

Прикладная биохимия и микробиология, 2022, T. 58, № 5, стр. 500-506
Ферментативно синтезированный полианилин, допированный ионами меди: физико-химические и антимикробные свойства
И. С. Васильева 1, Г. П. Шумакович 1, О. В. Морозова 1, А. И. Ярополов 1, *
1 Институт биохимии им. А.Н. Баха, Федеральный исследовательский центр
“Фундаментальные основы биотехнологии” Российской академии наук
119071 Москва, Россия
* E-mail: yaropolov@inbi.ras.ru
Поступила в редакцию 23.03.2022
После доработки 04.04.2022
Принята к публикации 27.04.2022
- EDN: GJDMRE
- DOI: 10.31857/S0555109922050154
Аннотация
Разработан ферментативный синтез интерполиэлектролитного комплекса полианилин (ПАНИ)/полистиролсульфонат натрия (ПСС), в котором ПАНИ допирован ионами Cu(II). Биокатализатором полимеризации анилина (АНИ) являлась грибная лакказа Trametes hirsuta, а окислителем – атмосферный кислород. Полученный комплекс ПАНИ-Cu/ПСС был исследован методами УФ-видимой и FTIR-ATR спектроскопии и рентгенофлуоресцентного анализа. Весовое содержание меди в ПАНИ-Cu/ПСС составляло ~8 вес. %. Минимальная ингибирующая концентрация (МИК) комплекса ПАНИ‑Cu/ПСС, блокирующего рост грамотрицательных (Escherichia coli) и грамположительным (Staphylococcus aureus) бактерий составляла 2.65 мг/мл и 0.66 мг/мл соответственно.
В последние десятилетия большое внимание уделяется гибридным системам и композитам на основе электропроводящего полианилина (ПАНИ), что связано с перспективностью их использования в различных областях [1–5]. Свойства ПАНИ можно изменять в широких пределах в зависимости от условий проведения синтеза [6, 7]. Обычно ПАНИ синтезируют химическим или электрохимическим окислением анилина (АНИ) в сильнокислой среде [1, 2, 8, 9]. Также описан ферментативный метод получения электропроводящего ПАНИ, отвечающий требованиям “белой” биотехнологии [1, 10–15]. С экологической точки зрения ферментативный синтез ПАНИ является хорошей альтернативой традиционным методам синтеза.
Перспективным катализатором окислительной полимеризации АНИ является лакказа, так как окислителем в реакции является кислород воздуха. Лакказа (п-дифенол:кислород оксидоредуктаза, КФ 1.10.3.2) относится к медь-содержащим оксидазам и катализирует окисление различных органических соединений, в результате которого образуются радикалы, вступающие в реакции сочетания с образованием олигомерных/полимерных продуктов [16–18].
Электрические и оптические свойства ПАНИ можно обратимо контролировать редокс реакциями или протонированием [19, 20]. Окислительное состояние ПАНИ может изменяться от полностью окисленного (пернигранилин) до полностью восстановленного (лейкоэмеральдин). Полуокисленное состояние основной цепи полимера называют эмеральдином [1, 2]. Электропроводящую форму ПАНИ, эмеральдиновую соль, можно получить как протонированием [1, 2, 21], так и допированием цепи полимера различными допантами, такими как кислоты Льюиса, йод, соли переходных металлов [22–26]. Допантами полимеров называют доноры или акцепторы электронов, которые при взаимодействии с основной цепью полимера приводят к образованию на ней зарядов, обуславливающих электропроводность полимера.
В ряде работ было показано, что химически синтезированный ПАНИ обладает антимикробными свойствами [27–32]. Эффективность химически синтезированного ПАНИ подавлять рост грамположительных и грамотрицательных бактерий и грибов впервые была описана в работе [27]: хлопчатобумажные ткани, покрытые электропроводящим ПАНИ, подавляли рост S. aureus на 95%, E. coli на 85% и грибов Candida albicans на 92%. Авторы связывали антибактериальные эффекты ПАНИ с электростатическим взаимодействием между молекулами полимера и клетками бактерий, что приводило к разрушению клеточных стенок и гибели клеток. Ши с соавт. [28] показали, что пленки поливинилового спирта, содержащие 1–10 вес. % ПАНИ полностью подавляли рост бактерий E. coli и S. aureus.
Известно, что медь и ее сплавы являются антимикробными материалами [33, 34]. Можно предположить, что ионы меди Cu2+ в качестве допанта основной цепи полимера могут усиливать антимикробные свойства ПАНИ.
Цель работы – провести ферментативный матричный синтез и изучить физико-химические и антибактериальные свойства полианилина, допированного ионами меди(II).
МЕТОДИКА
Лимонная кислота, NaH2PO4, KH2PO4, NaOH – производства “Riedel-de Haën” (Германия), CuSO4 · 5H2O, полистиролсульфонат (ПСС, 30 вес. %) “Sigma-Aldrich” (США) были использованы без дополнительной очистки. АНИ (“Sigma-Aldrich”, США) был очищен вакуумной перегонкой.
Лакказа была выделена из культуральной жидкости базидиального гриба Trametes hirsuta (Wulfen) Pilát (штамм T. hirsuta 56) согласно методу [35]. Фермент был гомогенен по данным ДДС-электрофореза и имел удельную активность 161 МЕ/мг белка. Активность фермента определяли спектрофотометрически, используя в качестве хромогенного субстрата 1 мМ раствор АБТС (λ = 420 нм; ε = 36000 M–1 см–1) в 0.1 М Na-цитратно-фосфатном буфере, рН 4.5. За единицу активности принимали количество фермента, катализирующего превращение 1 мкмоль АБТС за 1 мин при температуре 22°С. Концентрация белка, измеренная согласно методу [36], составляла 7.8 мг/мл.
Все растворы готовили с использованием воды, очищенной на установке Simplicity (“Millipore”, США).
Синтез ПАНИ/ПСС проводили следующим образом. К 10 мл раствора ПСС в 0.1 М цитратно-фосфатном буфере, рН 3.5 (концентрация 11 мМ на повторяющееся звено полимера), добавляли 10 мкл АНИ (концентрация 11 мМ) и перемешивали в течение 1 ч. Полимеризацию АНИ инициировали добавлением фермента, удельная активность которого в реакционной смеси была ~1 МЕ/мл. Реакцию проводили при комнатной температуре (21–22°С) и постоянном перемешивании со скоростью 400 об./мин на магнитной мешалке RT-10 (“IKA®-Werke GmbH & Co”, Германия) в течение 24 ч. Затем реакционную смесь диализовали против деионизированной воды для удаления низкомолекулярных соединений.
Дедопирование интерполимерного комплекса ПАНИ/ПСС проводили, добавляя к диализованному раствору 1 М NaOH до рН 10.0. Через 12 ч раствор диализовали против воды, рН которой доводили до значения 7.8 раствором NaOH. Для редопирования комплекса ионами меди к раствору добавляли 27.5 мг CuSO4 · 5H2O (молярное соотношение АНИ/CuSO4 · 5H2O = 1 : 1), перемешивали при комнатной температуре (21–22°С) на магнитной мешалке со скоростью 400 об./мин в течение 12 ч и диализовали против деионизированной воды.
УФ-видимые спектры регистрировали на спектрофотометре UV1240 mini (“Shimadzu”, Япония). Анализ образцов методом спектроскопии нарушенного полного внутреннего отражения в инфракрасной области с преобразованием Фурье (FTIR-ATR) проводили на спектрометре FT/IR-67000 (“Jasco”, Япония). Содержание меди в образцах определяли методом энергодисперсионной рентгеновской спектроскопии (EDX) с использованием сканирующего электронного микроскопа JSM-6510LV (“JEOL Ltd”, Япония), оснащенного энергодисперсионной приставкой сX-MAX (“Oxford Instruments”, Великобритания). Для определения содержания меди и исследований методом FTIR-ATR спектроскопии образцы высушивали при 60°С до постоянного веса.
Для исследований на антимикробную активность водные растворы комплексов были сконцентрированы упариванием при 37°C до 5.8 мг/мл (ПАНИ/ПСС) и 5.3 мг/мл (ПАНИ-Cu/ПСС). Минимальную ингибирующую рост концентрацию (МИК) синтезированных комплексов определяли по стандартной методике серийных (двукратных) разведений в питательной среде LB (Luria-Bertani, Miller, “Sigma”, США). В качестве тест-культур использовали грамположительные бактерии Staphylococcus aureus 209P и грамотрицательные бактерии Escherichia coli K-12. 200 мкл предварительно стерильно приготовленных сред LB, содержащих различные концентрации комплексов, вносили в лунки 96-луночного планшета в 3-кратной повторности для каждой концентрации. Затем в каждую лунку вносили 4 мкл клеток тест-культур стационарной фазы роста (1 сут) и инкубировали при 28°C и перемешивании на роторной качалке (150 об./мин). После 24 ч инкубации измеряли оптическую плотность каждой лунки относительно контролей (без инокулята) при 540 нм с использованием Ao Absorbance Microplate Reader (“Azure Biosystems”, UK). Рост микроорганизмов оценивали по изменению оптической плотности по сравнению с начальным значением (сразу после инокуляции среды). МИК определяли как наименьшую концентрацию соединения, подавляющую рост бактерий в течение 24 ч. Каждую культуру тестировали трижды.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Одна из ключевых проблем использования электропроводящего ПАНИ – плохие эксплуатационные характеристики полимера (нерастворимость, неплавкость, плохие механические свойства), которые препятствуют его широкому промышленному использованию. Одним из вариантов решения этой проблемы является синтез ПАНИ с использованием водорастворимой матрицы [1, 2, 37, 38]. В настоящей работе в качестве матрицы был использован ПСС.
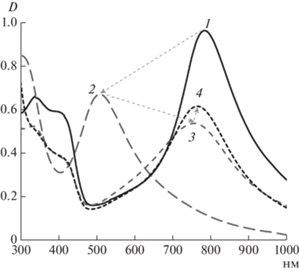
В результате катализируемой лакказой окислительной полимеризации АНИ на матрице ПСС был получен электропроводящий интерполиэлектролитный комплекс ПАНИ/ПСС, УФ-видимый спектр которого представлен на рис. 1 (кривая 1). На спектре ПАНИ/ПСС присутствовали полосы, характерные для электропроводящего ПАНИ: полоса в области 340 нм, соответствующая π–π* электронным переходам в ароматических кольцах, и две полосы поглощения (420 и 787 нм), указывающие на формирование полярона в структуре ПАНИ [39, 40]. При дедопировании комплекса эти полосы исчезали, и появлялась полоса поглощения в области 510 нм, соответствующая ПАНИ в неэлектропроводящей форме эмеральдинового основания (рис. 1, 2). При этом переходе цвет раствора менялся с зеленого на голубой. Так как дедопирование/допирование процесс обратимый, то после диализа на спектре снова появляются полосы, характерные для электропроводящей формы ПАНИ (рис. 1, 3). Однако при рН 7.8 основная полоса поглощения, соответствующая полярону, смещалась в более коротковолновую область (755 нм). При редопировании полимера ионами Cu2+ (рис. 1, 4) происходил сдвиг этой полосы до 764 нм, что являлось косвенным доказательством взаимодействия ионов меди с основной цепью ПАНИ.
Рис. 1.
УФ-видимые спектры ферментативно синтезированного комплекса ПАНИ/ПСС (1, рН 3.5), дедопированного комплекса (2, рН 10.0), (комплекса после диализа (3, рН 7.8) и редопированного ионами меди комплекса ПАНИ‑Cu/ПСС (4).
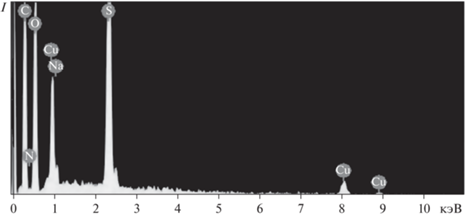
Исследование ПАНИ‑Cu/ПСС методом энергодисперсионной рентгеновской спектроскопии (рис. 2) показало, что содержание меди в комплексе, рассчитанное как среднее значение по четырем измерениям, составляло 1.66 ± 0.31 ат. % или 6.81 ± 1.18 вес. %.
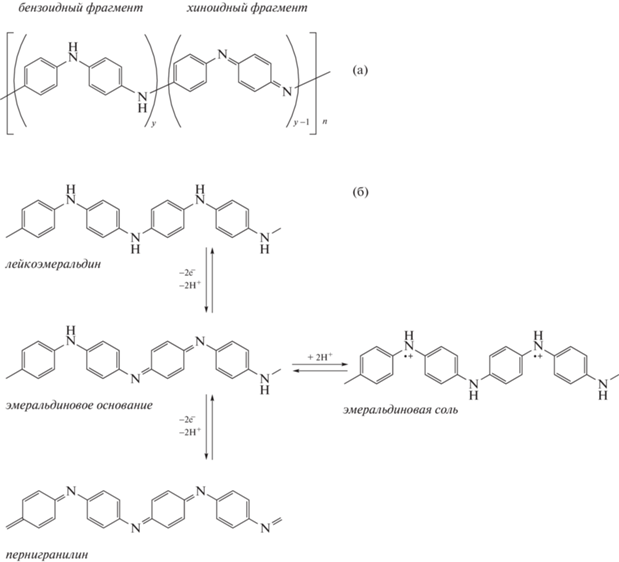
На рис. 3а представлена общая формула ПАНИ. Соотношениe фенилендиаминных (бензеноидных) и хиноиддииминных (хиноидных) структур в цепи ПАНИ обуславливает несколько возможных редокс-состояний полимера: лейкоэмеральдин (полностью восстановленное состояние), эмеральдин (полуокисленное состояние) и пернигранилин (полностью окисленное состояние). При кислотном допировании (рис. 3б) протоны взаимодействуют с иминными атомами азота, что приводит к образованию поликатиона. Положительные заряды, локализованные на атомах азота, повышают общую энергию полимерной системы, в результате чего происходит перераспределение электронной плотности и распаривание неподеленной электронной пары атомов азота. В цепи полимера появляются делокализованные электроны и возникает электронная проводимость, которая зависит от степени протонирования ПАНИ и от природы допирующей кислоты. Таким образом электропроводность ПАНИ обусловлена наличием в полимерной цепи катион-радикалов (поляронов), локализованных на атомах азота хиноиддииминных колец. Допирование цепи ПАНИ ионами переходных металлов относится к окислительному допированию. Основываясь на результатах экспериментов с солями переходных металлов, которые могут действовать как окислитель, Димитриев [25] предложил следующий механизм окислительного допирования эмеральдинового основания ПАНИ. Ионы переходных металлов окисляют атомы азота бензеноидных фрагментов; после этого восстановленные ионы металла координируются с иминными атомами азота хиноидных фрагментов. Затем происходит окисление восстановленных катионов металлов и восстановление иминных групп, что приводит к образованию допированной (электропроводящей) формы ПАНИ и окисленного катиона.
Рис. 3.
Общая формула ПАНИ (а): y = 1 – лейкоэмеральдин; y = 0.5 – эмеральдин и y = 0 – пернигранилин; схематическое изображение взаимопревращений различных редокс-состояний ПАНИ (лейкоэмеральдин, эмеральдин, пернигранилин) при кислотном допировании (б).
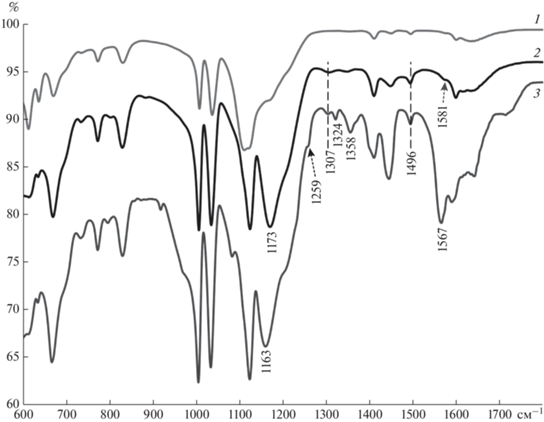
На рис. 4 приведены FTIR-ATR спектры матрицы ПСС (кривая 1) и комплексов ПАНИ/ПСС (кривая 2) и ПАНИ‑Cu/ПСС (кривая 3). Полосы в области 600–1150 см–1 соответствовали колебаниям связей в молекуле ПСС, что подтверждало образование комплекса ПАНИ/ПСС в результате ферментативной реакции. Наиболее существенные различия в спектрах ПАНИ/ПСС и ПАНИ-Cu/ПСС наблюдались в области 1150–1600 см–1. На обоих спектрах присутствовала полоса в области 1496 см–1, относящаяся к колебаниям С‒С связи бензеноидных фрагментов в структуре ПАНИ [41, 42]. В то же время интенсивность полосы поглощения, соответствующей колебаниям С=С связи хиноидных фрагментов, значительно увеличивалась при допировании ПАНИ ионами меди, а положение полосы смещалось сторону меньшего волнового числа (от 1581 см–1 в ПАНИ/ПСС до 1567 см–1 в ПАНИ-Cu/ПСС), что указывало на увеличение относительного количества хиноидных фрагментов в цепи ПАНИ [24]. Также происходил сдвиг полосы, соответствующей колебаниям структуры ‒NH+=, от 1173 до 1163 см–1. Кроме того на спектре ПАНИ-Cu/ПСС появлялись дополнительные полосы, соответствующие колебаниям связи С‒N рядом с хиноидным кольцом (1324 и 1358 см–1), и полоса, относящаяся к колебаниям связей катион-радикала C‒N•+ (1259 см–1). При допировании ПАНИ ионами меди увеличивалась интенсивность полосы, относящейся к колебаниям C‒N связи вторичных ароматических аминов в области 1307 см–1. Такие изменения в ИК-Фурье спектрах свидетельствовали о допировании основной цепи ПАНИ ионами меди.
Для антимикробных исследований водные растворы ПАНИ/ПСС и ПАНИ-Cu/ПСС были сконцентрированы до 5.8 и 5.3 мг/мл соответственно. Исследования показали, что матрица ПСС не обладала антимикробной активностью, а комплекс ПАНИ/ПСС, хотя и проявлял антимикробную активность в отношении S. aureus и E. coli, однако его МИК для обоих микроорганизмов выше, чем максимально достигнутая в исследовании концентрация (2.9 мг/мл). Необходимо отметить, что в литературе крайне мало данных о МИК “чистого” ПАНИ, что, по-видимому, связано с его плохой растворимостью. В статье [29] было показано, что МИК суспензии в питательной среде химически синтезированного ПАНИ для E. coli и S. aureus выше 10 мг/мл. Шалини с соавт. [31] показали, что МИК суспензии ПАНИ в диметилсульфоксиде для E. coli и S. aureus была 10 и 2.5 мг/мл соответственно.
Комплекс ПАНИ-Cu/ПСС показал более высокую ингибирующую активность, чем ПАНИ/ПСС. МИК ПАНИ-Cu/ПСС по отношению S. aureus составляла 0.66 мг/мл, а E. coli – 2.65 мг/мл. Поскольку в литературе отсутствуют данные о значениях МИК для композитов, в состав которых входит ПАНИ, допированный ионами меди, провести прямое сравнение с полученными нами результатами не представляется возможным. Однако можно сделать вывод, что ионы меди в составе комплекса ПАНИ-Cu/ПСС усиливали антимикробные свойства полимера и использование их в качестве допанта основной цепи ПАНИ перспективно для разработки антимикробных пленочных покрытий.
Авторы выражают благодарность Ю.А. Николаеву и Е.В. Демкиной за помощь в проведении микробиологических экспериментов. При проведении исследований использовалось оборудование Центра коллективного пользования “Промышленные биотехнологии” ФИЦ Биотехнологии РАН.
Работа выполнена при частичной финансовой поддержке Российского Фонда Фундаментальных исследований (проект № 19-08-00420).
Список литературы
Bhadra S., Khastgir D., Singha N.K., Lee J.H. // Prog. Polym. Sci. 2009. V. 34. № 8. P. 783–810.
Боева Ж.А., Сергеев В.Г. // Высокомолекулярные соединения. Сер. С. 2014. Т. 56. № 1. С. 153–164.
Zhang L., Du W., Nautiyal A., Liu Z., Zhang X.Y. // Sci. China Mater. 2018. V. 61. № 3. P. 303–352.
Singh P., Shukla S.K. // J. Mater. Sci. 2020. V. 55. № 4. P. 1331–1365.
Della Pina C., Falletta E. // Curr. Med. Chem. 2022. V. 29. № 2. P. 329–357.
Anand J., Palaniappan S., Sathyanarayana D.N. // Prog. Polym. Sci. 1998. V. 23. № 6. P. 993–1018.
Stejskal J., Gilbert R.G. // Pure Appl. Chem. 2002. V. 74. № 5. 857–867.
Huang W.S., Humphrey B.D., MacDiarmid A.G. // J. Chem. Soc., Faraday Trans. 1. 1986. V. 82. P. 2385–2400.
Syed A.A., Dinesan M.K. // Talanta 1991. V. 38. № 8. P. 815–837.
Liu W., Kumar J., Tripathy S., Senecal K.J., Samuelson L. // J. Am. Chem. Soc. 1999. V. 121. № 1. P. 71–78.
Cruz-Silva R., Romero-García J., Angulo-Sánchez J.L., Ledezma-Pérez A., Arias-Marín E., Moggio I., Flores-Loyola E. // Eur. Polym. J. 2005. V. 41. № 5. P. 1129–1135.
Streltsov A.V., Morozova O.V., Arkharova N.A., Klechkovskaya V.V., Staroverova I.N., Shumakovich G.P., Yaropolov A.I. // J. Appl. Polym. Sci. 2009. V. 114. № 2. P. 928–934.
Shumakovich G.P., Vasil’eva I.S., Morozova O.V., Khomenkov V.G., Staroverova I.N., Budashov I.A., Kurochkin I.N., Boyeva J.A., Sergeyev V.G., Yaropolov A.I. // J. Appl. Polym. Sci. 2010. V. 117. № 3. P. 1544–1550.
Отрохов Г.В., Морозова О.В., Васильева И.С., Шумакович Г.П., Зайцева Е.А., Хлупова М.Е., Ярополов А.И. // Успехи биологической химии. 2013. Т. 53. С. 355–386.
Walde P., Kashima K., Ćirić-Marjanović G. // Front. Bioeng. Biotechnol. 2019. V. 7. Article № 165. https://doi.org/10.3389/fbioe.2019.00165
Witayakran S., Ragauskas A.J. // Adv. Synth. Catal. 2009. V. 351. № 9. P. 1187–1209.
Hollmann F., Arends I. // Polymers 2012. V. 4. № 1. P. 759–793.
Bassanini I., Ferrandi E.E., Riva S., Monti D. // Catalysts. 2021. V. 11. № 1. Article № 26. https://doi.org/10.3390/catal11010026
MacDiarmid A.G., Epstein A.J. // Faraday Discuss. Chem. Soc. 1989. V. 88. P. 317–332.
Genies E.M., Boyle A., Lapkowski M., Tsintavis C. // Synth. Met. 1990. V. 36. № 2. P. 139–182.
Huang W.-S., MacDiarmid A.G., Epstein A.J. // J. Chem. Soc., Chem. Commun. 1987. V. 23. P. 1784–1786.
Chen S.A., Lin L.C. // Macromolecules 1995. V. 28. № 4. P. 1239–1245.
Bienkowski K., Kulszewicz-Bajer I., Genoud F., Oddou J.L., Pron A. // Synth. Met. 2003. V. 135–136. P. 159–160.
Dimitriev O.P. // Macromolecules 2004. V. 37. № 9. P. 3388–3395.
Dimitriev O. // Polym. Bull. 2003. V. 50. № 1–2. P. 83–90.
Izumi C.M.S., Ferreira A.M.D.C., Constantino V.R.L., Temperini M.L.A. // Macromolecules 2007. V. 40. № 9. P. 3204–3212.
Sheshardi D.T., Bhat N. // Indian J. Fibre Text. Res. 2005. V. 30. № 2. P. 204–206.
Shi N.L., Guo X.M., Jing H.M., Gong J., Sun C., Yang K. // J. Mater. Sci. Technol. 2006. V. 22. № 3. P. 289–290.
Gizdavic-Nikolaidis M.R., Bennett J.R., Swift. S., Easteal A.J., Ambrose M. // Acta Biomaterialia. 2011. V. 7. № 12. P. 4204–4209.
Kucekova Z.б Humpolicek P., Kasparkova V., Perecko T., Lehocký M., Hauerlandová I., Sáha P., Stejskal J. // Colloids Surf. B 2014. V. 116. P. 411–417.
Shalini A., Nishanthi R., Palani P., Jaisankar V. // Mater. Today: Proc. 2016. V. 3. № 6. P. 1633–1642.
Lashkenari M.S., Eisazadeh H. // Adv. Polym. Technol. 2017. V. 33. № S1. Article № 21466.https://doi.org/10.1002/adv.21466
Mittapally S., Taranu R., Parveen S. // Drug J. Delivery Ther. 2018. V. 8. № 6s. P. 411–419.
Borkow G., Gabbay J. // Cur. Med. Chem. 2005. V. 12. № 18. P. 2163–2175.
Горшина Е.С., Русинова Т.В., Бирюков В.В., Морозова О.В., Шлеев С.В., Ярополов А.И. // Прикл. биохимия и микробиология 2006. Т. 42. № 6. С. 558–563.
Ehresmann B., Imbault P., Well J.H. // Analyt. Biochem. 1973. V. 54. № 2. P. 454–463.
Liu W., Cholli A.L., Nagarajan R., Kumar J., Tripathy S., Bruno F.F., Samuelson L. // J. Am. Chem. Soc. 1999. V. 121. № 49. P. 11345–11355.
Walde P., Guo Z. // Soft Matter. 2011. V. 7. № 2. P. 316–331.
Yue J., Wang Z.H., Cromack K.R., Epstein A.J., MacDiarmid A.G. // J. Am. Chem. Soc. 1991. V. 113. № 7. P. 2665–2671.
Yuan G.L., Kuramoto N., Su S.H. // Synth. Met. 2002. V. 129. № 2. P. 173–178.
Trchová M., Šeděnková I., Tobolková E., Stejskal J. // Polym. Degrad. Stab. 2004. V. 86. № 1. P. 179–185.
Gomes E.C., Oliveira M.A.S. // Am. J. Polym. Sci. 2012. V. 2. № 2. P. 5–13.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология