Sixth-Century Justinianic Plague Victim // Molecular Biology and Evolution. 2016. V. 33. № 11. P. 2911–2923.
Frith J. The history of plague – Part 1. The three great pandemics // Journal of Military and Veterans’ Health. 2012. V. 20. № 2. P. 11–16.
Gamble L.H. et al. Finding Archaeological Relevance during a Pandemic and What Comes After // American Antiquity. 2021. V. 86. Iss. 1. P. 2–22.
Gilbert M.T.P. et al. Absence of Yersinia pestis-specific DNA in human teeth from five European excavations of putative plague victims // Microbiology. 2004. V. 150. № 2. P. 341–354.
Guellil M. et al. A genomic and historical synthesis of plague in 18th century Eurasia // Proceedings of the National Academy of Sciences. 2020. V. 117. № 45. P. 28328–28335.
Harbeck M. et al. Yersinia pestis DNA from Skeletal Remains from the 6th Century AD Reveals Insights into Justinianic Plague // PLoS Pathogens. 2013. V. 9. № 5. e1003349.
Hinnebusch B.J. et al. Role of Yersinia Murine Toxin in Survival of Yersinia pestis in the Midgut of the Flea Vector // Science. 2002. V. 296. Iss. 5568. P. 733–735.
Hinnebusch B.J., Jarrett C.O., Bland D.M. Molecular and Genetic Mechanisms That Mediate Transmission of Yersinia pestis by Fleas // Biomolecules. 2021. V. 11. № 2. P. 210.
Hull K.L. Pestilence and persistence: Yosemite Indian demography and culture in colonial California. Berkeley: University of California Press, 2009. 375 p.
Keller M. et al. Ancient Yersinia pestis genomes from across Western Europe reveal early diversification during the First Pandemic (541–750) // Proceedings of the National Academy of Sciences. 2019. V. 116. № 25. P. 12363–12372.
Kılınç G.M. et al. Human population dynamics and Yersinia pestis in ancient northeast Asia // Science Advances. 2021. V. 7. № 2. 4587.
McCormick M. Rats, Communications, and Plague: Toward an Ecological History // The Journal of Interdisciplinary History. 2003. V. 34. № 1. P. 1–25.
Morozova I. et al. New ancient Eastern European Yersinia pestis genomes illuminate the dispersal of plague in Europe // Philosophical Transactions of the Royal Society. B. 2020. V. 375. Iss. 1812. 20190569.
Müller R., Roberts C.A., Brown T.A. Complications in the study of ancient tuberculosis: Presence of environmental bacteria in human archaeological remains // Journal of Archaeological Science. 2016. V. 68. P. 5–11.
Namouchi A. et al. Integrative approach using Yersinia pestis genomes to revisit the historical landscape of plague during the Medieval Period // Proceedings of the National Academy of Sciences. 2018. V. 115. № 50. P. E11790–E11797.
Paabo S. Ancient DNA: extraction, characterization, molecular cloning, and enzymatic amplification // Proceedings of the National Academy of Sciences. 1989. V. 86. № 6. P. 1939–1943.
Ramirez D.A., Saka H.A., Nores R. Detection of Vibrio cholerae aDNA in human burials from the fifth cholera pandemic in Argentina (1886–1887 AD) // International Journal of Paleopathology. 2021. V. 32. P. 74–79.
Rascovan N. et al. Emergence and Spread of Basal Lineages of Yersinia pestis during the Neolithic Decline // Cell. 2019. V. 176. № 1–2. P. 295–305.
Rasmussen S. et al. Early Divergent Strains of Yersinia pestis in Eurasia 5,000 Years Ago // Cell. 2015. V. 163. № 3. P. 571–582.
Reuter S. et al. Parallel independent evolution of pathogenicity within the genus Yersinia // Proceedings of the National Academy of Sciences. 2014. V. 111. № 18. P. 6768–6773.
Schmid B.V. et al. Climate-driven introduction of the Black Death and successive plague reintroductions into Europe // Proceedings of the National Academy of Sciences. 2015. V. 112. № 10. P. 3020–3025.
Schuenemann V.J. et al. Targeted enrichment of ancient pathogens yielding the pPCP1 plasmid of Yersinia pestis from victims of the Black Death // Proceedings of the National Academy of Sciences. 2011. V. 108. № 38. P. E746–E752.
Scott S., Duncan C.J. Biology of plagues: evidence from historical populations. Cambridge: Cambridge University Press, 2001. 420 p.
Seguin-Orlando A. et al. No particular genomic features underpin the dramatic economic consequences of 17th century plague epidemics in Italy // Science. 2021. V. 24. Iss. 4. 102383.
Skoglund P. et al. Origins and Genetic Legacy of Neolithic Farmers and Hunter-Gatherers in Europe // Science. 2012. V. 336. Iss. 6080. P. 466–469.
Skoglund P. et al. Genomic Diversity and Admixture Differs for Stone-Age Scandinavian Foragers and Farmers // Science. 2014. V. 344. Iss. 6185. P. 747–750.
Spigelman M., Lemma E. The use of the polymerase chain reaction (PCR) to detect Mycobacterium tuberculosis in ancient skeletons // International Journal of Osteoarchaeology. 1993. V. 3. Iss. 2. P. 137–143.
Spyrou M.A. et al. Historical Y. pestis Genomes Reveal the European Black Death as the Source of Ancient and Modern Plague Pandemics // Cell Host & Microbe. 2016. V. 19. Iss. 6. P. 874–881.
Spyrou M.A. et al. Phylogeography of the second plague pandemic revealed through analysis of historical Yersinia pestis genomes // Nature Communications. 2019. V. 10. 4470.
Stenseth N.C. et al. Plague: Past, Present, and Future // PLoS Medicine. 2008. V. 5. № 1. e3.
Susat J. et al. Yersinia pestis strains from Latvia show depletion of the pla virulence gene at the end of the second plague pandemic // Scientific Reports. 2020. V. 10. № 1. 14628.
Valtueña A. et al. The Stone Age Plague and Its Persistence in Eurasia // Current Biology. 2017. V. 27. № 23. P. 3683–3691.
Vernot B. et al. Unearthing Neanderthal population history using nuclear and mitochondrial DNA from cave sediments // Science. 2021. V. 372. Iss. 6542. 590.
Wagner D.M. et al. Yersinia pestis and the Plague of Justinian 541–543 AD: a genomic analysis // The Lancet Infectious Diseases. 2014. V. 14. № 4. P. 319–326.
Yu H. et al. Paleolithic to Bronze Age Siberians Reveal Connections with First Americans and across Eurasia // Cell. 2020. V. 181. Iss. 6. P. 1232–1245.
Zavala E.I. et al. Pleistocene sediment DNA reveals hominin and faunal turnovers at Denisova Cave // Nature. 2021. V. 595. № 7867. P. 399–403.
Zhou D. et al. Genetics of Metabolic Variations between Yersinia pestis Biovars and the Proposal of a New Biovar, microtus // Journal of bacteriology. 2004. V. 186. № 15. P. 5147–5152.
Zhou D., Yang R. Molecular Darwinian Evolution of Virulence in Yersinia pestis // Infection and Immunity. 2009. V. 77. № 6. P. 2242–2250.
Zimbler D.L. et al. Early emergence of Yersinia pestis as a severe respiratory pathogen // Nature Communications. 2015. V. 6. 7487.
Дополнительные материалы отсутствуют.
Инструменты
Российская археология