

Прикладная биохимия и микробиология, 2021, T. 57, № 6, стр. 556-562
Полимеризация (+)-катехина в глубоком эвтектическом растворителе с использованием грибной лакказы: физико-химические свойства продуктов и ингибирование α-глюкозидазы
М. Е. Хлупова 1, О. В. Морозова 1, И. С. Васильева 1, Г. П. Шумакович 1, Е. А. Зайцева 2, В. А. Чертков 2, А. К. Шестакова 3, А. И. Ярополов 1, *
1 Институт биохимии им. А.Н. Баха, Федеральный исследовательский центр “Фундаментальные основы биотехнологии” Российской академии наук
119071 Москва, Россия
2 Московский государственный университет им. М.В. Ломоносова, химический факультет
119991 Москва, Россия
3 Государственный научно-исследовательский институт химии и технологии элементоорганических соединений
105118 Москва, Россия
* E-mail: yaropolov@inbi.ras.ru
Поступила в редакцию 26.05.2021
После доработки 18.06.2021
Принята к публикации 02.07.2021
Аннотация
Глубокие эвтектические растворители (ГЭР) являются альтернативой традиционным органическим растворителям для проведения ферментативных реакций между соединениями с плохой растворимостью. Проведена биокаталитическая полимеризация флавоноида (+)-катехина (КХ) с использованием лакказы из культуральной жидкости базидиального гриба Trametes hirsuta в ГЭР-буферной смеси (бетаин/глицерин 60 об. % – буфер 40 об. %). Подобраны условия синтеза олигомеров катехина (олигоКХ), растворимых в органических растворителях. По данным высокоэффективной жидкостной хроматографии олигоКХ имели среднечисленную молекулярную массу 10620 и 2540 г/моль с индексом полидисперсности 1.1 и 1.09, соответственно. Исследованы физико-химические свойства полученных олигомеров методами УФ-видимой, FTIR, H1 и C13 ЯМР спектроскопии. Полученные олигоКХ ингибировали активность α-глюкозидазы (IC50 ~ 8 мкг/мл).
Катехины относятся к растительным флавоноидам и обладают антиканцерогенными, антибактериальными, антиоксидантными, кардиопротекторными и хелатирующими свойствами, а также ингибируют активность различных ферментов [1–6]. Биологические свойства катехинов сильно зависят от их химической структуры. В отличие от полифенольных соединений с низкой молекулярной массой, их полимерные и олигомерные производные обладают улучшенными физиологическими характеристиками и более продолжительным временем жизни in vivo [7]. В литературе описано получение олигомеров/полимеров КХ ферментативной полимеризацией мономера с использованием лакказ и пероксидазы [8–10], фотополимеризацией [11] и HCl-катализируемой полимеризацией [12]. В связи с плохой растворимостью катехинов в водных растворах, их полимеризацию обычно проводят в водно-органических смесях. Катехины и их олигомеры проявляют ингибиторную активность по отношению к α-амилазе и α-глюкозидазе [8, 13, 14]. Эти ферменты играют значительную роль в гидролизе полисахаридов, способствуя уменьшению уровня глюкозы в крови и, таким образом, влияют на развитие сахарного диабета 2 типа – хронического заболевания для многих людей во всем мире. Одним из направлений лечения этого заболевания является уменьшение скорости разложения полисахаридов путем ингибирования активности α-амилазы и α‑глюкозидазы. Таким образом, олиго-/поликатехины, обладающие большей стабильностью по сравнению с мономером КХ, могут являться перспективной субстанцией для лечения диабета 2 типа.
Лакказа (п-дифенол:кислород оксидоредуктаза, КФ 1.10.3.2) относится к “голубым” оксидазам, катализирует окисление различных органических соединений, включая полифенолы, метокси-замещенные фенолы, полиамины молекулярным кислородом. При ферментативном окислении фенольных субстратов образуются радикальные продукты, которые вступают в реакции сочетания с образованием олигомерных/полимерных соединений [15, 16]. Этот фермент привлекает большое внимание в качестве катализатора для тонкого органического синтеза [17, 18] и может быть использован для полимеризации различных органических соединений.
В последние годы появился новый класс растворителей, названных глубокими эвтектическими растворителями (ГЭР) [19], которые имеют большое преимущество перед традиционными органическими растворителями, а именно, нетоксичность, невоспламеняемость, биодеградируемость и др. Они рассматриваются как “зеленые” растворители. Как правило, ГЭР получают простым термическим смешиванием как минимум двух компонентов, в результате которого образуется эвтектический раствор с температурой плавления ниже, чем у отдельных компонентов [19–21]. Компоненты ГЭР обычно представляют собой органические соединения, одно из которых является акцептором, а другое донором водородных связей. ГЭР или их смеси с буферным раствором могут использоваться в различных областях, в том числе для проведения биокаталитических реакций [22]. В литературе описано весьма ограниченное количество примеров использования оксидоредуктаз для синтеза соединений в ГЭР-буферных смесях [23, 24].
Цель работы – проведение окислительной полимеризации (+)-катехина с участием грибной лакказы Trametes hirsuta в ГЭР-буферной смеси и исследование структуры и свойств полученных продуктов.
МЕТОДИКА
Лимонная кислота, NaH2PO4, KH2PO4, NaOH – производства “Riedel-de Haën” (Германия), 2,2'‑азино-бис(3-этилбензтиазолин-6-сульфонат (АБТС), α-глюкозидаза из Saccharomyces cerevisiae, N-нитрофенил-α-D-глюкопиранозид (ПНФГ) – “Sigma-Aldrich” (США), тетрагидрофуран (ТГФ), диметилформамид (ДМФА), ацетонитрил, бетаин моногидрат “Acros Organics”(США), (+)-катехин (≥96%) – “Carl Roth GmbH” (Германия), диметилсульфоксид (ДМСО) – “Marbiopharm” (Россия), ДМСО-D6 (“Aldrich”), глицерин 99% “Panreac” (Испания), уксусная кислота высшей степени очистки производства “Химмед” (Россия) были использованы без дополнительной очистки.
Лакказа была выделена из культуральной жидкости базидиального гриба Trametes hirsuta (Wulfen) Pilát (штамм T. hirsuta 56) согласно методу [25]. Фермент был гомогенен по данным ДДС-электрофореза и имел удельную активность 163 МЕ/мг белка. Активность фермента определяли спектрофотометрически, используя в качестве хромогенного субстрата 1 мМ раствор АБТС (λ = 420 нм; ε = 36000 M–1 см–1) в 0.1 М Na-цитратно-фосфатном буфере, рН 4.5. За единицу активности принимали количество фермента, катализирующего превращение 1 мкмоль АБТС за 1 мин при температуре 22°С. Концентрация белка, измеренная согласно методу [26], составляла 7.8 мг/мл.
Все растворы готовили с использованием воды, очищенной на установке Simplicity (“Millipore”, США).
ГЭР бетаин-глицерин получали термическим смешиванием компонентов с молярным соотношением 1 : 2 при 45°С. Смесь ГЭР/буфер с соотношением 60 : 40 об. % готовили добавлением соответствующего объема 0.1 М Na-цитратно-фосфатного буфера, рН 4.5, к ГЭР.
Ферментативную полимеризацию КХ проводили следующим образом: 15 мг КХ растворяли в 1.8 мл ГЭР, добавляли 1.2 мл буферного раствора и интенсивно перемешивали. Концентрация КХ в реакционной среде была 17 мМ. Полимеризацию инициировали добавлением фермента (удельная активность в реакционной смеси 1.05 МЕ/мл). Синтез проводили в аэробных условиях при комнатной температуре (21–22°С) и постоянном перемешивании со скоростью 400 об./мин на магнитной мешалке RT-10 (“IKA®-Werke GmbH & Co”, Германия) в течение 24 ч. Затем прозрачный раствор разбавляли в 5 раз деионизированной водой, образовавшийся осадок отделяли центрифугированием (7000 g, 20 мин) с использованием центрифуги Eppendorf 5804 R (Австрия), многократно промывали 3%-ным раствором этанола, высушивали при 37°С до постоянного веса и использовали в дальнейших экспериментах. Выход продукта рассчитывали, как процентное отношение массы полученного олигоКХ к массе исходного мономера.
Среднечисленную молекулярную массу и индекс полидисперсности олигоКХ определяли методом гель-проникающей хроматографии на хроматографе высокого давления “Waters” (США), оснащенном колонкой Styragel HR 1Е, 300 × 7.8 мм (“Waters”, США), УФ- и рефрактометрическим детекторами. В качестве элюента использовали ТГФ, скорость потока 1 мл/мин, объем вводимой пробы 40 мкл. Калибровку системы проводили по полистирольным стандартам.
УФ-видимые спектры регистрировали на спектрофотометре UV1240 mini (“Shimadzu”, Япония), ИК спектры образцов в таблетках KBr на спектрометре Frontier FT-IR/FIR (“PerkinElmer Inc.”, США). Спектры ЯМР 1H и 13С регистрировали в ДМСО-D6 при температуре 303K на спектрометре AV-600 (“Bruker”, США) с рабочей частотой 600.03 МГц для ядер 1Н. Для отнесения сигналов ядер 1Н и 13С использовали данные двумерных экспериментов COSY, HSQC и HMBC [27]. Полученные параметры спектров мономера КХ ЯМР 1Н: 2.354 (1H, dd, J = 15.96; 8.12, H4b); 2.660 (1H, dd, J = 15.96; 5.36, H4a); 3.812 (1H, ddt, Jd = = 7.49; 8.12, Jt = 5.25, H3); 4.482 (1H, d, Jd = 7.49, H2); 4.836 (1H, d, Jd = 5.17, 3-OH); 5.691 (1H, d, Jd = = 2.29, H8); 5.889 (1H, d, Jd = 2.29, H6); 6.597 (1H, dd, Jd = 2.05, 8.02 H6'); 6.686 (1H, d, Jd = 8.02 H5'); 6.725 (1H, d, Jd = 2.05 H2’); 8.774 (1H, s, 4’-OH); 8.822 (1H, s, 3'-OH); 8.906 (1H, s, 7-OH); 9.139 (1H, s, 5-OH); ЯМР 13С: 27.91 (С4); 66.39 (С3); 81.07 (С2); 93.94 (С8); 95.21 (С6); 99.14 (С10); 114.60 (С2'); 115.15 (С5'); 118.48 (С6'); 130.69 (С1'); 144.90 (С3' & C4'); 155.42 (С9); 156.23 (С5); 156.52 (С7). Химические сдвиги приведены в м.д., константы спин-спинового взаимодействия (КССВ) – в Гц, s – синглет, d – дублет, dd – дублет дублетов, ddt – дублет дублетов триплетов.
Активность α-глюкозидазы определяли по гидролизу субстрата фермента ПНФГ, продукт гидролиза которого имеет максимум поглощения при 400 нм. Определение ингибирования α-глюкозидазы проводили по методу [28] с минимальными изменениями. В реакционную смесь: 100 мкл α-глюкозидазы (0.5 МЕ/мл) и 600 мкл 0.1 M фосфатного буфера (pH 6.9) вносили 50 мкл раствора исследуемого вещества в ДМСО в концентрации 31.25, 62.5, 125, 250, 500 и 1000 мкг/мл. Затем смесь инкубировали при 37°C в течение 15 мин. Реакцию инициировали добавлением 100 мкл 5 мМ раствора ПНФГ в 0.1 M фосфатном буфере (pH 6.9) и инкубировали при 37°C в течение 15 мин. Затем реакцию останавливали добавлением 400 мкл 0.2 М раствора Na2CO3 и измеряли поглощение при 400 нм при комнатной температуре. Ингибирование фермента рассчитывали по формуле:
где D0 – контроль (поглощение раствора без исследуемого вещества); D – поглощение раствора в присутствие КХ или олигоКХ. Влияние ДМСО на фермент учитывали в контроле. Определение ингибирующего действия мономера КХ проводили аналогично в диапазоне концентраций 1–20 мг/мл ДМСО.Эффективность ингибирования оценивали по значению IC50, которое определяли из графика зависимости ингибирования (%) от концентрации исследуемого вещества. IC50 – концентрация вещества, необходимая для ингибирования 50% активности фермента.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
На рис. 1 представлена структурная формула (+)-катехина, использованного в данной работе. Согласно литературным данным, ГЭР сохраняют свои свойства в составе водных смесей при содержании ГЭР выше 50 вес. %. При меньшем содержании ГЭР, смесь ГЭР/вода нужно рассматривать как водный раствор отдельных компонентов [29]. Ферментативную полимеризацию КХ проводили в смеси ГЭР/буфер, содержащей 40 об. % (~35 вес. %) буфера. Были подобраны условия полимеризации КХ (активность фермента в реакционной среде, время реакции) для получения продуктов полимеризации, растворимых в ацетонитриле, ДМСО, ТГФ и ДМФА. Обнаружено, что для получения растворимых в органических растворителях олигоКХ, активность лакказы в реакционной среде должна быть 1.05 МЕ/мл, а время синтеза – 24 ч. Выход продуктов в этих условиях составлял 32%. При увеличении активности фермента продукты полимеризации КХ были не растворимы в указанных выше растворителях, что препятствовало их исследованию.
По данным гель-проникающей хроматографии в результате ферментативной полимеризации КХ в смеси ГЭР/буфер образовывалось два продукта со среднечисленной молекулярной массой 10620 и 2540 г/моль и индексом полидисперсности 1.1 и 1.09 соответственно. Таким образом, в результате ферментативной полимеризации КХ в смеси ГЭР/буфер образуется 2 олигомера со степенью полимеризации 24 и 6.
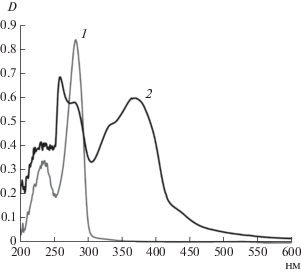
УФ-видимые спектры мономера КХ и олигоКХ в ДМСО представлены на рис. 2. Острый пик при 280 нм на спектре КХ соответствует π-π* переходам ароматических фрагментов мономера. Продукты ферментативной полимеризации КХ, растворимые в ДМСО, поглощают в широком диапазоне длин волн, вплоть до 600 нм. На спектре олигоКХ помимо максимума при 280 нм видны максимумы при 260, 330 и 370 нм.
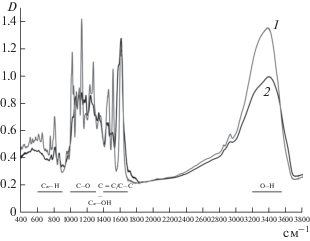
На рис. 3 представлены ИК-Фурье спектры КХ и олигоКХ. На спектре мономера (рис. 3, кривая 1) присутствуют полосы, соответствующие валентным колебаниям связи О–Н (широкий максимум в области 3200–3550 см–1), валентным колебаниям C=С и С–С связей ароматических колец (1430–1625 см–1) и С–O фенолов и простых эфиров (1100–1310 см–1), деформационным колебаниям С–ОН (1330–1390 см–1) и C–H (650–900 см–1) связей ароматических колец [30]. Характерные области адсорбции для этих групп отмечены на рис. 3 горизонтальными линиями. На спектре олигоКХ также присутствовали полосы, характерные для валентных колебаний групп О–Н, С=С, С–С и С–O (рис. 3, кривая 2), однако их интенсивность снижена по сравнению со спектром КХ. Наиболее существенные различия наблюдались в низкочастотной области спектров: на спектре олигоКХ отсутствовали полосы, соответствующие колебаниям связи Саром–Н. Можно предположить, что при ферментативной полимеризация КХ происходило замещение ароматических протонов.
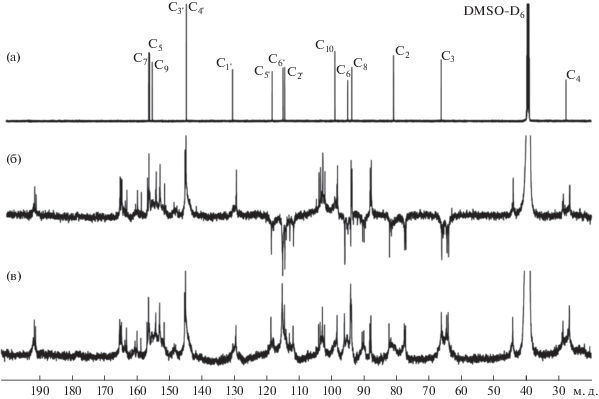
Для структурного анализа олигоКХ была использована спектроскопия ЯМР на ядрах 1Н и 13С (рис. 4, 5). Было проведено отнесение всех сигналов в спектрах ЯМР 1Н и 13С мономера КХ (нумерация атомов показана на рис. 1). Опорными служили сигналы протонов Н4a и H4b, имеющие характерную мультиплетность (рис. 4а). Для отнесения сигналов в спектрах ЯМР 1Н и 13C также использовали данные двумерных спектров ЯМР COSY, HSQC и HMBC. В настоящей работе с использованием двумерных спектров HMBC впервые было проведено отнесение сигналов протонов всех гидроксильных групп. Значения параметров спектров ЯМР мономера КХ приведены в экспериментальной части.
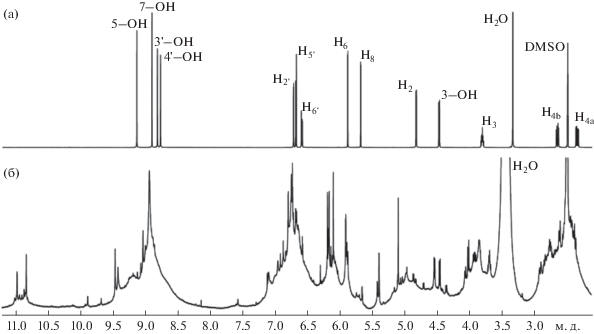
Рис. 5.
Спектры ЯМР 13С (a) 13C-{1H} мономера КХ, (б) 13C-APT олигоКХ (сигналы четвертичных атомов водорода и СН2-группы направлены вверх, а сигналы CH‑групп вниз) и (в) 13C-{1H} олигоКХ.
Следует отметить характерную особенность структуры КХ: кольцо С этой молекулы является неплоским. Наибольший выход из плоскости наблюдается для атомов углерода С2, С3 и С4. Согласно нашим оценкам, эти отклонения составляют от 0.2 до 0.4 Å. При этом протон Н4b, ароматическое кольцо В и гидроксильная группа 3-ОН занимают псевдоэкваториальное положение, а протоны Н2, Н3 и Н4a – псевдоаксиальное [31].
Сравнительный анализ спектров ЯМР 1H и 13C олигоКХ и мономера КХ показал, что всем сигналам в спектрах мономера соответствуют группы сигналов в спектрах олигоКХ, что свидетельствует о сохранении углеродного скелета КХ в процессе ферментативной олигомеризации. В спектре ЯМР 1Н олигоКХ (рис. 4б) видны уширенные сложные мультиплеты в областях 6.3–7.4 и 5.6–6.3 м.д., которые соотносятся с протонами ароматических колец В (H2', H5', H6') и А (Н6 и Н8), соответственно. При этом суммарная интегральная интенсивность протонов кольца А олигоКХ почти в 3 раза ниже, чем интенсивность протонов кольца В, хотя в мономере КХ общий интеграл протонов кольца А меньше общего интеграла протонов кольца В всего в 1.5 раза. В области 4.3–5.5 м.д. находятся сигналы протона Н2 и гидроксильного протона 3-ОН, а в диапазоне 3.6–4.3 м.д. – сигнал протона Н3. Интегральные интенсивности сигналов этих протонов также сильно снижены по сравнению со значениями для мономера КХ. В области 2.2–3.0 м.д. находятся сигналы протонов Н4а и H4b СН2-группы с интегралом, близким к значению для мономера. В слабопольной части спектра ЯМР 1Н олигоКХ наблюдались уширенные сигналы в области 8.0–11.3 м.д. с общей интегральной интенсивностью, соответствующей трем протонам, которые соответствуют фенольным гидроксилам. Важно отметить, что интенсивность уширенных сигналов протонов Н4а и H4b при С4 соответствует интенсивности двух протонов. Это однозначно свидетельствует о том, что в олигоКХ не происходит образование связей в положении С4. Однако в литературе ранее отмечалось, что образование димеров и тримеров КХ природного происхождения может происходить за счет окислительного кросс-сочетания с образованием связей между атомами С4 одной молекулы с атомом углерода С6 или С8 другой молекулы флавоноида [32]. Очевидно, что механизм “сшивания” молекул флавоноидов может существенно различаться в зависимости от условий реакций.
Анализ спектров ЯМР 1Н олигоКХ позволяет обоснованно предположить, что при ферментативной полимеризации сохраняется углеродный скелет молекулы КХ (кольца A, B и C), при этом один из протонов кольца А (H6 и/или H8) замещается на алкоксильную или арилоксильную группу, образованную из другой молекулы КХ. Также при отрыве атома водорода от 3-ОН группы кольца С под действием лакказы помимо образования алкокси-радикала происходит частичное окисление 3-ОН до карбонильной группы.
Анализ спектров ЯМР 13С (рис. 5) показал, что всем сигналам атомов углерода кольца В мономера КХ соответствуют уширенные интенсивные сигналы в спектре олигоКХ. При замещении протона в бензольном кольце арилокси- или алкокси-группой химические сдвиги всех атомов углерода в кольце должны заметно измениться (до 10 м.д.). Однако при ферментативной полимеризации КХ таких изменений в кольце В не наблюдается.
Сигналы углерода C2 и C3 в спектре олигоКХ (рис. 5б, 5в) представляют собой группы уширенных сигналов, занимающих довольно широкие области спектра 77–82 м.д. и 64–66 м.д. соответственно. Поскольку в процессе ферментативной полимеризации КХ не происходило замещение протонов кольца B, т.е. структура кольца В сохранялась, на изменение химических сдвигов C2, по-видимому, существенное влияние оказывали изменения при атоме C3. Сигналы углеродов С6 и С8 лежат в области 90.0–96.0 м.д. и имеют сильно пониженную для сигналов СН-групп интенсивность. При этом к четвертичному атому углерода С10 помимо основного сигнала при 98.3 м.д. относятся еще несколько сигналов при 88.3 м.д., 94.1 м.д. и широкий сигнал при 102.0–104.0 м.д. Такое разнообразие сигналов атома С10 олигоКХ обусловлено эффектами, связанными с замещением атомов водорода в кольце А, и частичным окислением 3-OH группы до карбонильной группы. Те же эффекты приводят к появлению многочисленных сигналов четвертичных углеродов в области 157.0–165.0 м.д. Эти сигналы относятся как к атомам углерода С5, С7, С9 кольца А, так и к сигналам четвертичных углеродов С6 и С8, возникающим в процессе замещения атома водорода на арилокси- или алкокси-группы. Подтверждением образования продукта, содержащего карбонильную группу при С3, служит характеристичный сигнал четвертичного углерода при 193.6 м.д. и появление сигнала СН2-группы при 44.0 м.д., который оказывается смещенным в слабое поле относительно основного сигнала при 26.0–29.0 м.д.
Таким образом, анализ спектров ЯМР 1Н и 13С олиго КХ и мономера КХ позволяет заключить, что при ферментативной полимеризации сохраняется углеродный скелет молекулы КХ, а в образовании активных радикальных частиц участвуют все пять гидроксильных групп мономера. Образующиеся радикалы атакуют углерод С6 и/или С8 кольца А другой молекулы КХ, в результате чего происходит замещение протона на алкокси- или арилокси- группу. Также при отрыве атома водорода от гидроксила в положении С3 под действием лакказы параллельно происходит окисление 3-OH группы до карбонила. Количество звеньев, содержащих карбонильную группу, в олигоКХ составляет 15–20% от общего количества мономерных звеньев.
Важно отметить, что механизм ферментативной полимеризации КХ, описанный в данной работе, подобен механизму лакказа-катализируемой полимеризации дигидрокверцетина в смеси ГЭР/буфер [33]. В обоих случаях это двухстадийный процесс. Первая стадия – это ферментативное окисление гидроксильных групп с образованием радикалов. Вторая стадия – радикальная атака активных частиц на ароматическую систему другой молекулы флавоноида. Однако, структура олигоКХ существенно отличается от структуры олигомера дигидрокверцетина. При полимеризации КХ атака происходит по атомам углерода С6 и С8 кольца A, в то время как при полимеризации дигидрокверцетина атака идет исключительно по атому углерода С6' кольца B. Согласно проведенным нами квантовохимическим расчетам, причиной этого являются существенные различия в электронной структуре этих флавоноидов. Наличие в молекуле дигидрокверцетина карбонильной группы в положении С4 существенно дестабилизирует энергию переходного состояния при радикальной атаке по атомам С6 и С8 дигидрокверцетина. В случае КХ этой дестабилизации нет, а согласованный эффект трех кислородсодержащих заместителей кольца А оказывается сильнее, чем аналогичный эффект двух заместителей кольца В, поэтому радикальная атака в КХ идет по кольцу А.
Синтезированные олигоКХ были протестированы на ингибирование активности α-глюкозидазы. Концентрация олигоКХ, необходимая для ингибирования 50% активности фермента (IC50) с использованием в качестве субстрата ПНФГ составляла ~8 мкг/мл, в то время как для мономера КХ IC50 – ~980 мкг/мл.
Таким образом, олигомеры катехина, синтезированные с использованием грибной лакказы T. hirsuta в ГЭР-буферной смеси, являются эффективными ингибиторами α-глюкозидазы и могут быть перспективной субстанцией для лечения сахарного диабета 2-го типа.
Работа выполнена при частичной финансовой поддержке Российского Фонда Фундаментальных исследований (проект № 20-08-00104а).
Список литературы
Vinson J.A. //Adv. Exp. Med. Biol. 1998. V. 439. P. 151–164.
Jankun J., Selman S.H., Swiercz R., Skrzypczak-Jankun E. // Nature. 1997. V. 387. № 6633. P. 561.
Bordoni A., Hrelia S., Angeloni C., Giordano E., Guarnieri C., Caldarera C.M., Biagi P.L. // J. Nutr. Biochem. 2002. V. 13. № 2. P. 103–111.
Nakagawa K., Ninomiya M., Okubo T., Aoi N., Juneja L.R., Kim M., Yamanaka K., Miyazawa T. // J. Agric. Food Chem. 1999. V. 47. № 10. P. 3967–3973.
Kumar S., Pandey A.K. // Scientific World J. 2013. V. 2013. Article № 162750.https://doi.org/10.1155/2013/162750
Latos-Brozio M., Masek A., Piotrowska M. // Biomacromolecules. 2021. V. 11. № 1. P. Article № 50.
Hagerman A.E., Riedl K.H., Jones G.A., Sovik K. N., Ritchard N.T., Hartzfeld P.W., Riedel T.L. // J. Agric. Food Chem. 1998. V.46. № 5. P. 1887–1892.
Jeon S.Y., Oh S.J., Kim E., Imm J.Y. // J. Agric Food Chem. 2013. V. 61. № 19. P. 4577–4584.
Kurisawa M., Chung J.E., Uyama H., Kobayashi Sh. // Macromol. Biosci. 2003. V. 3. № 12. P. 758–764.
Kurisawa M., Chung J.E., Kim Y.J., Uyama H., Kobayashi Sh. // Biomacromolecules. 2003. V. 4. № 3. P. 469–471.
Liang J.Y., Yang M.Y., Hu A., Chen L.Y. // J. Photochem. Photobiol. B: Biology. 2016, V. 165. P. 115–120.
Oliver S., Hook J.M., Boyer C. // Polym. Chem. 2017. V. 8. № 15. P. 2317–2326.
Kim T., Choi H.J., Eom S.H., Lee J., Kim T.H. // Bioorg. Med. Chem Letters. 2014. V. 24. № 6. P. 1621–1624.
Rasouli H., Hossein-Ghazvini S.M.-B., Adibi H., Khodarahmi R. // Food and Function. 2017. V. 8. № 5. P. 1942–1954.
Hollmann F., Arends I. // Polymers. 2012. V. 4. № 1. P. 759–793.
Bassanini I., Ferrandi E.E., Riva S., Monti D. // Catalysts. 2021. V. 11. № 1. P. 26.
Witayakran S., Ragauskas A.J. // Adv. Synth. Catal. 2009. V. 351. № 9. P. 1187–1209.
Mogharabi M., Faramarzi M.A. // Adv. Synth. Catal. 2014. V. 356. № 5. P. 897–927.
Abbott A., Boothby D., Capper G., Davies D., Rasheed R. // J. Am. Chem. Soc. 2004. V. 126. № 29. P. 9142–9147.
Smith E., Abbott A., Ryder K. // Chem. Rev. 2014. V. 114. № 21. P. 11060–11082.
Dai Y., Witkamp G.J., Verpoorte R., Choi Y. // Food Chem. 2015. V. 187. P. 14–19.
Cicco L., Dilauro G., Perna F., Vitale P., Capriati V. // Org. Biomol. Chem. 2021. V. 19. №12. P. 2558–2577.
Ünlü A., Prasad B., Anavekar K., Bubenheim P., Liese A. // Prep. Biochem. Biotechnol. 2017. V. 47. № 9. P. 918–924.
Altundağ A., Ünlü A.E., Takaç S. // J. Chem. Technol. Biotechnol. 2020. V. 96. № 4. P. 1107–1115.
Горшина Е.С., Русинова Т.В., Бирюков В.В., Морозова О.В., Шлеев С.В., Ярополов А.И. // Прикл. биохимия и микробиология. 2006. Т. 42. № 6. С. 638–644.
Ehresmann B., Imbault P., Well J.H. // Analyt. Biochem. 1973. V. 54. № 2. P. 454–463.
Chertkov V.A., Davydov D.V., Shestakova A.K. // Chem. Heterocycl. Compd. 2011. V. 47. № 1. P. 45–54.
Nazir N., Zahoor M., Ullah R., Ezzeldin E., Mostafa G.A.E. // Molecules. 2021. V. 26. № 1. Article № 137. https://doi.org/10.3390/molecules26010137
Hammond O.S., Bowron D.T., Elder K.J. // Angew. Chem. Int. 2017. V. 56. № 33. P. 9782–9785.
Ramos-Tejada M.M., Durán J.D.G., Ontiveros-Ortega A., Espinosa-Jimenez M., Perea-Carpio R., Chibowski E. // Colloids Surf. B: Biointerfaces. 2002. V. 24. № 3–4. P. 297–308.
Chertkov A.V., Pokrovskiy O.I., Shestakova A.K., Chertkov V.A. // Chem. Heterocycl. Compd. 2008. V. 44. № 5. P. 621–623.
Tarascou I., Baratheu K., Andre Y., Pianet I., Dufourc E.J., Fouguet E. // Eur. J. Org. Chem. 2006. V. 2006. № 23. P. 5367–5377.
Khlupova M., Vasil’eva I., Shumakovich G., Zaitseva E., Chertkov V., Shestakova A., Morozova O., Yaropolov A. // Catalysts. 2021. V. 11. № 5. Article № 639. https://doi.org/10.3390/catal11050639
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология