

Прикладная биохимия и микробиология, 2021, T. 57, № 6, стр. 613-620
Разработка иммунохроматографической тест-системы для выявления антибиотика клинафлоксацина и ее применение для контроля мёда
Е. А. Зверева 1, Д. В. Сотников 1, К. А. Беличенко 1, О. Д. Гендриксон 1, И. А. Шанин 1, А. В. Жердев 1, Б. Б. Дзантиев 1, *
1 Институт биохимии им. А.Н. Баха, Федеральный исследовательский центр
“Фундаментальные основы биотехнологии” Российской академии наук
119071 Москва, Россия
* E-mail: dzantiev@inbi.ras.ru
Поступила в редакцию 07.05.2021
После доработки 25.06.2021
Принята к публикации 02.07.2021
Аннотация
Для экспрессного контроля качества и безопасности продуктов питания разработана иммунохроматографическая тест-система, обеспечивающая выявление антибиотика клинафлоксацина. В тест-системе реализован непрямой формат конкурентного иммуноанализа, в котором с немодифицированными специфическими поликлональными антителами взаимодействуют антибиотик, потенциально содержащийся в тестируемой пробе, и конъюгат антибиотик-белок, иммобилизованный на рабочей мембране тест-полоски. Выявление образующихся на мембране иммунных комплексов происходит с помощью конъюгата антивидовых антител и наночастиц золота. Определены условия проведения иммунохроматографии, обеспечивающие достижение минимального предела обнаружения. Продолжительность анализа составляет 20 мин, пределы обнаружения клинафлоксацина при инструментальной и визуальной детекции – 0.3 и 10 нг/мл соответственно. Изучена селективность тест-системы по отношению к антибиотикам из класса фторхинолонов. Для контроля контаминации мёда клинафлоксацином и структурно близкими фторхинолонами установлен оптимальный режим пробоподготовки и подтверждена эффективность разработанной тест-системы.
Тенденцией последних десятилетий является увеличение объемов техногенных соединений, попадающих в окружающую среду в результате хозяйственной деятельности человека и представляющих угрозу для здоровья. Среди токсичных техногенных контаминантов особый интерес представляют антибиотики, негативно влияющие на ряд физиологических процессов, а также обусловливающие распространение антибиотикорезистентных штаммов патогенных микроорганизмов [1, 2]. Из-за этих угроз предпринимаются меры по контролю схем медицинского применения антибиотиков и ограничению их использования в животноводстве. Однако в связи с регулярно выявляемыми нарушениями нормативных требований для эффективной защиты населения необходим массовый контроль антибиотиков в потребительской продукции, прежде всего – в продуктах питания. Такой контроль должен включать как первичное скрининговое тестирование недорогими аналитическими средствами непосредственно в местах отбора проб, так и последующую подтверждающую идентификацию контаминанта в специализированных лабораториях [3, 4].
Показана эффективность иммунохимических методов для скринигового контроля антибиотиков, а для подтверждающего анализа – методов газовой и жидкостной хроматографии. Однако, несмотря на наличие ряда коммерчески доступных иммуноаналитических систем для выявления и оценки содержания антибиотиков, разработка новых методик по-прежнему остается востребованной задачей. Эта востребованность обусловлена несколькими факторами: ростом числа контролируемых соединений, тенденцией к методическому упрощению и повышению производительности скринингового тестирования, необходимостью сочетать иммуноанализ с предельно простой пробоподготовкой, вплоть до простого разведения исходных проб и использования необработанных экстрактов, что требует высокочувствительного выявления иммунных комплексов на последующих стадиях [4–9].
Дополнительные сложности в разработке и практическом применении методов иммунохимического выявления и оценки содержания антибиотиков связаны с их значительным структурным разнообразием, возможностью присутствия в тестируемой пробе нескольких соединений из одного и того же класса антибиотиков с минимальными структурными отличиями, но c разной токсичностью, и, соответственно, с разными уровнями предельно допустимого содержания [10, 11]. Распространенные в настоящее время и поддерживаемые наличием промышленных технологий виды иммуноанализа – иммуноферментный анализ (ИФА), иммунохроматографический анализ (ИХА) и др. – предполагают регистрацию одного интегрального сигнала, отражающего общее количество образовавшихся комплексов антиген-антитело, но не содержание в пробе конкретных иммунохимически активных соединений [10, 12]. Адекватное принятие решения о безопасности продукции на основании этого параметра требует детального учета специфичности антител, их перекрестной реактивности (ПР) по отношению к различным структурным аналогам целевого аналита, продуктам его модификации и частичной деструкции. Наличие нескольких антител к соединениям одного класса, отличающихся по ПР, существенно расширяет возможные варианты реализации иммуноанализа и повышает информативность результатов, получаемых при его проведении [7, 10, 13].
Среди различных классов антибиотиков задача поиска антител и разработки иммуноаналитических систем с разными спектрами ПР особенно важна для фторхинолонов. Группа фторхинолонов включает несколько десятков используемых антибиотиков, характеризующихся широким спектром токсических эффектов и поэтому контролируемых в продуктах питания [14, 15]. При этом нормативные требования ограничивают как суммарное содержание всех фторхинолонов, так и уровни нескольких конкретных соединений, причем их перечни, установленные в разных странах, неодинаковы [16–19]. В рамках данного исследования были оценены возможности антител, полученных против клинафлоксацина – одного из представителей фторхинолонов с циклопропильным радикалом в первом положении хинолонового ядра. Для разработки и характеристики был выбран ИХА – иммунохимический метод, активно применяемый для скринингового контроля вследствие минимальных требований к дополнительным реагентам и оборудованию. Нанесение иммунореагентов на тест-полоску для ИХА до проведения анализа сводит процедуру тестирования к погружению этой полоски в один или несколько растворов и последующей визуальной оценке наличия или инструментальной оценке интенсивности формирующихся на тест-полоске окрашенных зон [20, 21]. Апробация тест-системы состояла в изучении возможностей ее применения для контроля мeда – важного элемента здорового питания с децентрализованным производством и широким спектром контролируемых токсичных контаминант [22, 23].
Цель работы – создание иммунохроматографической тест-системы для экспрессного выявления антибиотика клинафлоксацина и его ближайших структурных аналогов, а также апробация данной тест-системы для контроля контаминации мёда.
МЕТОДИКА
Реактивы. В работе использовали клинафлоксацин (КЛИ), гареноксацин (ГАР), гатифлоксацин (ГАТ), данофлоксацин (ДАН), дифлоксацин (ДИФ), левофлоксацин (ЛЕВ), ломефлоксацин (ЛОМ), марбофлоксацин (МАР), моксифлоксацин (МОКС), надифлоксацин (НАД), налидиксовую кислоту (НАЛ), оксолиниевую кислоту (ОКСО), орбифлоксацин (ОРБ), пазуфлоксацин (ПАЗ), пефлоксацин (ПЕФ), пипемидовая кислоту (ПИП), руфлоксацин (РУФ), сарафлоксацин (САР), тосуфлоксацин (ТОС), флюмеквин (ФЛЮ), ципрофлоксацин (ЦИП), эноксацин (ЭНО), энрофлоксацин (ЭНРО), бычий сывороточный альбумин (БСА), овальбумин (ОВА), боргидрид натрия, глутаровый альдегид, детергент Triton Х-100, 3,3',5,5'-тетраметилбензидин и триэтиламин – все производства “Sigma-Aldrich”, (США), золотохлористоводородную кислоту (“Fluka”, США), антитела козы против иммуноглобулинов кролика (АКоКр), антитела кролика против иммуноглобулинов козы (АКрКо) (“Arista Biologicals”, США), антитела против иммуноглобулинов кролика, меченные пероксидазой (“Jackson ImmunoResearch”, Великобритания). Все вспомогательные реагенты (соли, кислоты, щелочи и органические растворители) были аналитической или химической чистоты.
Растворы для синтеза наночастиц золота (НЧЗ) и межмолекулярных конъюгатов готовили на воде, деионизированной с помощью установки Milli-Q (“Millipore”, США). Исходные растворы фторхинолонов готовили в метаноле (“Fluka”, США). ИФА проводили в 96-луночных полистироловых микропланшетах Costar 9018 (“Corning Costar”, США).
Получение поликлональных антител. Кроличьи поликлональные антитела против КЛИ (ПАт КЛИ) были получены в соответствии с методиками, представленными в [24]. Для иммунизации использовали самцов кроликов породы “Калифорнийский кролик” в возрасте 3 мес. Животных иммунизировали с интервалом 2 нед. свежеприготовленной эмульсией белкового конъюгата с полным адъювантом Фрейнда для первой иммунизации и с неполным адъювантом Фрейнда для последующих, смешивая компоненты в отношении 1 : 1 по объему. Забор крови проводили из краевой вены уха с помощью вакуумных пробирок с гелем и активатором свертывания. Сыворотку отделяли центрифугированием и выделяли фракцию IgG осаждением насыщенным раствором сульфата аммония при 4°C. Полученную фракцию растворяли в 50 мМ калий-фосфатном буфере, pH 7.4, содержавшем 100 мМ NaCl (ФБС), смешивали с глицерином 1 : 1 и хранили при –20°C.
Синтез конъюгата клинофлоксацина с овальбумином (КЛИ-ОВА). Конъюгат синтезировали в соответствии с методикой [25]. В колбу при перемешивании вносили 8 мл деионизированной воды, 5 мг ОВА, 2.2 мг КЛИ, 20 мкл триэтиламина и 23 мкл глутарового альдегида. Смесь инкубировали в темноте при постоянном перемешивании и комнатной температуре в течение часа, вносили 0.5 мл 0.22%-ного раствора боргидрида натрия и инкубировали еще 30 мин. Полученный конъюгат диализовали против ФБС и хранили при –20°C.
Конкурентный ИФА клинафлоксацина. В лунки микропланшета вносили по 100 мкл конъюгата КЛИ–ОВА в концентрации 1 мкг/мл в ФБС и проводили сорбцию в течение 16 ч при 4°C. После этого микропланшет четырехкратно промывали ФБС, содержащим 0.05% детергента Тритон Х-100 (ФБСТ), вносили в лунки по 50 мкл раствора КЛИ (от 10 мкг/мл до 0.01 нг/мл) в ФБСТ и добавляли по 50 мкл ПАт КЛИ в разведении 1 : 100 000. Микропланшет инкубировали в течение 60 мин при 37°C, четырехкратно промывали ФБСТ и вносили в лунки по 100 мкл антител против иммуноглобулинов кролика, меченных пероксидазой, в разведении 1 : 5000. После часовой инкубации при 37°C микропланшет четырехкратно промывали ФБСТ и определяли активность пероксидазной метки. Для этого во все лунки вносили по 100 мкл субстратного раствора (0.42 мM 3,3',5,5'-тетраметилбензидин и 1.8 мM H2O2 в 0.1 M цитратном буфере, pH 4.0), инкубировали при комнатной температуре и останавливали реакцию добавлением 1 M H2SO4 (50 мкл на лунку). Оптическую плотность (ОП) продукта реакции измеряли при 450 нм с использованием микропланшетного фотометра Zenyth 3100 (“Anthos Labtec Instruments”, Австрия).
Синтез наночастиц золота. Синтез НЧЗ проводили в соответствии с методикой, описанной в работе [26]. В коническую колбу с обратным холодильником вносили 48.75 мл деионизированной воды, 0.5 мл 1%-ного раствора золотохлористоводородной кислоты, доводили до кипения и добавляли 0.75 мл 1%-ного раствора цитрата натрия. Полученную смесь кипятили при перемешивании в течение 25 мин и охлаждали. Препараты НЧЗ хранили при 4°С.
Синтез конъюгатов НЧЗ с антителами. В соответствии с методикой [27] для определения концентрации ПАт, используемой при конъюгирования с НЧЗ, готовили серию водных растворов ПАт КЛИ и АКоКр (от 200 до 0.5 мкг/мл). В эппендорфах смешивали 1 мл НЧЗ (ОП520 = 1.0), 100 мкл каждого раствора антител и инкубировали 20 мин при перемешивании. После этого вносили в эппендорфы по 100 мкл 10%-ного раствора хлорида натрия, инкубировали 10 мин и измеряли ОП520. Для конъюгирования с НЧЗ были выбраны концентрации ПАт КЛИ 6 мкг/мл и АКоКр 8 мкг/мл.
Перед проведением конъюгирования антитела диализовали против 10 мМ трис-HCl буфера, рН 8.5, в течение 2 ч при 4°C. Раствор НЧЗ с ОП520 = 1.0 доводили до рН 8.5 с помощью 0.1 М карбоната калия, смешивали с антителами, инкубировали при комнатной температуре и перемешивании в течение 45 мин. После этого в смесь вносили БСА до конечной концентрации 0.25% и инкубировали 15 мин. Конъюгаты осаждали центрифугированием при 8500 g в течение 15 мин при 4°C и ресуспендировали в 10 мМ трис-буфере, рН 8.5, содержавшем 1% БСА, 1% сахарозы и 0.1% азида натрия (ТБСА). Полученные конъюгаты хранили при 4°С.
Изготовление иммунохроматографических тес-полосок. Для производства тест-полосок использовались нитроцеллюлозная мембрана CNPC с размером пор 15 мкм (СNPC-SS12), мембрана под образец GFB-R4, стекловолоконная мембрана PT-R7 и адсорбционная мембрана AP045 (“Advanced Microdevices”, Индия), а также нитроцеллюлозная мембрана HF090 (“Millipore”, США).
Тест-полоски для прямой схемы ИХА. Аналитическую зону тест-полоски формировали нанесением конъюгата КЛИ-ОВА (0.5 мг/мл), контрольную зону – АКоКр (0.15 мг/мл). Конъюгат ПАт КЛИ-НЧЗ в ТБСА, содержащем 0.05% Твин-20 (ТТБСА), наносили на мембрану PT-R7 в разведении, соответствующем ОП520 = 2.0.
Тест-полоски для непрямой схемы ИХА (полной). Аналитическую зону тест-полоски формировали нанесением конъюгата КЛИ-ОВА (0.5 мг/мл), контрольную зону – АКрКо (0.15 мг/мл). Конъюгат АКоКр-НЧЗ в ТТБСА наносили на мембрану PT-R7 в разведении, соответствующем ОП520 = 2.0.
Тест-полоски для непрямой схемы ИХА (укороченной). Аналитическую зону тест-полоски формировали нанесением конъюгата КЛИ-ОВА (0.6 мг/мл), контрольную зону – АКрКо (0.25 мг/мл). В данную комплектацию мембрана под образец и стекловолоконная мембрана не входят.
После нанесения иммунореагентов с помощью автоматического диспенсера IsoFlow (“Imagene Technology”, США) нитроцеллюлозную мембрану сушили 24 ч при комнатной температуре и 5 ч при 37°C. Высушенный и склеенный мембранный комплекс нарезали на тест-полоски шириной 3.0 мм с помощью автоматического гильотинного нарезчика Index Cutter-1 (“A-Point Technologies”, США).
Проведение ИХА. Прямая схема анализа. В лунку микропланшета вносили 100 мкл тестируемой пробы (растворы КЛИ разной концентрации в ФБСТ) и погружали тест-полоску в вертикальном положении. Через 15 мин контролировали результат анализа.
Непрямая схема анализа (полная). В лунку микропланшета вносили 50 мкл тестируемой пробы (растворы КЛИ разной концентрации в ФБСТ) и 50 мкл ПАт КЛИ в разведении 1 : 4000 в ФБСТ, инкубировали 5 мин, погружали тест-полоску в вертикальном положении и контролировали результат анализа через 15 мин.
Непрямая схема анализа (укороченная). В лунку микропланшета вносили 25 мкл пробы (растворы КЛИ разной концентрации в ФБСТ или экстракты мёда) и 25 мкл ПАт КЛИ в разведении 1 : 4000 в ФБСТ, инкубировали 5 мин и погружали тест-полоску в вертикальном положении. После 5 мин инкубации тест-полоску переносили в лунку с 40 мкл раствора АКоКр-НЧЗ с ОП520 = 2.0 и контролировали результат через 15 мин.
Регистрацию результатов ИХА проводили с помощью сканера CanoScan LiDE 90 (“Canon”, Япония). Оценку интенсивности окрашивания аналитических зон проводили, обрабатывая изображения с помощью программы TotalLab (“Nonlinear Dynamics”, Великобритания).
Приготовление экстрактов мeда. Экстракты получали по схемам, изложенным в табл. 2. Схема № 4 соответствовала рекомендациям производителей тест-систем RIDASCREEN ELISA (“R-Biopharm”, Германия).
Приготовление проб мёда с известным содержанием КЛИ. Для оценки полноты определения содержания КЛИ методом “добавлено – выявлено” к 1.0 г мёда добавляли стандартный раствор КЛИ в таком объеме, чтобы содержание КЛИ в экстракте составили 0.5, 1.0, 2.0, 5.0 нг/мл.
Обработка результатов ИФА и ИХА. Для аппроксимации полученных концентрационных зависимостей использовали программу Origin 8.5.1 (“OriginLab”, США) и четырехпараметрическую сигмоидную функцию y = (A – D)/(1 + (x/C)B) + D. В качестве предела обнаружения (для ИХА – инструментальный предел обнаружения) рассматривали концентрацию КЛИ, вызывающую 10%-ное ингибирование аналитического сигнала.
Специфичность ИХА оценивали по величине процента перекрестного реагирования (ПР) с другими представителями фторхинолонов, исходя из концентраций, вызывающих 50%-ное ингибирование аналитического сигнала (IC50):
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
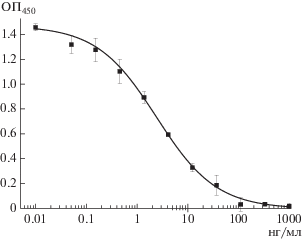
Получение и характеристика специфических иммунореагентов. Поликлональные кроличьи антитела против КЛИ в сочетании с конъюгатом КЛИ-ОВА тестировали методом ИФА и отбирали антитела, характеризовавшиеся минимальным пределом обнаружения КЛИ. Согласно полученной в ИФА концентрационной зависимости аналитического сигнала (ОП450) для лучшего препарата (рис. 1), предел обнаружения КЛИ составил 0.6 нг/мл, что свидетельствовало о возможности использования антител для разработки чувствительного иммунохроматографического анализа.
В качестве маркера в ИХА использовали наночастицы золота, которые синтезировали, восстанавливая цитратом натрия золотохлористоводородную кислоту [26]. Размерные характеристики НЧЗ определяли с помощью просвечивающей электронной микроскопии. Наночастицы имели сферическую форму и средний диаметр, равный 30 нм.
Были синтезированы конъюгаты с НЧЗ специфических ПАт КЛИ, а также антител козы против иммуноглобулинов кролика (АКоКр). Концентрацию антител для конъюгирования определяли на основании флоккуляционных кривых – зависимостей оптической плотности конъюгата от концентрации антител в растворе с высокой ионной силой [27]. Выбранные концентрации, соответствовавшие выходу флоккуляционных кривых на плато, то есть стабилизации коллоида белком при его потенциальной монослойной иммобилизации, составляли 6 мкг/мл для ПАт КЛИ и 8 мкг/мл для АКоКр.
Реализация прямой схемы конкурентного ИХА клинафлоксацина. Показано, что конкурентное взаимодействие ПАт КЛИ, конъюгированных с НЧЗ, со свободным КЛИ пробы и КЛИ-ОВА, иммобилизованным в аналитической зоне тест-полоски, отсутствовало – аналитическая зона окрашивалась независимо от концентрации КЛИ. При этом конъюгат ПАт КЛИ-НЧЗ хорошо взаимодействовал со свободным КЛИ и иммобилизованным КЛИ-ОВА в ИФА. Таким образом, в условиях проведения прямой схемы ИХА влияние КЛИ, содержащегося в пробе, на аналитический сигнал было недостаточным для выявления КЛИ. Поэтому схема прямого ИХА не использовалась в последующей работе.
Реализация непрямой схемы конкурентного ИХА клинафлоксацина. В отличие от прямого взаимодействия меченых антител и антигенных препаратов, данная схема предполагала использование конъюгата АКоКр-НЧЗ, который реагировал со специфическим комплексом, образованным в аналитической зоне (конъюгат КЛИ-ОВА + ПАт КЛИ), и обеспечивал возможность визуализации этого комплекса.
В тест-системе, в которой все иммунореагенты были нанесены на тест-полоску, достаточного для определения КЛИ количества детектируемых комплексов не образовывалось. Аналитическая зона слабо окрашивалась в первые минуты, после чего окраска исчезала независимо от концентрации антигена. Это могло быть связано с нестабильностью иммунных комплексов, образующихся в предложенной динамической системе.
Конкурентная детекция КЛИ была возможна только при использовании укороченных тест-полосок, в которых отсутствовали стекловолоконная мембрана и мембрана под образец. На первом этапе анализа КЛИ, содержавшийся в пробе, взаимодействовал со специфическими ПАт, после чего в смесь вносили тест-полоску для связывания непрореагировавших (незаблокированных антигеном) ПАт в аналитической зоне. На заключительном этапе тест-полоску переносили в раствор конъюгата АКоКр-НЧЗ и инкубировали для формирования на рабочей мембране окрашенных иммунных комплексов.
В данной системе для формирования контрольной зоны использовали поликлональные антитела кролика против иммуноглобулинов козы (АКрКо), которые эффективно связывали АКоКр-НЧЗ. В аналитической зоне тест-полоски был иммобилизован конъюгат КЛИ-ОВА.
Оптимизация условий проведения ИХА была направлена на достижение минимального предела определения КЛИ. Для этого варьировали концентрацию конъюгата КЛИ-ОВА в аналитической зоне, концентрацию специфических антител, продолжительность инкубации (см. табл. 1). Низкий предел обнаружения КЛИ в сочетании с интенсивным окрашиванием аналитической и контрольной зон достигались при нанесении АКрКо в концентрации 0.2 мг/мл, КЛИ-ОВА в концентрации 0.6 мг/мл и использовании ПАт КЛИ в разведении 1 : 2000. Выбранная концентрация конъюгата АКоКр-НЧЗ, соответствовавшая ОП520 = 2.0, обеспечивала минимальный расход конъюгата для количественно воспроизводимых значений интенсивности окрашивания обеих зон.
Таблица 1.
Оптимизация непрямой схемы ИХА клинафлоксацина
| Оптимизируемый параметр | Интервал изменений параметра | Оптимальное значение параметра |
|---|---|---|
| Концентрация КЛИ-ОВА в аналитической зоне | 0.3–1.0 мг/мл | 0.6 мг/мл |
| Разведение ПАт КЛИ | 1 : 1000–1 : 5000 | 1 : 2000 |
| Концентрация конъюгата АКоКр-НЧЗ | ОП520 = 1.0–4.0 | ОП520 = 2.0 |
| Продолжительность предынкубации | 2–8 мин | 5 мин |
Были протестированы два вида рабочих мембран – СNPC-SS12 и Millipore HF090. В дальнейшей работе использовали мембрану СNPC-SS12, поскольку на ней аналитическая зона окрашивалась более равномерно.
Таблица 2.
Иммунохроматографическое определение клинафлоксацина в мёде при различной пробоподготовке
| Схемы пробоподготовки для 1 г мёда | Результаты тестирования отрицательной (“–”) и положительной (“+”, 10 нг/мл КЛИ) проб мёда | |
|---|---|---|
| 1. | Перемешивание 10 мин с 2 мл ФБСТ |  |
| 2. | Перемешивание 10 мин с 4 мл ФБСТ |  |
| 3. | Перемешивание 10 мин с 9 мл ФБСТ |  |
| 4. | Перемешивание 10 мин с 2 мл воды и 2 мл этилацетата, центрифугирование 10 мин при 3000 g. Верхняя фракция отбирается, выпаривается при 60°C и перерастворяется в ФБСТ |  |
Проведена оптимизация продолжительности стадий анализа. Показано, что 5-минутная предынкубация ПАт КЛИ и пробы перед контактом с тест-полоской достаточна для образования стабильного иммунного комплекса в аналитической зоне. Сокращение этой стадии приводило к снижению интенсивности окрашивания аналитической зоны и ухудшению чувствительности.
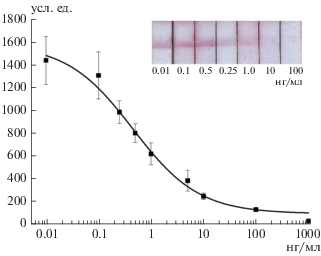
Таким образом, минимальная выявляемая концентрация КЛИ достигалась при продолжительности анализа 20 мин, включавшего предынкубацию ПАт КЛИ и пробы (5 мин), инкубации тест-полоски с этой смесью (5 мин) и с АКоКр-НЧЗ (10 мин). Калибровочная кривая ИХА (рис. 2) характеризовалась пределами обнаружения КЛИ при инструментальной и визуальной детекции, равными 0.3 и 10 нг/мл, соответственно.
Специфичность тест-системы. Тестирование разработанной иммунохроматографической системы для определения других фторхинолонов позволило выделить следующие группы по степени перекрестной реактивности:
– высокая (ПР > 40%): ПЕФ – 54%, ПИП – 51%, ЭНРО – 50%, ГАР – 41%, ДАН – 41%;
– средняя (от 10 до 40%): ГАТ – 23%, НАД – 22%, МОКС – 20%, РУФ – 18%, ЛОМ – 17%, МАР – 14%, САР – 12%, ОРБ – 12%, ЭНО – 11%;
– низкая (менее 10%): ФЛЮ – 8%, ОКСО – 7%, ЛЕВ – 5%, НАЛ – 5%, ПАЗ – 3%, ЦИП – 2%.
Для двух фторхинолонов (ТОС, ДИФ) достоверное влияние на связывание в изученных диапазонах концентраций не наблюдалось, то есть ПР не превышала 0.1%.
Отметим, что для всех фторхинолонов, содержавших циклопропильный радикал в первом положении хинолонового ядра, кроме ципрофлокасацина, величина ПР составляла не менее 20%, что позволило рассматривать разработанную тест-систему как средство интегрального выявления данной подгруппы фторхинолонов, включавшей КЛИ, ЭНРО, ГАР, ДАН, ГАТ и МОКС.
Определение КЛИ в мёде. Существующие методики пробоподготовки при иммунодетекции антибиотиков разных классов в мёде можно разделить на две основные группы: разбавление буферными растворами [28, 29] и экстракция органическими растворителями [30, 31].
Для выбора методики, эффективно сочетающейся с возможностями разработанной тест-системы, были рассмотрены варианты разбавления проб мёда ФБСТ в объемных соотношениях 1 : 2 (экстракт № 1), 1 : 4 (экстракт № 2) и 1 : 9 (экстракт № 3), а также экстракции аналита этилацетатом по методике, рекомендованной производителями иммуноферментных тест-систем на фторхинолоны RIDASCREEN ELISA (“R-Biopharm”, Германия) (экстракт № 4). Показано, что разработанная тест-система позволяла определять КЛИ в полученных экстрактах в интервале от 0.5 до 10 нг/мл (табл. 2).
Одна из основных характеристик ИХА – экспрессность. Поэтому была подобрана методика, которая позволяла проводить пробоподготовку быстро и эффективно, с использованием минимального числа стадий, а именно – разбавление проб мёда в ФБСТ в соотношении 1 : 2 (экстракт № 1, рис. 3). Дальнейшее уменьшение количества ФБСТ, вносимого в пробу, было нецелесообразно, поскольку в таких случаях разбавленный мёд сохранял высокую вязкость, что затрудняло движение пробы по рабочей мембране тест-полоски.
Рис. 3.
Иммунохроматографическое определение КЛИ (непрямая схема) в экстрактах мёда. Калибровочные графики в ФБСТ (1), экстрактах № 1 (2), № 2 (3) и № 3 (4).
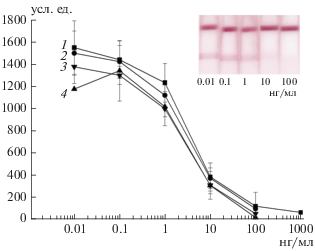
При оценке степени выявления КЛИ в тестируемых пробах были сопоставлены результаты, полученные с использованием двух калибровочных кривых – в ФБСТ и в разбавленном в соотношении 1 : 2 мёде. Как следует из табл. 3, оба подхода давали приемлемые результаты. Хотя получение калибровочной кривой в ФБСТ облегчало процесс тестирования, более точные величины наблюдались при работе со второй калибровочной кривой, отражавшей особенности матрикса проб.
Таблица 3.
Степень выявления клинафлоксацина в мёде методом ИХА
| Введено, нг/мл | Определено по калибровочному графику | |||
|---|---|---|---|---|
| в ФБСТ | в разбавленном (1 : 2) мeде | |||
| нг/мл | % | нг/мл | % | |
| 0.5 | 1.1 | 220 | 0.6 | 120 |
| 1.0 | 1.3 | 130 | 0.9 | 90 |
| 2.0 | 2.5 | 120 | 1.9 | 95 |
| 5.0 | 5.7 | 114 | 4.9 | 98 |
Таким образом, впервые разработана методика иммунохроматографического определения антибиотика клинафлоксацина, одного из потенциальных контаминантов мёда. Методика характеризовалась пределами инструментального обнаружения 0.3 нг/мл и визуальной детекции 10 нг/мл, существенно более низкими по сравнению с нормативными требованиями для фторхинолонов – до 100 нг/мл [16–19]. Широкая специфичность тест-системы позволяет использовать ее для выявления ряда фторхинолоновых антибиотиков – КЛИ, ЭНРО, ГАР, ДАН, ГАТ и МОКС.
Авторы выражают благодарность А.А. Рязанову (ФИЦ Биотехнологии РАН) за помощь в характеристике специфичности тест-системы.
Работа выполнена при финансовой поддержке Российского научного фонда (проект № 20-76-10033).
Список литературы
Bengtsson-Palme J., Kristiansson E., Larsson D.G.J. // FEMS Microbiol. Rev. 2018. V. 42. № 1. P. 80–68.
Kraemer S.A., Ramachandran A., Perron G.G. // Microorganisms. 2019. V. 7. № 6. Article 180. https://www.mdpi.com/2076-2607/7/6/180
Chen J., Ying G.G., Deng W.J. // J. Agric. Food Chem. 2019. V. 67. № 27. P. 7569–7586.
Ahmed S., Ning J.N., Peng D.P., Chen T., Ahmad I., Ali A., Lei Z.X., Shabbir M.A., Cheng G.Y., Yuan Z.H. // Food Agric. Immunol. 2020. V. 31. № 1. P. 268–290.
Wang Q., Zhao W.M. // Sens. Actuators B. 2018. V. 269. P. 238–256.
Anfossi L., Di Nardo F., Cavalera S., Giovannoli C., Baggiani C. // Biosensors (Basel). 2018. V. 9. № 1. Article 2. https://www.mdpi.com/2079-6374/9/1/2
Dzantiev B.B., Byzova N.A., Urusov A.E., Zherdev A.V. // TrAC Trends Anal. Chem. 2014. V. 55. P. 81–93.
Bishop J.D., Hsieh H.V., Gasperino D.J., Weigl B.H. // Lab on Chip. 2019. V. 19. № 15. P. 2486–2499.
Liu L., Yang D., Liu G. // Biosens. Biolectron. 2019. V. 136. P. 60–75.
Li Y.F., Sun Y.M., Ross C.B., Lei H.T., Gee S., Hammock B.D., Wang H., Wang Z., Sun X., Shen Y.D., Yang J.Y., Xu Z.L. // TrAC Trends Anal. Chem. 2017. V. 88. P. 25–40.
Majdinasab M., Mishra R.K., Tang X., Marty J.L. // TrAC Trends Anal. Chem. 2020. V. 127. Article 115883. https://www.sciencedirect.com/science/article/pii/ S0165993620301126
Wild D. The Immunoassay Handbook. Theory and Applications of Ligand Binding, ELISA and Related Techniques. 4th Ed. Amsterdam, the Netherlands: Elsevier Science. 2013. P. 29–242.
Shanin I.A., Zvereva E.A., Zherdev A.V., Godjevargova T., Eremin S. A. // Bulgar. Chem. Commun. 2019. V. 51. Special Issue D. P. 27–31.
Filloux C., Geniaux H., Buxeraud J. // Actual. Pharmaceut. 2019. V. 58. № 588. P. 53–55.
Drlica K., Hiasa H., Kerns R., Malik M., Mustaev A., Zhao X.L. // Curr. Topics Med. Chem. 2009. V. 9. № 11. P. 998–981.
Технический регламент Таможенного Союза “О безопасности пищевой продукции” ТР ТС 021/2011. М.: Центрмаг, 2021. 261 с.
Evaluation of Certain Contaminants in Food; Technical Report Series; Geneva, Switzerland: World Health Organization, 2011. P. 153–316.
Maximum Residue Limits (MRLs) and Risk Management Recommendations (RMRs) for Residues of Veterinary Drugs in Foods CX/MRL (2-2018); FAO/WHO. Codex Alimentarius, International Food Standards: Rome, Italy, 2018. P. 1–46.
Chinese National Food Safety Standard for Maximum Levels of Contaminants in Foods (GB 2762-2017). Beijing: National Health and Family Planning Commission, National Medical Products Administration of China. 2017. P. 1–19.
Pohanka M. // Int. J. Anal. Chem. 2021. V. 2021. Article 6685619. https://www.hindawi.com/journals/ijac/ 2021/6685619/
Parolo C., Sena-Torralba A., Bergua J.F., Calucho E., Fuentes-Chust C., Hu L.M., Rivas L., Alvarez-Diduk R., Nguyen E.P., Cinti S., Quesada-Gonzalez D., Merkoci A. // Nature Prot. 2020. V. 15. № 12. P. 3816–3788.
Zhang Y., Li X.Q., Li H.M., Zhang Q.H., Gao Y., Li X.J. // TrAC Trends Anal. Chem. 2019. V. 110. P. 356–344.
Fakhlaei R., Selamat J., Khatib A., Razis A.F.A., Sukor R., Ahmad S., Babadi A.A. // Foods. 2020. V. 9. № 11. Article 1538. https://www.mdpi.com/2304-8158/9/11/1538
Byzova N.A., Smirnova N.I., Zherdev A.V., Eremin S.A., Shanin I.A., Lei H.T., Sun Y., Dzantiev B.B. // Talanta. 2014. V. 119. P. 125–132.
Шанин И.А., Зверева Е.А., Ерёмин С.А., Свиридов О.В., Жердев А.В., Дзантиев Б.Б. // Прикл. биохимия и микробиология. 2019. Т. 55. № 5. С. 513–520.
Frens G. // Nat. Phys. Sci. 1973. V. 241. P. 20–22.
Byzova N.A., Zvereva E.A., Zherdev A.V., Eremin S.A., Dzantiev B.B. // Talanta. 2010. V. 81. № 3. P. 843–848.
Wang Z., Hu S., Bao H., Xing K., Liu J., Xia J., Lai W., Peng J. // J. Sci. Food Agric. 2021. V. 101. № 2. P. 684–692.
Guo L., Wu X., Liu L., Kuang H., Xu C. // Biotechnol. J. 2020. V. 15. № 1. Article 1900174. https://doi.org/10.1002/biot.201900174
Zhao L., Li J., Li Y., Wang T., Jin X., Wang K., Rahman E., Xing Y., Ji B., Zhou F. // Food Chem. 2017. V. 229. P. 439–444.
Wang S., Liu J., Yong W., Chen Q., Zhang L., Dong Y., Su H., Tan T. // Talanta. 2015. V. 131. P. 562–569.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология