

Прикладная биохимия и микробиология, 2021, T. 57, № 4, стр. 326-331
CRISPR-интерференция аденилатциклазы Mycobacterium tuberculosis
Н. И. Надолинская 1, *, М. В. Замахаев 1, М. С. Шумков 1, Д. К. Армянинова 1, Д. С. Карпов 2, А. В. Гончаренко 1
1 Институт биохимии им. А.Н. Баха, Федеральный исследовательский центр
“Фундаментальные основы биотехнологии” Российской академии наук
119071 Москва, Россия
2 Центр высокоточного редактирования и генетических технологий для биомедицины,
Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
* E-mail: nioriss@gmail.com
Поступила в редакцию 25.12.2020
После доработки 20.01.2021
Принята к публикации 22.02.2021
Аннотация
В работе описана модификация вектора pRH2521 системы pRH2502/pRH2521, используемой для CRISPR-dCas9-опосредованной РНК-интерференции микобактерий. Введенная модификация обеспечивала повышение эффективности клонирования спейсеров направляющих РНК. Способность изменённой системы pRH2502/pRH2521подавлять транскрипцию определенных генов проверена на примере генов аденилатциклаз Mycobacterium tuberculosis. Полученные результаты выявили ограничения использования системы pRH2502/pRH2521 для CRISPR-интерференции, связанные с вероятностью нахождения участка PAM в области промотора подавляемого гена.
Несмотря на усилия медицинского и научного сообщества, туберкулез (ТБ) остается основной причиной смерти среди инфекционных заболеваний. По данным ВОЗ, он ежедневно уносит жизни около 4000 человек [1], что сопоставимо с количеством ежедневных смертей от COVID-19 в пик пандемии коронавирусной инфекции в 2020 г. Функции почти четверти из 4000 генов Mycobacterium tuberculosis (Mtb) остаются неизвестными, хотя 5% из них предположительно значимы для роста [2] и могут быть потенциальными мишенями при разработке противотуберкулёзных препаратов и вакцин. Технология CRISPR (clustered regularly interspaced short palindromic repeats) – многофункциональный инструмент генной инженерии [3], созданный из бактериальной системы приобретенной устойчивости к фагам и плазмидам. CRISPR-ассоциированная нуклеаза образует комплекс с направляющей РНК (gRNA), предназначенной для распознавания интересующей целевой последовательности (протоспейсера) и расположенного рядом участка (мотива) PAM (protospacer adjacent motif), состоящего из 2–6 пар оснований ДНК. Результатом узнавания и связывания комплекса gRNA и CRISPR-ассоциированной нуклеазы с целевой последовательностью является ее двухцепочечный разрыв. В процессе репарации разрыва возможно направленное редактирование последовательности ДНК. Позже метод был адаптирован для направленного подавления экспрессии генов без изменения последовательности ДНК. Специфичное подавление транскрипции с использованием CRISPR было названо CRISPR-интерференцией (CRISPRi) [4–6].
Для осуществления CRISPR-Cas9 интерференции нуклеаза Cas9 была лишена эндонуклеазной активности путем замены двух аминокислот (D10A и H841A) в никазных доменах RuvC и HNH исходного белка [5]. Поскольку ДНК-связывающая активность dCas9 не была затронута, комплекс этой нуклеазы с направляющей РНК может обратимо связывать ДНК-мишень, вследствие чего происходит блокировка транскрипции целевых генов. Наличие индуцибельного промотора делает регуляцию системы CRISPRi более тонкой и точной [7]. В результате CRISPRi-опосредованная репрессия генов может достигать 99% и приводить к фенотипическим изменениям, сходным с нокаутированием гена [8]. Такой подход позволяет изучать функции неизвестных генов, не прибегая к длительному и сложному получению делеций.
Оптимизированная для микобактерий система CRISPR-dCas9-опосредованной РНК-интерференции [9] включает вектор для экспрессии dCas9 pRH2502 и вектор pRH2521 для клонирования спейсеров, кодирующих направляющую РНК для нацеливания на интересующий ген. Авторы работы [9] продемонстрировали подавление экспрессии нескольких генов Mtb и возможность модуляции степени ингибирования транскрипции. Ключевым моментом, влияющим на эффективность подавления экспрессии, оказалась близость протоспейсера к промоторному участку.
Методически CRISPR-интерференция с помощью системы pRH2502/pRH2521 включает следующие этапы. Первый – это клонирование подобранных направляющих РНК в вектор pRH2521, затем последовательная трансформация в Mtb вектора pRH2502 с dCas9 (получение клеток Mtb-pRH2502), трансформация вектора с pRH2521-gRNA в Mtb-pRH2502 (получение клеток Mtb-pRH2502/ pRH2521-gRNA). Далее следует индукция CRISPR-интерференции и анализ результатов с помощью ПЦР в реальном времени. Оказалось, что эффективность клонирования спейсеров направляющей РНК в вектор pRH2521 довольно низка. В связи с этим, нами была поставлена задача улучшения эффективности клонирования путeм модификации вектора pRH2521. Модифицированная система была использована для оценки возможности подавления с ее помощью транскрипции генов аденилатциклаз (АС) микобактерий, представляющих значительный научный интерес с позиций их влияния на физиологию Mtb.
Циклический 3', 5'-AMP (cAMP) является одним из наиболее широко распространённых вторичных мессенджеров бактерий, архей и эукариот [10].
У микобактерий АС, осуществляющие синтез сАМР, кодируются необычно большим количеством различных генов. При этом передача сигналов сАМР у Mtb не связана с классической системой катаболитной репрессии, характерной для многих бактерий [11]. Уровень сАМР у туберкулeзной палочки практически не зависит от количества глюкозы в среде культивирования и регулирует иные метаболические пути, а также взаимодействие патогена с хозяином [12].
Ранее нами было показано [13], что гиперэкспрессия гена АС Rv2212 препятствует переходу Mtb в покоящееся состояние. Клетки, гиперэкспрессирующие АСRv2212, демонстрировали значительно более высокую скорость роста в легких и селезeнке инфицированных мышей по сравнению с контрольным штаммом и в отличие от контрольного штамма приводили к гибели ТБ-устойчивых мышей [13]. Вклад остальных генов АС в вирулентность Mtb в настоящее время неизвестен.
Цель работы – модификация системы pRH2502/ pRH2521, используемой для CRISPR-dCas9-опосредованной РНК-интерференции микобактерий, и оценка ее возможностей для CRISPR-интерференции генов АС Rv2212, Rv1320с, Rv1319с, Rv1318с.
МЕТОДИКА
Векторы для экспрессии dСas9 и sgRNA. С целью создания системы для CRISPR/Cas-нокдауна были приобретены векторы (“Addgene”, Великобритания), оптимизированные для работы с микобактериями [9]: плазмида pRH2502, несущая dCas9Spy из Streptococcus pyogenes, и плазмида для направляющих РНК для Cas9 pRH2521.
Модификация вектора pRH2521 для оптимизации клонирования спейсеров направляющих РНК. Для получения вектора pRH2521-α с плазмиды pCRCT был амплифицирован ген α-пептида β-галактозидазы lacZα. ПЦР-продукт гидролизовали по BbsI-сайтам и клонировали в вектор pRH2521, предварительно гидролизованный BbsI. В результате была создана плазмида, позволяющая отбирать колонии бактерий, содержащие вставку спейсеров, с помощью бело-голубой селекции в присутствии субстрата β-галактозидазы X-Gal. Колонии клеток, несущих пустой вектор pRH2521-α, окрашивались в синий цвет, а колонии бактерий, содержащие вставку спейсеров, теряли α-пептид и в присутствии хромогенного субстрата β-галактозидазы X‑Gal не имели синего окрашивания. Названия и последовательности праймеров приведены в табл. 1, сайт BbsI выделен жирным шрифтом.
Таблица 1.
Олигонуклеотиды и праймеры, использованные в исследовании
| Название | Последовательность | Амплифицируемый участок |
|---|---|---|
| UpBbsLacZα | ATTTCTACGGGAATGTCTTCTCAG CCGCTACAGGGCGCGTCCCAT | Ген α-пептида β-галактозидазы |
| LowBbsLacZα | AGTAGAAAAACGAGTCTTCTGGAA AGCGGGCAGTGAGCGCAA | |
| gRNA Rv2212 | CTTGGACTTCGACGCCCTCG | Направляющая РНК для гена Rv2212 |
| gRNA Rv1320c | GCCACCACTCGACACTTGCC | Направляющая РНК для гена Rv1320c |
| UpRT2212 | GCGGGCCGGCTTGCTCACCTACCT | Ген Rv2212 |
| LowRT2212 | CGGCGTTCGGCTTGCACCATCTCT | |
| UpRT1320 | CCGCATTGGGTGTCGTTCG | Ген Rv1320с |
| LowRT1320 | TGAGCCAGTAGGTGCCCAGTATGA | |
| UpRT1319 | CCGACGGGCGCAACACCTT | Ген Rv1319с |
| LowRT1319 | ACGCCCCGCCGATATCCCACAGAA | |
| UpRT1318 | TCCCGAAGACAAGGCACTGG | Ген Rv1318с |
| LowRT1318 | CCTGGCACTCGGGCATTTCGT T |
Подбор спейсеров направляющих РНК для генов аденилатциклаз Mtb. Для подбора спейсеров gRNA использовали ресурс http://grna.ctegd.uga.edu/, в который по просьбе авторов настоящей работы был внесен геном Mtb. Были подобраны направляющие для нокдауна выбранных генов АС с использованием эндонуклеазы Cas9. Последовательности спейсеров направляющих РНК приведены в табл. 1.
Подавление экспрессии генов оперона Rv1320c, Rv1319c, Rv1318c осуществляли с помощью одной общей направляющей gRNA.
Клонирование спейсеров направляющих РНК для генов аденилатциклаз Mtb. Спейсеры направляющих РНК подбирали по принципу максимальной близости к старту транскрипции, насколько это позволяло наличие в целевой области PAM-мотива (рис. 1).
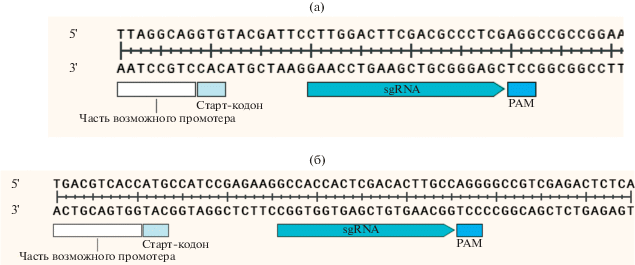
Для клонирования спейсеров направляющих РНК был проведён дизайн олигонуклеотидов, которые, отжигаясь друг на друга, образовывали липкие концы, совместимые с липкими концами вектора pRH2521-α, образующиеся после его гидролиза эндонуклеазой рестрикции BbsI (рис. 2).
Рис. 2.
Спейсер направляющей РНК для подавления экспрессии гена Rv2212 (а) и генов оперона Rv1320c-Rv1319c-Rv1318c (б).
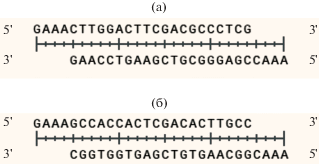
Бактериальные штаммы, среды, условия культивирования. В исследовании использованы бактериальные штаммы Escherichia coli DH5α и M. tuberculosis H37Rv.
Клетки E. coli DH5α выращивали в среде Лурия-Бертани (LB) с добавлением антибиотиков, когда это было необходимо (50 мкг/мл канамицина (Km) и/или 50 мкг/мл гигромицина (Hg). Mtb культивировали в средах Миддлбрука 7H9/7Н10 с добавлением ростовой добавки OADC (“Himedia”, Индия) и 0.025% тилоксапола и, при необходимости, антибиотиков (20 мкг/мл Km и/или 25 мкг/мл Hg). Культивирование микобактерий проводили в соответствии с рекомендациями Т. Пэриш [14].
Векторы системы CRISPRi трансформировали в Mtb последовательно (pRH2502, затем pRH2521-α-gRNA) методом электропорации, которую проводили на приборе Gene Pulser Xcell (“Bio-Rad”, США) согласно протоколу [14].
После электропорации Mtb вектором pRH2502 через 4 нед. были получены единичные колонии на среде с Km. Клетки с плазмидой pRH2502 были выращены в жидкой среде и трансформированы плазмидами pRH2521-α-gRNA. Через 4 нед. на среде с Km и Hg были получены колонии, содержащие пару векторов pRH2502/pRH2521-α-gRNA. Далее трансформанты подращивали на жидкой среде 2–3 нед., после чего синтез направляющих РНК и экспрессию dCas9 индуцировали добавлением тетрациклина в концентрации 20 нг/мл.
Подготовка образцов для ПЦР в реальном времени. После 24 ч индукции клеточную биомассу собирали и с помощью коммерческого набора RNeasy Mini Kit (“Qiagen”, Германия) выделяли тотальную РНК, которую затем очищали от примесей ДНК путём обработки ДНКазой I Ambion DNase I RNase free (“Thermo Scientific”, США), не содержащей РНКаз. Удаление примесей ДНК подтверждали ПЦР. Подготовленные образцы РНК служили матрицей для синтеза кДНК с использованием рассеянной затравки. Полученную кДНК использовали в качестве матрицы для ПЦР в реальном времени.
ПЦР в реальном времени. Для проведения ПЦР в реальном времени использовали наборы iQ SYBR Green Supermix Sample (“Bio-Rad”, США). Реакции осуществляли с использованием прибора “Mini Opticon Real-time PCR System” (“Applied Biosystems”, США) согласно протоколу производителя. В качестве референсного гена использовали ген 16S рРНК. Эксперименты проводили в трех технических повторах. Специфические праймеры, использованные в реакции, представлены в табл. 1.
Обработку результатов qRT-PCR проводили с использованием программы LinRegPCR v. 2017.0 (Heart Failure Research Center, Нидерланды), анализ полученных данных осуществляли методом 2–∆∆Ct.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Модификация вектора pRH2521 для оптимизации клонирования спейсеров направляющих РНК. Двухплазмидная система pRH2521/pRH2502 исходно была разработана для CRISPR-интерференции (CRISPR-нокдауна) генов микобактерий [9]; соответствующие модификации и свойства векторов описаны выше. Однако, оказалось, что эффективность клонирования спейсеров направляющей РНК в сайты рестрикции BbsI вектора pRH2521 была низкой, менее 10%, что затрудняло отбор клонов, несущих плазмиду со спейсерами. Для облегчения задачи клонирования спейсеров направляющей РНК в вектор pRH2521 по сайтам BbsI нами был клонирован ген α-пептида β-галактозидазы lacZα. В результате колонии клеток, несущих пустой вектор pRH2521-α, окрашивались в синий цвет, а колонии бактерий, содержащих вектор со вставкой спейсеров, теряли α-пептид и в присутствии X-Gal – хромогенного субстрата β-галактозидазы – не имели синей окраски. Такая модификация вектора pRH2521 позволила легко отбирать целевые клоны с помощью бело-синей селекции.
Оценка эффективности подавления транскрипции генов Rv2212, Rv1318, Rv1319, Rv1320 с помощью системы pRH2502/pRH2521-α CRISPRi. В результате проведения CRISPRi с помощью системы pRH2502/pRH2521-α удалось добиться подавления экспрессии гена Rv2212 на 43% (рис. 3). Гены Rv1320с, Rv1319с, Rv1318с находятся в составе единого оперона и транскрибируются в направлении от Rv1320с к Rv1318с. Транскрипцию генов оперона Rv1320-Rv1319-Rv1318 с помощью выбранной направляющей РНК подавить не удалось. Такой результат, видимо, связан с низким уровнем экспрессии исследуемого оперона в условиях in vitro. Нельзя исключать, что экспрессия оперона в условиях инфицированных макрофагов будет выше и, соответственно, можно будет добиться подавления экспрессии выбранных генов, а также наблюдать фенотипические эффекты такого подавления.
Рис. 3.
Относительная экспрессия (отн. ед.) генов аденилатциклаз Rv2212 и оперона Rv1320c-Rv1319c-Rv1318c в условиях активации системы pRH2502/pRH2521-α при индукции тетрациклином (Tet+). В качестве контроля использован уровень экспрессии в отсутствие индукции тетрациклином(Tet–). Представлены средние значения ± стандартное отклонение.
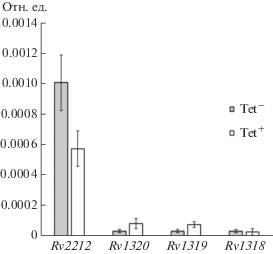
Авторы системы pRH2502/pRH2521 сообщали о подавления уровня экспрессии от 10–50% и до 80–90%. Это было сделано на генах inhA, dfrA, wag31 и ftsZ, первые два из которых представляют собой мишени для изониазида и метотрексата соответственно [15, 16], а два других участвуют в формировании морфологии клеток [17, 18]. Специфика выбора мишеней интерференции позволила кроме детекции подавления экспрессии наблюдать фенотипические проявления работы CRISPRi.
Более эффективное снижение генной экспресии, возможно, могло бы быть достигнуто при выборе в качестве протоспейсера участка ДНК, комплементарного промоторной области анализируемых генов, однако для ингибируемых генов аденилатциклаз расположение промотора требует уточнения. Другим ограничением метода явилась необходимость присутствия в непосредственной близости от протоспейсера определенного PAM (NGG). В экспериментах Сингха с соавт. [9] были исследованы разные варианты мишеней (захватывающие 35 элемент промотора, участок 5'-нетранслируемой области между сайтом начала транскрипции и кодоном инициации трансляции и последовательность на 5'-конце белок-кодирующей области ДНК). По результатам исследования авторы сделали вывод о важности выбора протоспейсера вблизи промоторной области. К сожалению, найти подходящие направляющие РНК в функционально различных участках матрицы оказывается возможным не для всех генов. Аналогичным образом, не для всех генов известна область промотора. И, кроме того, в узкой области желаемого места отжига направляющей РНК далеко не всегда можно обнаружить PAM, что также является ограничением метода и, по-видимому, одной из причин неудачного подавления оперона Rv1320-Rv1319-Rv1318.
Полученные нами результаты свидетельствуют о том, что проведенная модификация вектора pRH2521 для оптимизации клонирования спейсеров направляющих РНК позволяет легко осуществлять селекцию целевых клонов. Система pRH2502/pRH2521-α может быть эффективно использована для CRISPR-интерференции, но с ограничениями, которые определяются возможностью нахождения PAM в области картированного промотора.
Работа выполнена при частичной поддержке РФФИ (грант № 1901500149A).
Список литературы
Global Tuberculosis Report.World Health Organization, 2020. Geneva.
Zhang Y., Ioerger T., Huttenhower C., Long J., Sassetti C., Sacchettini J. et al. // PLoS Pathog.2012. V. 8. № 9. e1002946.
Zhang F., Wen Y., Guo X. // Human Molecular Genetics. 2014. V. 23. № R 1. P. R40–R46.
Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. // Science (New York). 2012. V. 337. № 6096. P. 816–821.
Qi L., Larson M., Gilbert L., Doudna J., Weissman J., Arkin A., Lim W. // Cell. 2013. V. 152. № 5. P. 1173–1183.
Larson M., Gilbert L., Wang X., Lim A., Weissman J., Qi L. // Nat. Protoc. 2013. V. 8. P. 2180–2196.
Ehrt S., Schnappinger D. // Future Microbiol. 2006. V. 1. № 2. P. 177–184.
Cleto S., Jensen J., Wendisch V., Lu T. // ACS Synthetic Biology. 2016. V. 5. № 5. P. 375–385.
Singh A., Carette X. et al. // Nucleic Acids Research 2016. V. 44. № 18. P. e143.
Botsford J., Harman J. // Microbiol. Rev. 1992. V. 56. № 1. P. 100–122.
De Carvalho L., Fischer S., Marrero J., Nathan C., Ehrt S., Rhee K. // Chem. Biol. 2010.V. 17. № 10. P. 1122–1131.
Bai G., Schaak D., McDonough K. // FEMS Immunol. Med. Microbiol. 2009. V. 55. № 1. P. 68–73.
Shleeva M., Kondratieva T., Demina G., Rubakova E., Goncharenko A., Apt A., Kaprelyants A. // Front. Cell. Infect. Microbiol. 2017. V. 7. № 370.
Mycobacteria Protocols. / Ed. T. Parish, D. Roberts. N.Y.: Media Springer Science + Business, 2015. 103 p.
Larsen M., Vilchèze C. et al. // Mol. Microbiol. 2002. V. 46. № 2. P. 453–66.
White E., Ross L., Cunningham A., Escuyer V. // FEMS Microbiol Lett. 2004. V. 232. № 1. P. 101–105.
Kang C., Nyayapathy S., Lee J., Suh J., Husson R. // Microbiology (Reading). 2008.V. 154. (Pt 3). P. 725–735.
Dziadek J., Rutherford S., Madiraju M., Atkinson M., Rajagopalan M. // Microbiology (Reading). 2003. V. 149. (Pt 6). P. 1593–1603.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология