

Прикладная биохимия и микробиология, 2023, T. 59, № 1, стр. 46-55
Новые ингибиторы панкреатической α-амилазы из Rhaponticum uniflorum
Д. Н. Оленников 1, *, Н. И. Кащенко 1
1 Институт общей и экспериментальной биологии СО РАН
670047 Улан-Удэ, Россия
* E-mail: olennikovdn@mail.ru
Поступила в редакцию 30.05.2022
После доработки 26.07.2022
Принята к публикации 02.09.2022
- EDN: CUYJJV
- DOI: 10.31857/S0555109923010063
Аннотация
Основной стратегией лечения сахарного диабета является контроль постпрандиального уровня глюкозы, в связи с чем α-амилаза поджелудочной железы, гидролизующая сложные углеводы, является важной ферментативной мишенью для научных исследований. В настоящем исследовании водный экстракт и его этилацетатная фракция (ЭАФ) из семян Rhaponticum uniflorum оказывали выраженный ингибиторный эффект на активность панкреатической α-амилазы человека. В результате хроматографического разделения из ЭАФ впервые были выделены и охарактеризованы 16 метаболитов, включая 4-О-, 5-О-, 3,4-ди-О-, 3,5-ди-О-, 4,5-ди-О-кофеилхинная кислота, 6-гидроксилютеолин 7-О-глюкозид, раунозид В, лютеолин 7-О-(6"-О-кофеил)-глюкозид, лютеолин, картамозид, картамогенин, трахелозид, изоферулоил-серотонин, 20-гидроксиэкдизон, 2-дезокси-20-гидроксиэкдизон и новое природное соединение, которое представляло собой картамогенин 4-О-(6"-О-ацетил)-β-D-глюкопиранозид (6"-О-ацетил-картамозид). Данные количественной ВЭЖХ указывали на различное распределение индивидуальных компонентов между эндоспермом и кожурой семени. Исследование влияния соединений на активность панкреатической α-амилазы человека показало, что некоторые флавоноиды, кофеилхинные кислоты, лигнаны и производные серотонина оказывали выраженное ингибиторное действие. Полученные результаты подтверждают вывод о том, что семена Rhaponticum uniflorum могут быть полезным природным источником для разработки средств, ингибирующих α-амилазу.
Сахарный диабет является серьезной проблемой здравоохранения во всем мире, лечение которого без каких-либо побочных эффектов по-прежнему остается большой проблемой. Постпрандиальная гипергликемия, связанная с нарушением углеводного обмена, считается наиболее опасным фактором, вызывающим возникновение и постепенное ухудшение течения сахарного диабета [1]. Ингибиторы панкреатической α-амилазы, фермента играющего ключевую роль в расщеплении сложных углеводов, рассматриваются в качестве эффективных средств для профилактики и лечения таких метаболических расстройств как диабет и ожирение [2]. Растения являются важным источником химических соединений, способных подавлять активность α-амилазы и оказывать положительный биологический эффект на организм человека. В традиционной восточной медицине для лечения диабета применяли различные растительные средства, в том числе сибирский вид Rhaponticum uniflorum (L.) DC. (Leuzea uniflora (L.) Holub, Stemmacantha uniflora (L.) Dittrich) семейства Compositae (Asteraceae), отвары семян которого использовались для лечения мочеизнурения [3]. Данные литературы указывают на присутствие в корнях и траве R. uniflorum экдистероидов [4], полисахаридов [5], сесквитерпенов [6], флавоноидов [4] и гидроксициннаматов [7], однако метаболиты семян R. uniflorum ранее не изучались. Продолжая поиски растительных ингибиторов α-амилазы [8], представляло интерес определить химические соединения – носители биологического эффекта семян R. uniflorum для их дальнейшего практического применения в качестве потенциальных противодиабетических агентов.
Цель работы – исследование химического состава отвара семян R. uniflorum, выделение основных соединений и определение их ингибиторного влияния на активность панкреатической α-амилазы человека.
МЕТОДИКА
Общие экспериментальные условия. Семена R. uniflorum были собраны в Прибайкальском районе (Республика Бурятия, Россия) и высушены в микроволновой вакуумной камере Муссон-1 (“ПК Ингредиент”, Россия) до влажности <5%. Образец сырья хранится в гербарии Института общей и экспериментальной биологии СО РАН (№ BU-COM-0920/29-428).
Для колоночной хроматографии использовали полиамид, нормально- (SiO2) и обращено-фазовый силикагель (ОФ-SiO2), сефадекс LH-20, оксид алюминия (Al2O3) (“Sigma-Aldrich”, Сент-Луис, США). Спектрофотометрические исследования проводили на спектрофотометре СФ-2000 (“ОКБ Спектр”, Россия).
Масс-спектры регистрировали на TQ-масс-спектрометре LCMS-8050 (“Shimadzu”, Япония) [7], спектры ЯМР – на спектрометре VXR 500S (“Varian”, США). Препаративную ВЭЖХ осуществляли на жидкостном хроматографе LC-20 Prominence (“Shimadzu”), снабженном колонкой Shim-pak PREP-ODS (20 × 250 мм, d – 15 мкм) и фотодиодным детектором SPD-M30A (“Shimadzu”), при скорости – 1.0 мл/мин и температуре колонки 20°С.
Экстракция и выделение соединений из семян R. uniflorum. Измельченное сырье (1 кг) экстрагировали водой (1 : 15, 90°С) трижды, после чего водный экстракт упаривали досуха в вакууме (320 г). Экстракт обрабатывали последовательно гексаном, этилацетатом и бутанолом при температуре кипения в аппарате Соклета до истощения, что приводило к получению гексановой (20 г), этилацетатной (ЭАФ, 55 г) и бутанольной фракций (125 г). Фракцию ЭАФ далее анализировали методом хромато-масс-спектрометрии (ВЭЖХ с диодно-матричным и масс-спектрометрическим детектированием, ВЭЖХ-ДМД-МС) на TQ-масс-спектрометре LCMS-8050 (“Shimadzu”, Япония) в условиях, описанных ранее [7]. Для выделения индивидуальных соединений ЭАФ (50 г) разделяли методом колоночной хроматографии на полиамиде (1.5 кг; элюент вода – фракция А, 60%-ный этанол – фракция В, 0.5%-ный аммиак в 90%-ном этаноле – фракция С). Фракцию А (25 г) хроматографировали на колонке с Al2O3 (2 × 50 см, хлороформ–метанол 100 : 0 → 70 : 30), а затем на колонке с ОФ-SiO2 (1 × 20 см, вода–ацетонитрил 95 : 5 → 70 : 30), что привело к выделению 20-гидроксиэкдизона (10 г) и 2-дезокси-20-гидроксиэкдизона (140 мг).
Для разделения фракций В (15 г) и С (18 г) применяли колоночную хроматографию на Сефадексе LH-20 (2 × 90 см) с элюцией в градиенте метанол–вода 90 : 10 → 0 : 100, в результате чего были получены 10 фракций, которые далее хроматографировали на колонке с SiO2 (2 × 40 см, этилацетат–этанол 100 : 0 → 70 : 30), а затем на колонках с ОФ-SiO2 (1 × 20 см, вода–ацетонитрил 95 : 5 → → 50 : 50) и Сефадексе LH-20 (1 × 60 см, метанол–вода–уксусная кислота 90 : 5 : 5 → 20 : 75 : 5). Для дополнительной очистки применяли препаративную ВЭЖХ, используя воду и ацетонитрил в качестве элюентов I и II, соответственно (программа элюирования: 0–40 мин 5–30% I в II, 40–90 мин 30–45% I в II, 90–120 мин 45–58% I в II). В результате из фракции В были выделены 6-гидроксилютеолин 7-О-глюкозид (1.5 г), трахелозид (1.0 г), картамозид (5.5 г), лютеолин (80 мг) и картамогенин (900 мг), а из фракции С – 6"-О-ацетил-картамозид (40 мг), 4-О-кофеилхинная кислота (80 мг), 5-О-кофеилхинная кислота (8.2 г), 3,4-ди-О-кофеилхинная кислота (55 мг), 3,5-ди-О-кофеилхинная кислота (4.5 г), 4,5-ди-О-кофеилхинная кислота (45 мг), раунозид В (1.2 г), лютеолин 7-О-(6"-О-кофеил)-глюкозид (1.5 г) и изоферулоил-серотонин (820 мг). Идентификацию выделенных соединений осуществляли по данным УФ, ЯМР спектроскопии и масс-спектрометрии [4–7, 9].
6"-О-Ацетил-картамозид {картамогенин 4-О-(6"-О-ацетил-)-β-D-глюкопиранозид, 15}. C29H34O12, УФ-спектр (МеОН, λmax, нм): 324. 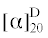 –5.3 (c 0.7, MeOH). HR-ESI-MS, m/z: 573.421 ([M–H]–; расчетное значение 573.557 для C29H33O12). ESI-MS, m/z: положительная ионизация – 613 [M + K]+, 597 [M + Na]+, 575 [M + H]+, 571 [(M + K)–C2H2O]+, 555 [(M + Na)–C2H2O]+, 551 [{(M + H) + + H2O)}–C2H2O]+, 533 [(M + H)–C2H2O]+, 371 [(M + H)–C2H2O–C6H10O5]+, 353 [(M + H)–C2H2O– C6H10O5–H2O]+, 247, 219; отрицательная ионизация – 619 [(M–H) + HCOOH]–, 609 [(M–H) + + 2H2O]–, 577 [{(M–H)–C2H2O} + HCOOH]–, 573 [M–H]–, 567 [{(M–H)–C2H2O} + 2H2O]–, 531 [(M–H)–C2H2O]–, 369 [(M–H)–C2H2O–C6H10O5]–. Спектр ЯМР 1Н (500 Гц, 300 К, МеОН-d4, δ, м.д.): картамогенин: 7.20 (1Н, д, J = 2.0, Н-2), 6.83 (1Н, д, J = 7.8, Н-5),
7.18 (1Н, дд, J = 7.8, 2.0, Н-6), 7.53 (1Н, д, J = 2.0, Н-7), 6.70 (1Н, д, J = 2.0,
Н-2'), 7.26 (1Н, д, J = 8.0, Н-5'), 6.75 (1Н, дд, J = 8.0, 2.0, Н-6'), 3.01 (1Н, дд,
J = 14.0, 5.6, Н-$7_{{\text{A}}}^{'}$), 3.78 (1Н, дд, J = = 14.0, 9.0, Н-$7_{{\text{B}}}^{'}$), 4.11 (1Н, м, Н-8'), 4.41 (1Н, дд, J = 9.0, 7.0, Н-$9_{{\text{A}}}^{'}$), 4.27 (1Н, дд, J = 9.0, 2.0, Н-$9_{{\text{B}}}^{'}$), 3.85 (3H, с, OCH3), 3.75 (3H, с, OCH3), 3.70 (3H, с, OCH3); 4-О-глюкоза: 5.05 (1Н, д, J = 7.5, Н-1"), 3.53 (1Н, м, Н-2"), 3.50 (1Н, м, Н-3"), 3.38
(1Н, м, Н-4"), 3.85 (1Н, м, Н-5"), 4.50 (1Н, дд, J = 12.0, 2.0, Н-
–5.3 (c 0.7, MeOH). HR-ESI-MS, m/z: 573.421 ([M–H]–; расчетное значение 573.557 для C29H33O12). ESI-MS, m/z: положительная ионизация – 613 [M + K]+, 597 [M + Na]+, 575 [M + H]+, 571 [(M + K)–C2H2O]+, 555 [(M + Na)–C2H2O]+, 551 [{(M + H) + + H2O)}–C2H2O]+, 533 [(M + H)–C2H2O]+, 371 [(M + H)–C2H2O–C6H10O5]+, 353 [(M + H)–C2H2O– C6H10O5–H2O]+, 247, 219; отрицательная ионизация – 619 [(M–H) + HCOOH]–, 609 [(M–H) + + 2H2O]–, 577 [{(M–H)–C2H2O} + HCOOH]–, 573 [M–H]–, 567 [{(M–H)–C2H2O} + 2H2O]–, 531 [(M–H)–C2H2O]–, 369 [(M–H)–C2H2O–C6H10O5]–. Спектр ЯМР 1Н (500 Гц, 300 К, МеОН-d4, δ, м.д.): картамогенин: 7.20 (1Н, д, J = 2.0, Н-2), 6.83 (1Н, д, J = 7.8, Н-5),
7.18 (1Н, дд, J = 7.8, 2.0, Н-6), 7.53 (1Н, д, J = 2.0, Н-7), 6.70 (1Н, д, J = 2.0,
Н-2'), 7.26 (1Н, д, J = 8.0, Н-5'), 6.75 (1Н, дд, J = 8.0, 2.0, Н-6'), 3.01 (1Н, дд,
J = 14.0, 5.6, Н-$7_{{\text{A}}}^{'}$), 3.78 (1Н, дд, J = = 14.0, 9.0, Н-$7_{{\text{B}}}^{'}$), 4.11 (1Н, м, Н-8'), 4.41 (1Н, дд, J = 9.0, 7.0, Н-$9_{{\text{A}}}^{'}$), 4.27 (1Н, дд, J = 9.0, 2.0, Н-$9_{{\text{B}}}^{'}$), 3.85 (3H, с, OCH3), 3.75 (3H, с, OCH3), 3.70 (3H, с, OCH3); 4-О-глюкоза: 5.05 (1Н, д, J = 7.5, Н-1"), 3.53 (1Н, м, Н-2"), 3.50 (1Н, м, Н-3"), 3.38
(1Н, м, Н-4"), 3.85 (1Н, м, Н-5"), 4.50 (1Н, дд, J = 12.0, 2.0, Н-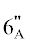 ), 4.25 (1Н, дд, J = 12.0, 5.4, Н-
), 4.25 (1Н, дд, J = 12.0, 5.4, Н-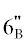 ); 6"-О-ацетил: 1.61 (3H, с, CH3CO). Спектр ЯМР 13С (125 Гц, 300 К, МеОН-d4, δ, м.д.): картамогенин: 128.5 (C, C-1), 116.9 (CH, C-2), 150.7 (C, C-3), 149.5 (C,
C-4), 113.1 (CH, C-5), 125.4 (CH, C-6), 138.7 (CH, C-7), 129.8 (C, C-8), 174.3 (C,
C-9), 132.3 (C, C-1'), 114.3 (CH, C-2'), 149.2 (C, C-3'), 151.2 (C, C-4'), 115.3 (CH,
C-5'), 121.8 (CH, C-6'), 38.0 (CH2, C-7'), 41.5 (CH, C-8'), 71.5 (CH2, C-9'); 4-О-глюкоза: 102.2 (CH, C-1"), 74.5 (CH, C-2"), 77.7 (CH, C-3"), 71.2 (CH, C-4"), 75.0
(CH, C-5"), 64.8 (CH, C-6"); 6"-О-ацетил: 19.7 (CH3, CH3CO), 170.5 (C, CH3CO).
); 6"-О-ацетил: 1.61 (3H, с, CH3CO). Спектр ЯМР 13С (125 Гц, 300 К, МеОН-d4, δ, м.д.): картамогенин: 128.5 (C, C-1), 116.9 (CH, C-2), 150.7 (C, C-3), 149.5 (C,
C-4), 113.1 (CH, C-5), 125.4 (CH, C-6), 138.7 (CH, C-7), 129.8 (C, C-8), 174.3 (C,
C-9), 132.3 (C, C-1'), 114.3 (CH, C-2'), 149.2 (C, C-3'), 151.2 (C, C-4'), 115.3 (CH,
C-5'), 121.8 (CH, C-6'), 38.0 (CH2, C-7'), 41.5 (CH, C-8'), 71.5 (CH2, C-9'); 4-О-глюкоза: 102.2 (CH, C-1"), 74.5 (CH, C-2"), 77.7 (CH, C-3"), 71.2 (CH, C-4"), 75.0
(CH, C-5"), 64.8 (CH, C-6"); 6"-О-ацетил: 19.7 (CH3, CH3CO), 170.5 (C, CH3CO).
Картамозид (картамогенин 4-О-β-D-глюкопиранозид, 12). Спектр ЯМР 1Н (500 Гц, 300 К, МеОН-d4, δ, м.д.): 4-О-глюкоза: 5.01 (1Н, д, J = 7.6, Н-1"), 3.50 (1Н, м, Н-2"), 3.47 (1Н, м, Н-3"), 3.35
(1Н, м, Н-4"), 3.43 (1Н, м, Н-5"), 3.58 (1Н, дд, J = = 12.1, 1.9, Н-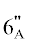 ), 3.40 (1Н, дд, J = 12.1, 5.6, Н-
), 3.40 (1Н, дд, J = 12.1, 5.6, Н-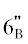 ). Спектр ЯМР 13С (125 Гц, 300 К, МеОН-d4, δ, м.д.): 4-О-глюкоза: 102.4 (CH, C-1"), 74.5 (CH, C-2"), 77.9 (CH, C-3"), 70.8 (CH, C-4"), 76.2
(CH, C-5"), 60.9 (CH, C-6").
). Спектр ЯМР 13С (125 Гц, 300 К, МеОН-d4, δ, м.д.): 4-О-глюкоза: 102.4 (CH, C-1"), 74.5 (CH, C-2"), 77.9 (CH, C-3"), 70.8 (CH, C-4"), 76.2
(CH, C-5"), 60.9 (CH, C-6").
Гидролиз. Для осуществления кислотного гидролиза навеску 6"-О-ацетил-картамозида (5 мг) нагревали с 2 М ТФУ (4 мл) при 100°С в течение 2 ч, далее гидролизат упаривали в вакууме досуха. Сухой остаток растворяли в 50%-ном этаноле (2 мл) и пропускали раствор через полиамидный картридж (3000 мг, “Capital Analytical”, Великобритания), элюируя последовательно водой (50 мл; элюат I) и 70%-ным этанолом (100 мл; элюат II). Для выявления присутствия моносахаридов порцию элюата I дериватизировали 3-метил-1-фенил-2-пиразолин-5-оном и анализировали методом ВЭЖХ, как описано ранее [10]. Для определения принадлежности моносахаридов к D- и L-ряду в элюате I использовали метод восстановительного аминирования с L-триптофаном [11] с последующим анализом методом ВЭЖХ [10]. Гидролиз с 0.5%-ной NaOH проводили как описано ранее [12].
Биологическая активность. Влияние экстрактов, фракций и индивидуальных соединений на активность α-амилазы изучали спектрофотометрическим методом [8] с использованием α-амилазы поджелудочной железы человека (400 ед./мл; “Lee Biosolutions”, США), α-амилазы слюнной железы человека (тип IX-A, 3000 ед./мг белка; “Sigma-Aldrich”) и α-амилазы поджелудочной железы свиней (тип I-A, 1000 ед./мг белка; “Sigma-Aldrich”). Акарбоза использовалась в качестве положительного контроля. Ингибиторная активность выражалась величиной IC50 (концентрация, вызывающая 50% ингибирование активности фермента) в мкг/мл, которую определяли графически после построения зависимости ингибиторной активности от концентрации.
Для ВЭЖХ-микрофракционирования использовали условия хроматографического анализа, указанные в работе [7], при которых отдельные фракции собирались каждые 30 с. После этого элюаты концентрировали досуха и растворяли в 50 мкл 50%-ного метанола, добавляли 50 мкл воды, 2.5 мл 2%-ной суспензии крахмала, окрашенного ремазол-бриллиантовым синим R (“Sigma-Aldrich”), 500 мкл α-амилазы поджелудочной железы человека (0.4 ед./мл), инкубировали 50 мин при 37°C. Оптическую плотность пробы определяли при длине волны 620 нм [8]. Элюаты с наиболее выраженным ингибированием фермента предотвращали образование синего комплекса, в противоположность неактивным пробам, которые давали интенсивное окрашивание. Отсутствие активности определялось для фракции со временем удерживания 0.5–1.0 мин.
Тест на летальность. Цисты Artemia salina (50 мг; “Арсал”, Россия) инкубировали при 25°С в 1 л искусственной морской воды (“Sigma-Aldrich”), в которую через 24 ч вносили 15 мл 0.06%-ной суспензии дрожжей и продолжали инкубацию еще 48 ч. Для анализа на одну пробу отбирали 10 живых личинок Artemia salina, которых помещали в 5 мл искусственной морской воды, содержащей исследуемое вещество (20–2000 мкг/мл) или 0.9%-ный раствор NaCl (контроль). Через 24 ч проводили подсчет живых личинок, после чего рассчитывали показатель 50%-ной летальности с применением программы MediCalc (“MedCalc Software Ltd”, Бельгия).
Статистический анализ проводили с использованием однофакторного дисперсионного анализа (ANOVA). Значимость различий средних определяли с помощью многорангового теста Дункана. Отличия при р < 0.05 считались статистически значимыми.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Исследование влияния водного отвара (ВО) R. uniflorum на активность трех α-амилаз млекопитающих показало, что он демонстрировал наибольшее ингибирование α-амилазы поджелудочной железы человека – IC50 397.11 мкг/мл (табл. 1). После экстракции ВО различными растворителями были получены фракции, из которых наиболее активной оказалась этилацетатная фракция (IC50 96.25 мкг/мл), для исследования которой было проведено ее разделение с применением хромато-масс-спектрометрии (ВЭЖХ-ДМД-МС) и колоночной хроматографии.
Таблица 1.
Показатели 50%-ного ингибирования активности (IC50, мг/мл) α-амилазы млекопитающих препаратами из R. uniflorum (мг/мл ± SD)
| Препарат | α-Амилаза | ||
|---|---|---|---|
| поджелудочная железы человека | поджелудочная железы свиней | слюнные железы человека | |
| Водный отвар (ВО) | 397.11 ± 12.42* | >500 | 495.63 ± 15.84* |
| Гексановая фракция ВО | >500 | >500 | >500 |
| Этилацетатная фракция ВО | 96.25 ± 2.98* | 163.14 ± 5.05* | 126.03 ± 3.52* |
| Бутанольная фракция ВО | 363.15 ± 11.25* | >500 | 402.56 ± 11.65* |
| Акарбоза (вещество сравнения) | 57.34 ± 1.61 | 32.65 ± 0.97 | 81.16 ± 2.59 |
Согласно данным ВЭЖХ-ДМД-МС в этилацетатной фракции семян R. uniflorum было выявлено присутствие 16 соединений (1–16), идентификацию которых осуществляли по результатам определения хроматографической подвижности (рис. 1а), УФ-, масс-спектров (табл. 2) в сравнении с известными соединениями и данными литературы, а также после выделения и анализа спектров ЯМР (рис. 2).
Рис. 1.
Хроматограмма (ВЭЖХ-ДМД) этилацетатной фракции семян Rhaponticum uniflorum при 240 и 330 нм (а) и ингибиторная активность ВЭЖХ-элюатов в отношении панкреатической α-амилазы (б). Номера пиков соединений соответствуют обозначениям в табл. 2.
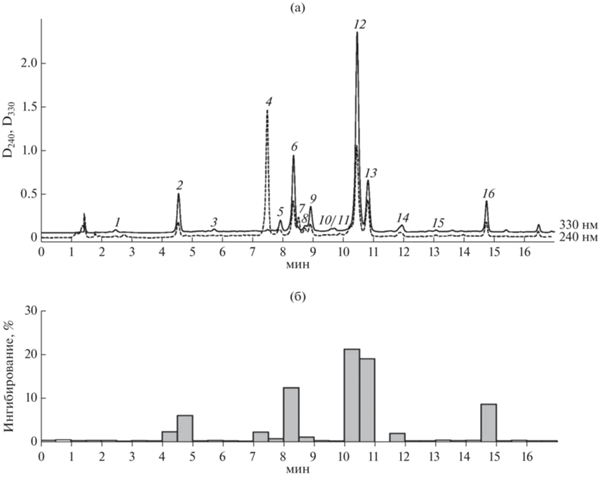
Таблица 2.
Хроматографическая подвижность (tR), молекулярная формула, данные УФ- (λmax) и масс-спектров соединений 1–14, 16 из семян R. uniflorum
| № | Соединение | tR, мин | Формула | УФ, λmax, нм | Данные масс-спектров, m/z | |||
|---|---|---|---|---|---|---|---|---|
| положительная ионизация | отрицательная ионизация | |||||||
| [M + H]+ | дополнительные ионы | [M–H]– | дополнительные ионы | |||||
| 1 | 4-О-Кофеилхинная кислота | 2.48 | C16H18O9 | 326 | 355 | – | 353 | 191 |
| 2 | 5-О-Кофеилхинная кислота | 4.51 | C16H18O9 | 326 | 355 | – | 353 | 707, 191 |
| 3 | 6-Гидроксилютеолин 7-О-глюкозид | 5.79 | C21H20O12 | 271, 345 | 465 | 303 | 463 | 301 |
| 4 | 20-Гидроксиэкдизон | 7.47 | C27H44O7 | 241 | 481 | 519, 503, 463, 445, 427, 409, 371, 347, 329, 303, 301 | 479 | 525, 515 |
| 5 | 3,4-Ди-О-кофеилхинная кислота | 7.92 | C25H24O12 | 325 | 517 | – | 515 | 563, 353, 191 |
| 6 | 3,5-Ди-О-кофеилхинная кислота | 8.39 | C25H24O12 | 325 | 517 | – | 515 | 563, 353, 191 |
| 7 | 2-Дезокси-20-гидроксиэкдизон | 8.52 | C27H44O6 | 243 | 465 | 503, 487, 447, 429, 411 | 463 | 509, 499 |
| 8 | 4,5-Ди-О-кофеилхинная кислота | 8.72 | C25H24O12 | 325 | 517 | – | 515 | 563, 353, 191 |
| 9 | Трахелозид | 8.90 | C27H34O12 | 280 | 551 | 589, 573, 389 | 549 | 595, 387 |
| 10 | Раунозид В | 9.58 | C30H26O13 | 253, 283, 347 | 595 | 303 | 593 | 463, 301 |
| 11 | Лютеолин 7-О-(6′′-О-кофеил)-глюкозид | 9.67 | C30H26O14 | 270, 295, 336 | 611 | 287 | 609 | 447, 285 |
| 12 | Картамозид | 10.45 | C27H32O11 | 325 | 533 | 571, 555, 551, 371, 353, 247, 219 | 531 | 577, 567, 369 |
| 13 | Изоферулоил-серотонин | 10.86 | C20H20N2O4 | 292, 314 | 353 | 391, 375, 177 | 351 | 397, 175 |
| 14 | Лютеолин | 11.93 | C15H10O6 | 266, 345 | 287 | 285 | – | |
| 16 | Картамогенин | 14.79 | C21H22O6 | 331 | 371 | 353, 247, 219 | 369 | – |
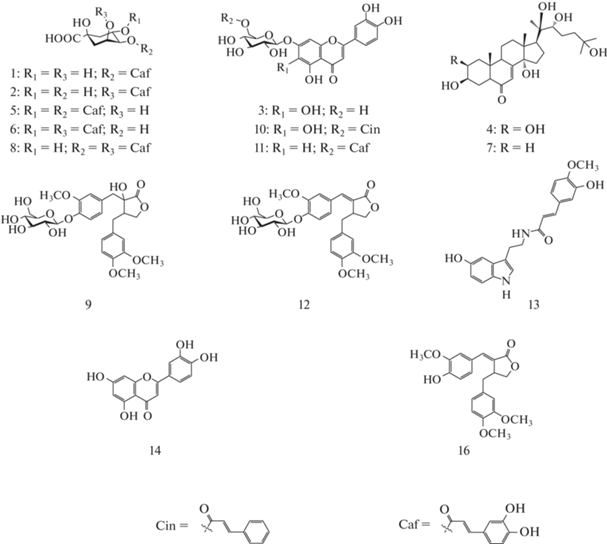
Рис. 2.
Структурные формулы соединений, обнаруженных в семенах R. uniflorum: 1 – 4-О-кофеилхинная кислота, 2 – 5-О-кофеилхинная кислота, 3 – 6-гидроксилютеолин 7-О-глюкозид, 4 – 20-гидроксиэкдизон, 5 – 3,4-ди-О-кофеилхинная кислота, 6 – 3,5-ди-О-кофеилхинная кислота, 7 – 2-дезокси-20-гидроксиэкдизон, 8 – 4,5-ди-О-кофеилхинная кислота, 9 – трахелозид, 10 – раунозид В, 11 – лютеолин 7-О-(6"-О-кофеил)-глюкозид, 12 – картамозид, 13 – изоферулоил-серотонин, 14 – лютеолин, 16 – картамогенин.
Компоненты 1, 2, 5, 6 и 8 обладали близким УФ-профилем, типичным для производных кофейной кислоты [13], а характер масс-спектров указывал на присутствие фрагмента хинной кислоты ацилированного одним (1, 2) или двумя (5, 6, 8) ее остатками [4]. Учитывая хроматографическую подвижность компонентов в сравнении с веществами-референтами, указанные соединения были идентифицированы как 4-О-кофеилхинная (1), 5-О-кофеилхинная (2), 3,4-ди-О-кофеилхинная (5), 3,5-ди-О-кофеилхинная (6) и 4,5-ди-О-кофеилхинная кислоты (8).
Близким к кофеилхинным кислотам УФ-профилем обладали компоненты 12 и 16. В масс-спектре отрицательной ионизации соединения 12 присутствовали сигналы депротонированной частицы (m/z 531) и ее фрагмента, обусловленного удалением гексозильного остатка (m/z 369). В спектре положительной ионизации были отмечены сигналы частиц с m/z 247 и 219, характерных для лигнановых производных типа 7,8-дидегидроарктигенина [9]. После выделения и анализа спектров ЯМР соединение 12 было идентифицировано как картамозид (картамогенин-4-О-глюкозид), ранее выделенный из семян Rhaponticum carthamoides [9] и впервые обнаруженный у R. uniflorum. Соединение 16 было определено как агликон картамозида – картамогенин. Близким к 12 масс-спектральным профилем обладал компонент 9, молекулярная масса которого была на 18 а.е.м. больше, что характерно для производных гидроксиарктигенина [9]. Хроматографические и спектральные параметры 9 и лигнанового гликозида трахелозида (трахелогенин-4-О-глюкозид) были идентичны, что позволило впервые выявить его присутствие в R. uniflorum. Данное соединение часто встречается в семенах различных видов Compositae, в том числе и R. carthamoides [9].
Соединение 13 обладало типичным для производных изоферуловой кислоты спектром поглощением с λmax у 292 и 314 нм, а также масс-спектральным профилем, отмеченным ранее для эфиров N-цинамоил-серотонинов [9]. После выделения и дополнительной ЯМР спектральной характеристики 13 было идентифицировано как N-транс-изоферулоил-серотонин, впервые обнаруженный в R. carthamoides [9]. Ранее в R. uniflorum данное соединение выявлено не было.
Два соединения 4 и 7 были идентифицированы как экдистероиды по характерному для данной группы соединений УФ- и масс-спектрометрическому профилю. Компоненты содержали в масс-спектрах положительной ионизации набор сигналов, отнесенных к протонированной частице и частицам аддуктов с ионами Na+ и K+, а также набор сигналов, вызванных постепенным удалением воды боковой группы и др. После сравнения с данными известных веществ 4 и 7 были идентифицированы как 20-гидроксиэкдизон и 2-дезокси-20-гидроксиэкдизон, соответственно. Оба соединения ранее были обнаружены в траве и корнях R. uniflorum [7].
Соединения 3, 10, 11 и 14 были определены как флавоноиды ввиду характерного для производных флавона типа поглощения в УФ-области спектра [13]. После анализа полученных данных с таковыми известных соединений компоненты были определены как 6-гидроксилютеолин 7-О-глюкозид (3), раунозид В (10), лютеолин 7-О-(6"-О-кофеил)-глюкозид (11) и лютеолин (14), которые ранее были выявлены в листьях и цветках R. uniflorum [4], но обнаружены в семенах этого вида впервые.
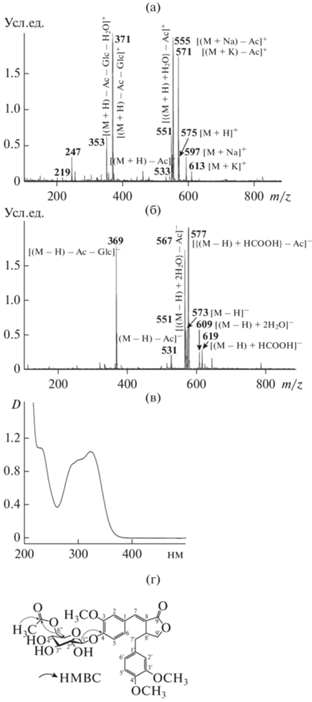
Соединению 15 соответствовала молекулярная формула C29H34O12 по данным масс-спектрометрии и спектроскопии ЯМР 13С. В масс-спектре положительной ионизации присутствовали сигналы протонированного иона (m/z 575) и частиц, образованных последовательным удалением фрагмента ацетильной группы (m/z 575 → 533) и остатка гексозы (m/z 533 → 371), что так же наблюдалось в спектре отрицательной ионизации (рис. 3а, 3б). Спектр поглощения был близок к спектрам лигнановых гликозидов (рис. 3в). После гидролиза с 0.5%-ным NaOH наблюдалось образование картамозида, а гидролиз с 2 М ТФУ приводил к образованию картамогенина и D-глюкозы. Спектры ЯМР 1Н и 13С были близки к таковым картамозида (картамогенин 4-О-β-D-глюкопиранозид) за исключением присутствия дополнительных сигналов ацетильной группы в спектре 1Н (δН 1.61) и 13С (δС 19.7, 170.5) м.д. Сдвиг в слабое поле сигналов Н-6" (δН 3.40, 3.58 → 4.25, 4.50) и С-6" (δН 60.9 → 64.8) в сравнении с картамозидом указывал на присутствие заместителя по положению С-6" глюкопиранозы, которым оказалась ацетильная группа, на что указывали корреляции в спектре гетероядерной многосвязной корреляционной спектроскопии HMBC (δН/δС 3.40, 3.58/170.5). Таким образом, соединение 15 представляло собой картамогенин 4-О-(6"-О-ацетил)-β-D-глюкопиранозид или 6"-О-ацетил-картамозид (рис. 3г), являющийся новым природным соединением.
Рис. 3.
Масс-спектры (а – положительная ионизация, б – отрицательная ионизация; Ac – ацетил, Glc – глюкоза), спектр поглощения (в) и структура (г) 6"-О-ацетил-картамозида (соединение 15).
Сведения о количественном распределении отдельных соединений в частях семени R. uniflorum указывали на то, что для эндосперма характерно накопление кофеилхинных кислот (8.89 мг/г), лигнанов (34.85 мг/г) и экдистероидов (27.20 мг/г), в то время как в кожуре наблюдалось аккумуляция флавоноидов (9.78 мг/г) и производных серотонина (2.03 мг/г) (табл. 3). Ранее сходный характер распределения был выявлен в семенах R. carthamoides, для которых наибольшее содержание лигнанов трахелозида и картамозида было установлено в эндосперме, а ферулоил-серотонина – в кожуре семян [14].
Таблица 3.
Концентрация индивидуальных соединений в эндосперме и кожуре семян R. uniflorum и показатель ингибирования панкреатической α-амилазы человека (IC50)
| Соединение | Концентрация, мг/г сухой массы | IC50, мкг/мл | |
|---|---|---|---|
| в эндосперме | в кожуре | ||
| Кофеилхинные кислоты | |||
| 4-О-Кофеилхинная кислота | 0.46 ± 0.01 | <0.10 | 125.32 ± 3.70* |
| 5-О-Кофеилхинная кислота | 1.48 ± 0.03 | 0.77 ± 0.02 | 67.11 ± 2.01* |
| 3,4-Ди-О-кофеилхинная кислота | 0.57 ± 0.01 | <0.10 | 61.02 ± 1.89* |
| 3,5-Ди-О-кофеилхинная кислота | 6.12 ± 0.12 | 4.01 ± 0.08 | 41.27 ± 1.19* |
| 4,5-Ди-О-кофеилхинная кислота | 0.26 ± 0.00 | <0.10 | 26.84 ± 0.75* |
| Флавоноиды | |||
| 6-Гидроксилютеолин 7-О-глюкозид | 3.05 ± 0.06 | 3.19 ± 0.07 | 28.14 ± 0.84* |
| Раунозид В | 0.63 ± 0.01 | 2.39 ± 0.04 | 10.50 ± 0.30* |
| Лютеолин 7-О-(6′′-О-кофеил)-глюкозид | 0.71 ± 0.02 | 2.57 ± 0.05 | 14.83 ± 0.45* |
| Лютеолин | < 0.10 | 1.07 ± 0.02 | 25.67 ± 0.77* |
| Лигнаны | |||
| Трахелозид | 5.48 ± 0.09 | 2.32 ± 0.04 | >500 |
| Картамозид | 28.62 ± 0.65 | 3.41 ± 0.07 | 85.63 ± 2.09* |
| 6′′-О-Ацетил-картамозид | <0.10 | <0.10 | 159.16 ± 4.77* |
| Картамогенин | 0.75 ± 0.02 | 2.27 ± 0.04 | 30.32 ± 0.46* |
| Производные серотонина | |||
| Изоферулоил-серотонин | <0.10 | 2.03 ± 0.03 | 50.02 ± 1.45* |
| Экдистероиды | |||
| 20-Гидроксиэкдизон | 26.95 ± 0.56 | 6.12 ± 0.14 | >500 |
| 2-Дезокси-20-гидроксиэкдизон | 0.25 ± 0.00 | <0.10 | >500 |
Проведенные исследования биологической активности индивидуальных соединений из семян R. uniflorum показали, что наиболее эффективными ингибиторами панкреатической α-амилазы человека были раунозид В (IC50 10.50 мкг/мл) и лютеолин 7-О-(6"-О-кофеил)-глюкозид (IC50 14.83 мкг/мл), активность которых была выше таковой акарбозы (табл. 3). Оба флавоноида содержали свободную орто-дигидрокси-группировку в кольце В агликона и кофеильном фрагменте, что является структурным фактором, повышающим ингибиторное влияние соединений на α-амилазу [15]. Среди кофеилхинных кислот и лигнанов наибольшая эффективность была выявлена для 4,5-ди-О-кофеилхинной кислоты (IC50 26.84 мкг/мл) и картамогенина (IC50 30.32 мкг/мл).
Активность изоферулоил-серотонина была близка к таковой акарбозы, а экдистероиды и трахелозид слабо ингибировали активность α-амилазы поджелудочной железы человека (IC50 > 500 мкг/мл). Применение ВЭЖХ-микрофракционирования с последующим анализом активности полученных элюатов показало, что наибольший вклад в суммарную активность отвара семян R. uniflorum вносило присутствие картамозида, изоферулоил-серотонина, 3,5-ди-О-кофеилхинной кислоты, картамогенина и 5-О-кофеилхинной кислоты (до 86% от суммарной активности пробы), что обусловлено высоким содержанием указанных соединений в препарате (рис. 1б).
С использованием теста на летальность с Artemia salina была определена токсичность выделенных соединений, составившая >1000 мкг/мл для всех веществ, что позволило предположить относительную безопасность при их применении.
Научные сведения, касающиеся эффективности ингибирования α-амилазы фенольными соединениями растений, в большей своей части получены в результате использования ферментов бактериального (Bacillus licheniformis, B. amyloliquefaciens), грибного (Aspergillus oryzae) происхождения, а также α-амилазы поджелудочной железы свиньи и слюнной железы человека [2]. Эксперименты, проведенные с применением α-амилазы поджелудочной железы человека немногочисленны, однако известно, что бисдеметоксикуркумин [16], дегидродиэвгенол B [17] и ругозин D [8] характеризуются наибольшей эффективностью ингибирования фермента со значениями IC50 7.70, 9.68 и 30.84 мкг/мл соответственно. Однако, следует отметить, что токсичность куркуминоидов, производных эвгенола и эллаготаннинов группы ругозина, определенная методом с Artemia salina, составляла <1000 [18], <1 [19] и <100 мкг/мл [20] соответственно, что характеризовало эти соединения как более токсичные в сравнении с метаболитами семян R. uniflorum.
Проведенные исследования впервые показали, что водный отвар семян R. uniflorum является эффективным ингибитором панкреатической α-амилазы человека, что обусловлено высоким содержанием фенольных соединений различных структурных типов. Наибольшим ингибиторным действием обладали раунозид В, лютеолин 7-О-(6"-О-кофеил)-глюкозид, лютеолин, 4,5-ди-О-кофеилхинная кислота, 6-гидроксилютеолин 7-О-глюкозид и картамогенин, активность которых установлена впервые. Эти соединения могут быть использованы для создания новых потенциальных антидиабетических средств, получаемых из растений.
Исследование выполнено при поддержке Министерства науки и высшего образования Российской Федерации в рамках научного проекта № 121030100227-7.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Alam S., Sarker M.M.R., Sultana T.N., Chowdhury M.N.R., Rashid M.A., Chaity N.I. et al. // Front. Endocrinol. 2022. V. 13. № 800714. https://doi.org/10.3389/fendo.2022.800714
Sales P.M., Souza P.M., Simeoni L.A., Magalhães P.O., Silveira D. // J. Pharm. Pharm. Sci. 2009. V. 15. P. 141–183. https://doi.org/10.18433/j35s3k
Баторова С.М., Яковлев Г.П., Асеева Т.А. Справочник лекарственных растений традиционной тибетской медицины. Новосибирск: Наука, 2003. 291 с.
Olennikov D.N., Kashchenko N.I. // Chem. Nat. Comp. 2019. V. 55. P. 256–264. https://doi.org/10.1007/s10600-019-02662-2
Olennikov D.N. // Chem. Nat. Comp. 2018. V. 54. P. 751–754. https://doi.org/10.1007/s10600-018-2462-4
Olennikov D.N. // Chem. Nat. Comp. 2019. V. 55. P. 157–159. https://doi.org/10.1007/s10600-019-02642-6
Shantanova L.N., Olennikov D.N., Matkhanov I.E., Gulyaev S.M., Toropova A.A., Nikolaeva I.G., Nikolaev S.M. // Pharmaceuticals. 2021. V. 14. № 1186. https://doi.org/10.3390/ph14111186
Olennikov D.N., Chemposov V.V., Chirikova N.K. // Plants. 2021. V. 10. № 2525. https://doi.org/10.3390/plants10112525
Harmatha J., Buděšínský M., Vokáč K., Pavlik M., Grüner K., Laudová V. // Collect. Czech. Chem. Commun. 2007. V. 72. P. 334–346. https://doi.org/10.1135/cccc20070334
Olennikov D.N., Chirikova N.K., Kashchenko N.I., Gornostai T.G., Selyutina I.Y., Zilfikarov I.N. // Int. J. Mol. Sci. 2017. V. 18. № 2579. https://doi.org/10.3390/ijms18122579
Akabane M., Yamamoto A., Aizawa S., Taga A., Kodama S. // Analyt. Sci. 2014. V. 30. P. 739–743. https://doi.org/10.2116/analsci.30.739
Olennikov D.N., Chirikova N.K. // Chem. Nat. Comp. 2019. V. 55. P. 1032–1038. https://doi.org/10.1007/s10600-019-02887-1
Olennikov D.N., Chirikova N.K., Kashchenko N.I., Nikolaev V.M., Kim S.-W., Vennos C. // Front. Pharmacol. 2018. V. 9. № 756. https://doi.org/10.3389/fphar.2018.00756
Sólyomváry A., Mervai Z., Molnár-Perl I., Boldizsár I. // Nat. Prod. Res. 2014. V. 28. P. 732–739. https://doi.org/10.1080/14786419.2013.879473
Tadera K., Minami Y., Takamatsu K., Matsuoka T. // J. Nutr. Sci. Vitaminol. 2006. V. 52. P. 149–153. https://doi.org/10.3177/jnsv.52.149
Dandekar P.D., Kotmale A.S., Chavan S.R., Kadlag P.P., Sawant S.V., Dhavale D.D., Kumar A.R. // ACS Omega. 2021. V. 6. P. 1780–1786. https://doi.org/10.1021/acsomega.0c00617
Ponnusamy S., Zinjarde S., Bhargava S., Rajamohanan P.R., Ravikumar A. // Food Chem. 2012. V. 135. P. 2638–2642. https://doi.org/10.1016/j.foodchem.2012.06.110
García A.L.L., Olaya M.Q.J.H., Sierra A.J.I. // Rev. Cubana Plant. Med. 2017. V. 22. P. 1–14.
Cansian R.L., Vanin A.B., Orlando T., Piazza S.P., Puton B.M.S., Cardoso R.I. et al. // Braz. J. Biol. 2017. V. 77. P. 155–161. https://doi.org/10.1590/1519-6984.12215
Yamasaki T., Sato M., Mori T., Mohamed A.S.A., Fujii K., Tsukioka J. // J. Nat. Toxins. 2002. V. 11. P. 165–171.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология