

Прикладная биохимия и микробиология, 2021, T. 57, № 4, стр. 315-325
Динитрозильные комплексы железа с глутатионовыми лигандами перехватывают пероксинитрит и защищают гемоглобин от окислительной модификации
О. В. Космачевская 1, Э. И. Насыбуллина 1, К. Б. Шумаев 1, Л. В. Чумикина 1, Л. И. Арабова 1, Н. В. Яглова 2, С. С. Обернихин 2, А. Ф. Топунов 1, *
1 Институт биохимии им. А.Н. Баха, Федеральный исследовательский центр
“Фундаментальные основы биотехнологии” Российской академии наук
119071 Москва, Россия
2 ФГБНУ НИИ морфологии человека
117418 Москва, Россия
* E-mail: aftopunov@yandex.ru
Поступила в редакцию 27.01.2021
После доработки 15.02.2021
Принята к публикации 22.02.2021
Аннотация
Динитрозильные комплексы железа с глутатионовыми лигандами (GS-ДНКЖ) являются физиологической формой оксида азота (NO), которая депонирует и переносит NO в организме. Эти комплексы могут действовать как антиоксиданты и антирадикальные агенты. Показано, что GS-ДНКЖ защищают молекулу гемоглобина (Hb) от окислительной модификации пероксинитритом. Они препятствуют образованию карбонильных производных Hb, окислению триптофановых и тирозиновых остатков, деградации гемовой группы, а также образованию межбелковых сшивок. Также эти комплексы ингибируют окисление дигидрородамина пероксинитритом, образующимся при декомпозиции 3-морфолиносиндонимина. В некоторых случаях антиоксидантное действие GS-ДНКЖ сопоставимо с таковым восстановленного глутатиона. Полученные результаты позволяют считать GS-ДНКЖ перехватчиками пероксинитрита и протекторами гемоглобина.
Динитрозильные комплексы железа (ДНКЖ) представляют собой физиологическую форму оксида азота (NO) в организме [1–3]. Формирование этих комплексов было зарегистрировано в организме человека, животных, растений и бактерий. В зависимости от того, какое соединение служит лигандом ДНКЖ, они могут быть связанными с белками или с низкомолекулярными веществами. В живых системах лигандами низкомолекулярных ДНКЖ, как правило, являются тиолсодержащие соединения: цистеин и глутатион [2–4]. ДНКЖ с тиольными лигандами функционируют в живых организмах в качестве доноров NO, NO+ и Fe–(NO)2 [2, 3, 5].
Низкомолекулярные ДНКЖ с тиольными лигандами имеют общую формулу [(RS–)2Fe+(NO+)2] и существуют в живых организмах в равновесии с белок-связанными ДНКЖ, которые формируются при участии тиольных и некоторых других групп белков [2, 3]. Это равновесие определяется редокс-условиями, а именно состоянием системы глутатиона. Снижение пула восстановленного глутатиона (GSH) сдвигает равновесие в сторону более стабильных связанных с белками комплексов. По мере окисления SH-групп комплексы постепенно димеризуются с образованием биядерных ДНКЖ, которые более устойчивы к окислению [2, 3]. Биядерные ДНКЖ в присутствии высокой концентрации GSH снова переходят в моноядерную форму.
В различных системах in vitro и in vivo было показано, что NO может действовать и как антиоксидант, и как прооксидант. Прооксидантное действие NO проявляется в его способности образовывать пероксинитрит (ONOO−) в диффузно-контролируемой реакции c супероксидом [6, 7]. При распаде ONOO− образуются гидроксильный радикал (•OH) и диоксид азота $\left( {{\text{NO}}_{{\text{2}}}^{ \bullet }} \right),$ которые в значительной степени определяют его токсические свойства [8, 9].
В крови источником супероксидного анион-радикала являются клетки иммунной системы – макрофаги, которые в ответ на появление патогенных микроорганизмов производят его в больших количествах [10]. Супероксид может образовываться и в самих эритроцитах в условиях гипоксии, когда при низком pO2 гемоглобин (Hb) активно отдает кислород [11]. В свою очередь NO может проникать в эритроцит с помощью свободной диффузии или продуцироваться внутри самой клектки в реакции восстановления деоксигенированным Hb ионов нитрита [12], а также NO-синтазой эритроцитов [13].
Антиоксидантные свойства NO проявляются в его способности перехватывать свободные радикалы: ${\text{O}}_{{\text{2}}}^{{ \bullet - }},$ •OH, Tyr•, RS•, ${\text{NO}}_{{\text{2}}}^{ \bullet },$ ROO•. В реакции NO с алкоксильными и алкилпероксильными радикалами образуются нитрозированные и нитрованные липиды. Благодаря этому обрываются цепные реакции перекисного окисления липидов [14]. Метаболиты NO – ДНКЖ также проявляют антиоксидантные свойства. Ранее было показано, что низкомолекулярные ДНКЖ с глутатионовыми лигандами (GS-ДНКЖ) перехватывают супероксидный радикал [15–17] и восстанавливают оксоферрильную форму миоглобина (Mb-FeIV = O) до нетоксичной метформы [18], а ДНКЖ, связанные с гемоглобином, защищают его тиольные группы от окисления [15, 16]. В этих экспериментах в качестве инициаторов окисления использовали пероксид водорода и гидропероксид трет-бутила. Кроме того, GS-ДНКЖ защищают эритроциты от HOCl-индуцированного гемолиза [19]. В ходе взаимодействия ДНКЖ с органическими свободными радикалами происходит необратимая деструкция комплексов и образование интермедиатов – нитроорганических соединений [18].
Основной мишенью пероксинитрита в эритроцитах является Hb [20]. Реакция NO с Hb конкурирует с реакцией NO с ${\text{O}}_{{\text{2}}}^{{ \bullet - }}$ [10]. При этом в результате взаимодействия ONOO− с гемоглобином происходит его окислительная модификация [7], которая сопровождается образованием карбонильных производных, дисульфидных и дитирозиновых сшивок, а также окислением железа в гемовой группе до состояния оксоферрила, образованием радикалов на порфирине и разрывом порфиринового кольца.
Цель работы – изучение влияния GS-ДНКЖ на образование карбонильных производных, дисульфидных и дитирозиновых сшивок, а также на окисление железа в гемовой группе до состояния оксоферрила, образование радикалов на порфирине и разрыв порфиринового кольца в Hb под действием пероксинитрита.
МЕТОДИКА
В работе были использованы следующие реактивы: метгемоглобин из эритроцитов быка; 3-морфолиносиндонимин (SIN-1), дигидрородамин, восстановленный глутатион (GSH), пиридин, дитионит Na, диэтилентриаминопентауксусная кислота (ДТПА), трихлоруксусная кислота (ТХУ), диметилсульфоксид (ДМСО), 2,4-динитрофенилгидразин (2,4-ДНФГ), ПААГ, Na-ДДС, Трис, HEPES, NaNO2, FeSO4, – “Sigma-Aldrich” (USA); 3H-нафтол[2,1-b]пиран-s-карбоновая кислота (ThioGlo1) – “Calbiochem” (США); 4-гидрокси-(2,2,6,6-тетраметилпиперидин-1-ил)оксил (4-гидрокси-TEMPO) – “Oxis” (США).
ДНКЖ с глутатионовыми лигандами получали, смешивая растворы FeSO4 и GSH в молярном соотношении 1 : 2 в сосуде Тунберга в атмосфере NO [15–17]. Концентрацию ДНКЖ рассчитывали по интегральной интенсивности сигнала электронного парамагнитного резонанса (ЭПР) комплексов, используя в качестве стандарта спиновую метку 4-гидрокси-TEMPO. Препараты ДНКЖ хранили при температуре жидкого азота (–196°С).
Пероксинитрит синтезировали по методике, описанной в работе [21], быстро смешивая 0.6 М раствор NaNO2 и 0.6 М раствор H2O2 в 0.7 М HCl, и стабилизировали 0.9 М NaOH. Непрореагировавший H2O2 удаляли, добавляя порошкообразный MnO2, после чего раствор фильтровали. Концентрацию ONOO− определяли по характерной полосе поглощения при 302 нм (ε = 1.67 мМ–1 см–1).
Определение белковых карбонилов. Количественную оценку карбонильных производных Hb проводили по методу [22] в модификации Ю.В. Абаленихиной и М.А. Фоминой [23]. Суть метода заключалась в том, что карбонильные группы (альдо- и кето-) образуют ковалентные аддукты с 2,4-ДНФГ, которые регистрируются спектрофотометрически (Е) при 13 различных длинах волн. Количество образовавшихся 2,4-динитрофенилгидразонов рассчитывали по формуле:
SОМБ = S1 + S2, где S1 – альдегидные группы, S2 – кетонные группы.
Образцы готовили следующим способом. К 0.1 мл 0.15 мМ раствора Hb в 20 мМ К-фосфатном буфере (рН 6.8) добавляли пероксинитрит до конечных концентраций 0.4, 0.6, 1.0, 1.6, 2.6, 4.2 мМ и инкубировали при комнатной температуре в течение 10 мин. Реакцию останавливали добавлением метионина до конечной концентрации 25 мМ. GS-ДНКЖ и GSH добавляли к раствору Hb в молярном соотношении 3 : 1 и 6 : 1 соответственно. Для изучения влияния металлов на ход реакции к растворам белка добавляли комплексон железа – ДТПА до конечной концентрации 2.5 мМ.
К раствору белка добавляли 0.5 мл раствора 2,4-ДНФГ в 2 М HCl и инкубировали в течение часа. Белок осаждали добавлением 0.5 мл 20%-ного раствора ТХУ. Через 10 мин образцы центрифугировали при 3000 g 15 мин. Для удаления несвязавшегося 2,4-ДНФГ осадок белка трижды отмывали 0.4 мл смеси этилового спирта и этилацетата (1 : 1). Полученный осадок подсушивали на воздухе и перед измерением растворяли в 1 мл 8 М раствора мочевины.
Спектры оптического поглощения регистрировали на UV-VIS спектрофотометре Carу 300 (“VarianBio”, США) при комнатной температуре в кювете с длиной оптического пути 1 см при скорости сканирования 600 нм/мин. Перед измерением образцы разбавляли в 5 раз.
Определение содержания гемовой группы. Концентрацию гема в растворе Hb определяли с помощью пиридигемохромного метода [24]. К 15 мкл каждого образца Hb, приготовленных как описано в пункте 1 данного раздела, добавляли 135 мкл воды и 450 мкл 30%-ного щелочного раствора пиридина. Непосредственно перед измерением раствор восстанавливали дитионитом натрия. Оптическое поглощение восстановленного комплекса гема с пиридином измеряли при 556 и 539 нм. Расчет концентрации гема осуществляли по разнице А556–А539, учитывая, что Ε556–539 = 4.3 мМ–1 см–1.
Определение SH-групп в низкомолекулярных и белковых тиолах. Количественную оценку свободных SH-групп проводили с помощью тиол-специфичного реагента ThioGlo1, который образует флуоресцирующий аддукт [25]. Для анализа восстановленных белковых SH-групп образцы готовили следующим образом: к 5 мкл раствора Hb (0.03 мМ), приготовленного как описано выше, добавляли 5 мкл 0.25 мМ ThioGlo1 в ДМСО и инкубировали 4 мин. Затем 10 мкл полученного раствора вносили в спектрофлуориметрическую кювету, содержащую 490 мкл 20 мМ К-фосфатного буфера (рН 6.8).
Флуоресценцию регистрировали на спектрофлуориметре Shimadzu RF-5301 PC (Япония) при высокой чувствительности (по обозначению прибора), ширине щели возбуждающего и испускающего света 3 и 5 нм соответственно. Длина волны возбуждающего света составляла 379 нм, испускающего – 500 нм.
Определение состояния триптофановых и тирозиновых остатков. Состояние триптофановых и тирозиновых остатков в молекуле Hb изучали с помощью флуоресцентной спектроскопии. Образцы белка были приготовлены, как описано в пункте “Определение белковых карбонилов”. Для селективного возбуждения автофлуоресценции триптофана использовали длину волны 295 нм. Флуоресценцию регистрировали при 330 нм. Спектрофотометрическую детекцию 3,3'-дитирозинов осуществляли при длине волны возбуждения 325 нм и испускания 400 нм [26]. Образцы Hb перед измерением разбавляли в 25 раз при измерении флуоресценции тритпофана и 12.5 раз при измерении флуоресценции дитирозинов. Регистрацию флуоресценции проводили в кварцевых оптических кюветах с длиной оптического пути 1 см на спектрофлуориметре Shimadzu RF-5301 PC (Япония) при высокой чувствительности и средней скорости сканирования. Ширина щели возбуждающего света составляла 10 нм, поглощающего – 5 нм.
Электрофорез в ПААГ с Na-ДДС. Электрофорез проводили в блоках 13%-ного ПААГ размером 15 × 15 × 2 мм по методу Лэммли [27]. Образцы гемоглобина, приготовленные как описано в разделе “Определение белковых карбонилов”, предварительно диализовали в 20 мМ К-фосфатном буфере (рН 6.8) для отделения низкомолекулярных тиолов (GSH и GS-ДНКЖ), а затем добавляли буфер для образцов в соотношении 1 : 1, прогревали в течение 5 мин при 95°С и наносили на гель. Буфер для образцов был приготовлен на основе 0.1 М трис-HCl буфера (рН 6.8) с 4% Na-ДДС, 0.2% бромфенолового синего и 20% глицерина. Для создания восстановительных условий в буфер добавляли 3% дитиотреитола. На гель наносили по 15 мкл раствора белка. В качестве электродного буфера использовали 0.2 М Трис-глициновый буфер, рН 8.3, содержащий 0.1% ДДС.
Электрофорез проводили при 4°С и I = 50 мА, U = 150 B. Необходимые параметры электрофореза обеспечивались источником питания “Эльф-4” (Россия). После окончания процесса разделения белков гель фиксировали и окрашивали раствором Кумасси бриллиантового синего R-250.
Окисление дигидрородамина пероксинитритом. Способность GS-ДНКЖ перехватывать пероксинитрит изучали в реакции окисления дигидрородамина пероксинитритом, образующимся при распаде SIN-1 [28]. При окислении дигидрородамина образуется родамин с максимумом поглощения при 500 нм. GSH или GS-ДНКЖ вносили в реакционную смесь через 5 с после начала реакции. Состав реакционной смеси: 0.4 мМ дигидрородамина, 0.4 мМ SIN-1, 0.08 мМ GSH или GS-ДНКЖ. Кинетику восстановления записывали в 1 мм кювете в течение 10 мин при комнатной температуре на UV-VIS спектрофотометре Carу 300 (“VarianBio”, США).
Все эксперименты были выполнены не менее чем в трех повторностях. На графиках представлены средние величины. Статистическая обработка данных проводилась исходя из 3–4 аналитических повторностей в режиме Excel. Данные представлены как среднее значение ± стандартное отклонение.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
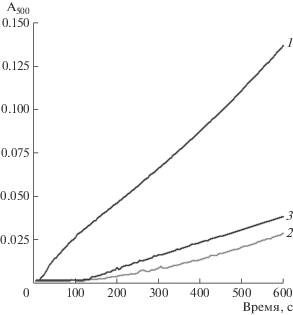
Влияние ДНКЖ на окисление дигидрородамина пероксинитритом. Способность GS-ДНКЖ перехватывать ONOO− была исследована в экспериментах по окислению дигидрородамина пероксинитритом, образующимся в ходе декомпозиции SIN-1. Поскольку при спонтанном распаде SIN-1 в присутствии кислорода одновременно образуются ${\text{O}}_{{\text{2}}}^{{ \bullet - }}$ и NO, это вещество часто используют для моделирования образования ONOO− в живых системах [28]. На рис. 1 представлены кинетики окисления дигидрородамина пероксинитритом. Как видно, GS-ДНКЖ ингибировали реакцию окисления. Еще в большей степени препятствовал окислению дигидрородамина GSH, что подтвердило его способность выступать в качестве эффективной ловушки пероксинитрита.
Рис. 1.
Кинетика окисления дигидрородамина пероксинитритом, образующимся при декомпозиции SIN-1 (1), а также при внесении GSH (2) или GS‑Д-НКЖ (3): 1 – контроль, состав реакционной смеси (см. Методика).
Образование карбонильных производных гемоглобина. Ранее нами было показано, что пероксинитрит может взаимодействовать с ДНКЖ, связанным с Hb [15]. Таким образом, способность низкомолекулярных (GS-ДНКЖ) и белковых ДНКЖ перехватывать пероксинитрит послужила предпосылкой для проведения исследований по окислительной модификации гемоглобина.
Карбонильные производные представляют собой необратимую стабильную форму неспецифического окисления аминокислотных остатков. ДНКЖ оказывали выраженное ингибирующее действие на образование карбонилов в присутствии пероксинитрита. Они защищали белок в широком диапазоне концентраций ONOO− от 0.38 до 4.20 мМ, что соответствовало молярному соотношению Hb : ONOO− от 1 : 2.5 до 1 : 28 (для тетрамера) (рис. 2а). При низких концентрациях пероксинитрита GSH стимулировал формирование карбонильных производных Hb, но начиная с концентрации 2.6 мМ – ингибировал (рис. 2а). Эти результаты показывают, что протекторное действие GS-ДНКЖ обусловлено не только их глутатионовыми лигандами.
Рис. 2.
Образование карбонильных производных (отн. ед.) гемоглобина под действием пероксинитрита без ДТПА (а) и в его присутствии (б). Варианты опыта: 1 – Hb, 2 – Hb + GSH, 3 – Hb + GS-ДНКЖ. (Состав реакционной смеси (мМ): Hb – 0.15, GS‑Д-НКЖ – 0.5, GSH – 1.0, ДТПА – 2.0).
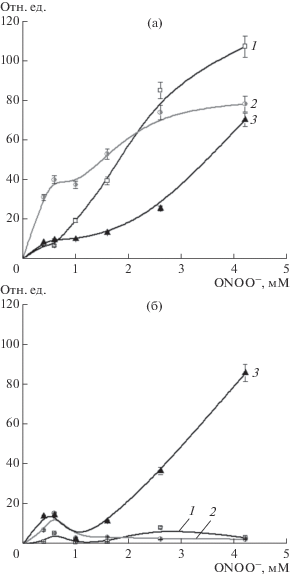
Кривые, отражающие зависимость образования карбонильных производных от концентрации окислителя, в присутствии GS-ДНКЖ имели двухфазный характер: при низких концентрациях ONOO− образование карбонильных производных происходило медленно, а начиная с концентрации 1.6 мМ – интенсивно. Вероятно, это связано с высвобождением железа из распавшихся ДНКЖ, которое далее участвовало в генерации свободных радикалов в реакциях Фентона и Габера-Вайса:
(1)
$2{\text{O}}_{2}^{{\centerdot - }} + 4{{{\text{H}}}^{ + }} \to ~{{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}},$(2)
${\text{F}}{{{\text{e}}}^{{3 + }}} + {\text{O}}_{2}^{{\centerdot - }} \to {\text{F}}{{{\text{e}}}^{{2 + }}} + {{{\text{O}}}_{2}},$(3)
${\text{F}}{{{\text{e}}}^{{2 + }}} + {{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{2}} \to {\text{F}}{{{\text{e}}}^{{3 + }}} + {\text{H}}{{{\text{O}}}^{\centerdot }} + {\text{O}}{{{\text{H}}}^{ - }}.$Для проверки этого предположения были поставлены эксперименты в присутствии ДТПА, который образует с ионами свободного железа комплекс, не участвующий в реакциях Фентона и Габера–Вайса. Как в контроле, так и в варианте с GSH этот хелатор практически полностью ингибировал образование карбонилов (рис. 2).
Напротив, введение ДТПА в реакционную среду, содержащую GS-ДНКЖ, незначительно стимулировало формирование карбонильных призводных (рис. 2б). Скорее всего, этот эффект обусловлен трансформацией глутатионовых ДНКЖ в комплексы, содержащие ДТПА в качестве лигандов.
Деградация гемовой группы гемоглобина. Основной мишенью действия ONOO¯ на гемопротеиды является гемовая группа. Деградации гема предшествует стадия образования оксоферрил-формы (гем-FeIV = O), в образовании которой ключевую роль играет гемовое железо, катализирующее разрыв O–O связи в комплексе гем-FeIII-ONOO– [29].
В экспериментах GSH и GS-ДНКЖ препятствовали разрушению гемовой группы (рис. 3). В контрольном варианте доля разрушенного гема прямо зависела от концентрации окислителя (рис. 3a). При концентрации ONOO− 4.2 мМ разрушалось 50% гемов, в то время как в присутствии GS-ДНКЖ (в соотношении 1:3) – только 25% (рис. 3a). GSH также оказывал протекторное действие на гемовую группу, однако оно было менее выражено, чем у GS-ДНКЖ (рис. 3a). Во всех вариантах добавление ДТПА в реакционную среду препятствовало распаду гемовой группы, что указывало на участие примесных ионов железа в окислительной деструкции гема (рис. 3б).
Рис. 3.
Разрушение гемовой группы гемоглобина под действием пероксинитрита без ДТПА (а) и в его присутствии (б). Варианты опыта: 1 – Hb; 2 – Hb + GSH; 3 – Hb + GS-ДНКЖ. Состав реакционной смеси как на рис. 2.
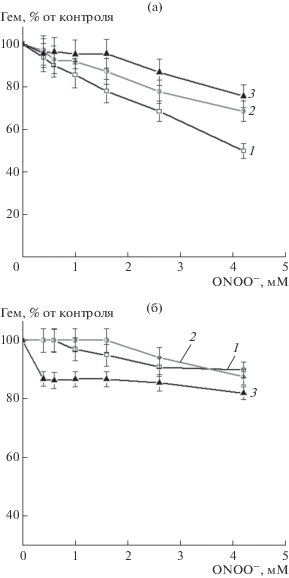
Защитное действие GSH по отношению к гему можно объяснить его способностью восстанавливать Hb-FeIV = O и радикалы Cys-93β, образуемые в результате внутримолекулярного переноса электронов, на оксоферрил-форму [30].
Защитное действие GS-ДНКЖ может быть связано с различными аспектами их функционирования. В работе [18] был предложен механизм защитного действия GS-ДНКЖ, заключающийся в формировании при взаимодействии этих комплексов с оксоферильной формой гемоглобина промежуточного интермедиата, который далее может распадается или регенерироваться до исходных ДНКЖ (4).
(4)
$\begin{gathered} {\text{Hb - F}}{{{\text{e}}}^{{{\text{IV}}}}} = {\text{O}} + {{({\text{G}}{{{\text{S}}}^{--}})}_{2}}--{\text{F}}{{{\text{e}}}^{ + }}{\kern 1pt} --{\kern 1pt} {{\left( {{\text{N}}{{{\text{O}}}^{ + }}} \right)}_{2}} \to \\ \to \,\,{\text{Hb - F}}{{{\text{e}}}^{{{\text{III}}}}} + {{\left( {{\text{G}}{{{\text{S}}}^{--}}} \right)}_{2}}{\kern 1pt} --{\kern 1pt} {\text{F}}{{{\text{e}}}^{{2 + }}}{\kern 1pt} --{\kern 1pt} \left( {{\text{N}}{{{\text{O}}}^{{\text{ + }}}}} \right){\text{ONO}}. \\ \end{gathered} $GS-ДНКЖ могут непосредственно реагировать с пероксинитритом. В этом случае может возникать интермедиат, содержащий связанный ONOO–, и происходить окисление глутатионовых лигандов. Вместе с тем, антиоксидантное действие ДНКЖ может быть обусловлено восстановлением радикалов порфирина и оксоферрильной формы Hb оксидом азота, выделяющимся из ДНКЖ [18, 31].
Окисление триптофановых и тирозиновых остатков в гемоглобине. При взаимодействии Hb с пероксинитритом продуцируются свободные радикалы тирозина, триптофана и цистеина. В качестве окислителя выступает гидроксильный радикал и диоксид азота [8, 32], а также оксоферрильная форма гема [32]. При этом в реакции одноэлектронного окисления остатков тирозина формируются феноксильные радикалы [32, 33], судьба которых может быть различной. С одной стороны, в реакции двух феноксильных радикалов образуется димер 3,3'-дитирозин [26]. С другой стороны, в реакции остатков тирозина и комплексов гемового железа с пероксинитритом (FeIII-ONOO−) образуется 3-нитротирозин [34]. 3-нитротирозин также образуется в реакции феноксильного радикала тирозина с ${\text{NO}}_{{\text{2}}}^{ \bullet }$ или NO, в последнем случае через такой интермедиат, как нитрозотирозин [35, 36]. Наличие металлсвязывающих центров в белке ускоряет реакцию нитрования тирозина [20, 37]. Предполагается, что переходные металлы, в том числе железо гема, катализируют гетеролитический раcпад ONOO− с образованием нитрониум-катиона $\left( {{\text{NO}}_{{\text{2}}}^{ + }} \right),$ являющегося сильным нитрующим агентом [34, 37].
Было проведено спектрофлуориметрическое исследование тирозиновых остатков Hb поcле обработки пероксинитритом. В контрольном варианте флуоресценция возрастала с увеличением концентрации ONOO− (рис. 4а), что могло происходить благодаря образованию 3,3'-дитирозина, который характеризуется максимумом испускания флуоресценции при 400 нм [26]. GSH и GS-ДНКЖ ингибировали образование дитирозинов примерно в равной степени (рис. 4а).
Рис. 4.
Образование дитирозинов (а) в Hb под действием пероксинитрита, изменение флуоресценции триптофановых остатков Hb под действием пероксинитрита (б). Интенсивность флюоресценции, отн. ед. Варианты опыта: 1 – Hb; 2 – Hb + GSH; 3 – Hb + + GS-ДНКЖ. Состав реакционной смеси как на рис. 2.
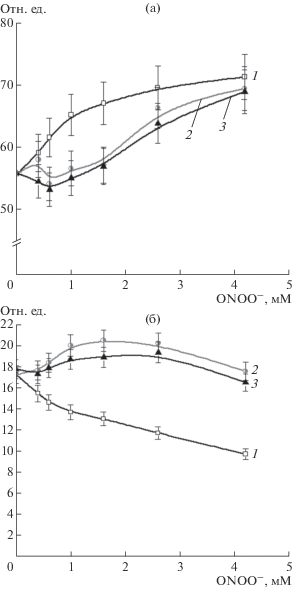
Этот факт объясняется действием глутатиона, который является эффективным перехватчиком ${\text{NO}}_{{\text{2}}}^{ \bullet }$ (k = 5.3 × 106 М–1 с–1) [38]. К тому же GSH может непосредственно восстанавливать пероксинитрит и радикалы тирозина [39]. Глутатион восстанавливает пероксинитрит по одноэлектронному механизму, образуя тиильный радикал (GS•), восстанавливающий кислород до супероксида. Последний реагирует с ${\text{NO}}_{{\text{2}}}^{ \bullet }$ с образованием пероксинитрата (O2NOOH/O2NOO–), не взаимодействующего с тирозином [40]. Действие GS-ДНКЖ можно объяснить образованием 3-нитротирозина. При этом нельзя исключить участие железа в катализе реакции нитрования [37]. Вместе с тем, хотя нитрование тирозина является кинетически более предпочтительным, чем образование его димеров, оба эти процесса могут происходить одновременно [36].
Известно, что нитрозильные комплексы с переходными металлами, в том числе ДНКЖ с нетиольными лигандами, могут трансформироваться в интермедиаты, содержащие связанный пероксинитрит [41–43]. Установлено, что эти интермедиаты способны вызывать нитрование ароматических молекул. Подобные нестабильные промежуточные комплексы могут формироваться и при взаимодействии ONOO− с GS-ДНКЖ. В реакции пероксинитрита с NO-лигандами ДНКЖ может образовываться нитрозилпероксинитрит (ONOONO) [44]:
(5)
$\begin{gathered} {{\left( {{\text{GS}}} \right)}_{2}}{\kern 1pt} - {\kern 1pt} {\text{Fe}}{\kern 1pt} - {\kern 1pt} {{\left( {{\text{NO}}} \right)}_{2}} + {\text{ONO}}{{{\text{O}}}^{ - }} \to \\ \to \,\,{{\left( {{\text{GS}}} \right)}_{2}}{\kern 1pt} - {\kern 1pt} {\text{Fe}}{\kern 1pt} - {\kern 1pt} \left( {{\text{ONOONO}}} \right)\left( {{\text{NO}}} \right). \\ \end{gathered} $Известно также, что нитрозилпероксинитрит может участвовать в нитровании дофамина [45].
Еще одной аминокислотой, являющейся мишенью действия пероксинитрита, является триптофан [46]. В молекуле Hb есть шесть остатков триптофана, из которых в первую очередь окисляется Trp15β, тогда как окисление Trp37β происходит при нарушении нативной структуры белка [47].
На следующем этапе была изучена зависимость интенсивности испускания флуоресценции триптофана при 327 нм от концентрации ONOO−. В контрольном варианте интенсивность флуоресценции дозозависимо уменьшалась (рис. 4б), что свидетельствовало об окислении или нитровании триптофановых остатков. В присутствии GSH и GS-ДНКЖ происходило незначительное возрастание флуоресценции относительно исходного уровня (рис. 4б), но начиная с концентрации ONOO− 1.6 мМ флуоресценция снижалась. Это можно объяснить тем, что при определенных концентрациях окислителя белок претерпевает конформационные перестройки, затрагивающее окружение остатка триптофана, что сказывается на автофлуоресценции.
Кроме этого, возможно образование нитротриптофана в реакции пероксинитрита с триптофаном. Эта реакция может происходить с участием пероксиазотистой кислоты или активированного интермедиата ONOOH*, образующегося из транс-пероксиазотистой кислоты, причем переходные металлы стимулируют нитрование триптофана [48]. Предполагают, что аналогично тирозину, интермедиатом в процессе нитрования триптофана является свободный радикал этой аминокислоты – тритофанил-радикал [49]. На важную роль гемовой группы в этих процессах указывает тот факт, что остатки 6- и 4-нитротриптофанов обнаружены при обработке пероксинитритом окси- и метгемоглобина, но не апогемоглобина.
Окисление тиоловых групп гемоглобина. Известно, что в Hb тирозиновые радикалы могут окислять остатки цистеина в реакции внутримолекулярного переноса электрона [33]. При этом образуются тиильные радикалы. Гемоглобин человека содержит два цистеина в бета-субъединицах тетрамера (Cys-93β), наиболее восприимчивых к окислению.
В проведенных экспериментах заметного окисление SH-групп Hb под действием ONOO− не происходило (рис. 5а). Незначительное (~20%) уменьшение интенсивности флуоресценции тиольного аддукта с ThioGlo1 было только в контроле (рис. 5а). Однако наблюдалось пероксинитрит-зависимое окисление низкомолекулярных тиолов (GSH и GS-ДНКЖ) в реакционной смеси с Hb (рис. 5б). Это может быть объяснено тем, что эти вещества, перехватывая ONOO−, защищают SH-группы Hb от окисления.
Рис. 5.
Окисление пероксинитритом SH-групп гемоглобина (% к контролю), освобожденного от низкомолекулярных тиолов (GSH и GS-ДНКЖ) с помощью диализа (а), и низкомолекулярных тиолов (% к контролю) в растворе гемоглобина (б). Варианты опыта: 1 – Hb; 2 – Hb + GSH; 3 – Hb + GS-ДНКЖ.
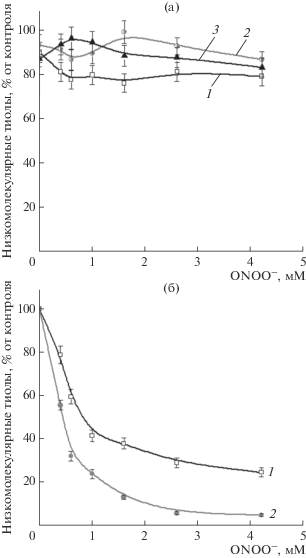
Как известно, низкомолекулярные тиолы подвергаются окислению пероксинитритом по одно- и двухэлектроному механизму [50, 51]. Окисление по двухэлектронному механизму включает нуклеофильную атаку тиола протонированной формой пероксинитрита (ONOOH). В результате образуются нитрит и сульфеновая кислота (R-SOH), которая может вступать в реакцию с другим тиолом с образованием дисульфида (R-SS-R). По одноэлектронному механизму реагируют пероксиазотистая кислота и производные от нее свободные радикалы (•OH и ${\text{NO}}_{{\text{2}}}^{ \bullet }$). В реакции с тиолат-анионом образуются тиильные радикалы (R-S•), которые инициируют зависимую от кислорода радикальную цепную реакцию [52]. Тиильные радикалы также рекомбинируют с другими R-S• или ${\text{NO}}_{{\text{2}}}^{ \bullet }$ с образованием дисульфидов или нитротиолов RS-NO2 [51].
Образование межбелковых сшивок. Окисление белков пероксинитритом приводит к возникновению межмолекулярных ковалентных связей (сшивок) и формированию агрегатов белка. Это может происходить из-за образования радикалов аминокислотных остатков (тирозина, цистеина, триптофана) в результате окисления оксоферрильной формой гема и различными свободными радикалами [29, 36].
Образование межсубъединичных сшивок и агрегатов Hb под действием ONOO− регистрировали с помощью денатурирующего электрофореза в 13% ПААГ с Na-ДДС в восстановительных условиях. Анализ электрофореграмм показал, что в контрольном варианте во всем диапазоне концентраций ONOO− происходило дозозависимое образование высокомолекулярных агрегатов Hb (рис. 6). При этом агрегация была настолько сильной, что часть белка не проникала в концентрирующий гель. В присутствии GSH и GS-ДНКЖ видимая агрегация происходила при концентрации ONOO− 2.6 мМ и 4.2 мМ и в гораздо меньшей степени, чем в контрольном образце (рис. 6). Эти результаты согласуются с данными по окислению остатков тирозина, триптофана и цистеина (см. выше).
Рис. 6.
Электрофорез гемоглобина в 13%-ном ПААГ с ДТТ при разной концентрации пероксинитрита в реакционной смеси: 1 – 0; 2 – 0.4; 3 – 0.6; 4 – 1.0; 5 – 1.6; 6 – 2.6; 7 – 4.2 мМ. Варианты опыта: Hb, Hb + GSH; Hb + GS-ДНКЖ. Состав реакционной смеси как на рис. 2.
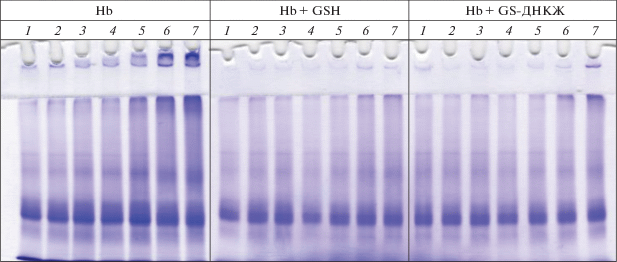
Таким образом, полученные результаты показали, что GS-ДНКЖ, так же, как и GSH, препятствуют образованию межбелковых сшивок, что еще раз подтверждает способность ДНКЖ выступать в роли антиоксидантов, предотвращающих агрегацию субъединиц Hb под действием активных форм кислорода и азота [16].
Продукция пероксинитрита усиливается при многих патологических состояниях (воспаления, сердечно-сосудистые и нейродегенеративные заболевания) [6]. Пероксинитрит, образованный макрофагами в сосудистом русле, транспортируется в эритроцит простой диффузией или с помощью мембранного белка полосы 3 (Band3), достигая значительных концентраций внутри клетки [53]. Гемоглобин является эффективной ловушкой пероксинитрита. Расчеты показали, что 80% пероксинитрита реагирует с Hb, который катализирует его изомеризацию в нитрат [29]. Быстрой элиминации перкосинитрита в эритроците способствуют высокая концентрация Hb (около 5 мМ) и высокая константа скорости реакции второго порядка (k = 2 × 104 М–1 с–1) [29, 53]. В результате этой реакции может происходить окислительная модификация гемоглобина.
В условиях гипоксии повреждающее действие пероксинитрита усиливается высокими уровнями CO2, который катализирует образование свободнорадикальных продуктов из пероксинитрита. В тоже время гипоксия благоприятствует формированию в эритроцитах ДНКЖ, которые могут не только служить формой транспорта NO за пределы клетки, но и оказывать протекторное [15, 16] и модулирующее действие на белки [4, 54]. Поскольку в эритроцитах высокая концентрация глутатиона (около 5 мМ) в них вероятно образование GS-ДНКЖ.
Взаимодействие тиолсодержащих ДНКЖ с пероксинитритом было исследовано в работах [16, 55]. Было показано, что пероксинитрит окисляет GS-ДНКЖ по двухэлектронному механизму с константой скорости третьего порядка k = (1.8 ± ± 0.3) × 107 М–2 с–1 [55]. В ходе этой реакции образуются интермедиаты – комплексы, содержащие связанный с железом пероксинитрит ((GS)2–Fe–(ONOO)(NO) (рис. 7) [16, 42, 55], или нитрозилпероксинитрит, как показано в реакции 5. Эти интермедиаты нестабильны и распадаются с высвобождением нерадикальных продуктов (${\text{NO}}_{{\text{2}}}^{ - },$ ${\text{NO}}_{{\text{3}}}^{ - }$) (рис. 7) или участвуют в нитровании биомолекул, чаще всего остатков тирозина [34, 42]. Глутатионовые лиганды, входящие в состав упомянутых выше интермедиатов, окисляются по двухэлектронному механизму без образования тиильных радикалов [18]. Это выгодно отличает GS-ДНКЖ от глутатиона, который в реакции с ONOO– дает широкий спектр свободнорадикальных продуктов [51]:
(6)
${\text{GSH}} + {\text{ONO}}{{{\text{O}}}^{ - }} \to {\text{G}}{{{\text{S}}}^{ \bullet }}~ + \,{\text{NO}}_{2}^{\centerdot } + {\text{O}}{{{\text{H}}}^{ - }},$(7)
${\text{GSH}} + {\text{O}}{{{\text{H}}}^{\centerdot }} \to {\text{G}}{{{\text{S}}}^{\centerdot }} + {{{\text{H}}}_{{\text{2}}}}{\text{O}},$(8)
${\text{G}}{{{\text{S}}}^{\centerdot }} + {{{\text{O}}}_{2}} \to {\text{GSO}}{{{\text{О}}}^{\centerdot }},$(9)
${\text{GSO}}{{{\text{О}}}^{\centerdot }} + {\text{GSH}} \to {\text{G}}{{{\text{S}}}^{\centerdot }} + {\text{GSOОH}},$(10)
${\text{G}}{{{\text{S}}}^{\centerdot }} + {\text{G}}{{{\text{S}}}^{--}} \to {\text{GSS}}{{{\text{G}}}^{{\centerdot --}}},$(11)
${\text{GSS}}{{{\text{G}}}^{{\centerdot --}}} + {{{\text{O}}}_{2}} \to {\text{GSSG}} + {\text{O}}_{2}^{{\centerdot - }}.$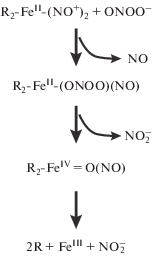
Антиоксидантные свойства ДНКЖ на первый взгляд кажутся парадоксальными, поскольку комплексы содержат редокс-активное двухвалентное железо, которое в реакции с пероксидами образует гидроксильный радикал, окисляющий белки, мембранные липиды и нуклеиновые кислоты. Однако к настоящему времени доказана способность низкомолекулярных ДНКЖ с тиольными лигандами купировать свободнорадикальные реакции и защищать липиды и белки от окислительного повреждения [15–19]. Полученные в нашей работе результаты являются еще одним подтверждением этой способности ДНКЖ.
Полученные результаты позволяют сделать вывод, что низкомолекулярные GS-ДНКЖ перехватывают пероксинитрит и защищают гемоглобин от окислительной модификации. Это объясняет показанное в работах [56, 57] кардиопротекторное действие GS-ДНКЖ в условиях ишемии, в том числе при искусственном кровообращении.
Список литературы
Hsiao H.Y., Chung C.W., Santos J.H., Villaflores O.B., Lu T.T. // Dalton Trans. 2019. V. 48. P. 9431–9453.
Vanin A.F. // Nitric Oxide. 2016. V. 54. P. 15–29.
Vanin A.F. Dinitrosyl Iron Complexes as a “Working Form” of Nitric Oxide in Living Organisms // Cambridge Scholars Publishing. 2019. 265 p.
Lewandowska H., Brzóska K., Meczyńska–Wielgosz S., Rumianek K., Wójciuk G., Kruszewski M. // Postepy Biochem. 2010. V. 56. P. 298–304.
Vanin A.F., Tronov V.A., Borodulin R.R. // Cell Biochem. Biophys. 2021. V. 79. № 1. P. 93–102.
Pacher P.L., Beckman J.S., Liaudet L. // Physiol. Rev. 2007. V. 87. P. 315–424.
Su J., Groves J.T. // Inorg. Chem. 2010. V. 49. № 14. P. 6317–6329.
Bartesaghi S., Radi R. // Redox Biology. 2018. V. 14. P. 618–625.
Abalenikhina Yu.V., Kosmachevskaya O.V., Topunov A.F. // Appl. Biochem. Microbiol. 2020. V. 56. № 6. P. 611–623.
Prolo C., Álvarez M.N., Ríos N., Peluffo G., Radi R., Romero N. // Free Radic. Biol. Med. 2015. V. 87. P. 346–355.
Balagopalakrishna C., Manoharan P.T., Abugo O.O., Rifkind J.M. // Biochemistry. 1996. V. 35. P. 6393–6398.
Huang Z., Shiva S., Kim-Shapiro D.B., Patel R.P., Ringwood L.A., Irby C.E., Huang K.T., Ho C., Hogg N., Schechter A.N., Gladwin M.T. // J. Clin. Invest. 2005. V. 115. № 8. P. 2099–2107.
Cortese-Krott M.M., Kelm M. // Redox Biol. 2014. V. 2. P. 251–258.
Padmaja S., Huie R.E. // Biochem. Biophys. Res. Commun. 1993. V. 195. P. 539–544.
Shumaev K.B., Gubkin A.A., Serezhenkov V.A., Lobysheva I.I., Kosmachevskaya O.V., Ruuge E.K., Lankin V.Z., Topunov A.F., Vanin A.F. // Nitric Oxide. 2008. V. 18. P. 37–46.
Shumaev K.B., Kosmachevskaya O.V., Timoshin A.A., Vanin A.F., Topunov A.F. // Methods Enzymol. 2008. V. 436. P. 445–461.
Shumaev K.B., Dudylina A.L., Ivanova M.V., Pugachenko I.S., Ruuge E.K. // Biofactors. 2018. V. 44. № 3. P. 237–244.
Shumaev K.B., Petrova N.E., Zabbarova I.V., Vanin A.F., Topunov A.F., Lankin V.Z., Ruuge E.K. // Biochemistry (Moscow). 2004. V. 69. № 5. P. 569–574.
Shumaev K.B., Gorudko I.V., Kosmachevskaya O.V., Grigoryeva D.V., Panasenko O.M., Vanin A.F., Topunov A.F., Terekhova M.S., Sokolov A.V., Cherenkevich S.N., Ruuge E.K. // Oxid. Med. Cell. Longev. 2019. V. 2019. e2798154. https://doi.org/10.1155/2019/2798154
Pietraforte D., Salzano A.M., Marino G., Minetti M. // Amino Acids. 2003. V. 25. № 3–4. P. 341–350.
Beckman J.S. // Methods Enzymol. 1994. V. 233. P. 229–240.
Schmorak J., Lewin M. // Anal. Chem. 1961. V. 33. P. 1403–1405.
Абаленихина Ю.В., Фомина М.А. // Казанский медицинский журн. 2014. Т. 95. № 4. С. 553–557.
Riggs A. // Methods Enzymol. 1981. V. 76. P. 5–29.
Hoff S., Larsen F.H Andersen M.L., Lund M.N. // Analyst. 2013. V. 138. P. 2096–2103.
Davies K.J., Delsignore M.E. // J. Biol. Chem. 1987. V. 262. № 20. P. 9908–9913.
Laemmli U.K. // Nature. 1970. V. 227. № 5259. P. 680–685.
Singh R.J., Hogg N., Joseph J., Konorev E., Kalyanaraman B. // Arch. Biochem. Biophys. 1999. V. 361. № 2. P. 331–339.
Romero N., Radi R., Linares E., Augusto O., Detweiler C.D., Mason R.P., Denicola A. // J. Biol. Chem. 2003. V. 278. № 45. P. 44049–44057.
Augusto O., Lopez de Menezes S., Linares E., Romero N., Radi R., Denicola A. // Biochemistry. 2002. V. 41. P.14323–14328.
Gorbunov N.V., Osipov A.N., Day B.W., Zayas-Rivera B., Kagan V.E., Elsayed N.M. // Biochemistry. 1995. V. 34. P. 6689–6699.
Radi R. // Acc. Chem. Res. 2012. V. 46. P. 550–559.
Bhattacharjee S., Deterding L.J., Jiang J., Bonini M.G., Tomer K.B., Ramirez D.C., Mason R.P. // J. Am. Chem. Soc. 2007. V. 129. № 44. P. 13493–13501.
Schopfer M.P., Mondal B., Lee D.-H., Sarjeant A.A.N., Karlin K.D. // J. Am. Chem. Soc. 2009. V. 131. № 32. P. 11304–11305.
Gunther M.R., Sturgeon B.E., Mason R.P. // Toxicology. 2002. V. 177. № 1. P. 1–9.
Radi R. // Acc. Chem. Res. 2013. V. 46. № 2. P. 550–559.
Campolo N., Bartesaghi S., Radi R. // Redox Rep. 2014. V. 19. № 6. P. 221–231.
Ford E., Hughes M.N., Wardman P. // Free Radic. Biol. Med. 2002. V. 32. P. 1314–1323.
Folkes L.K., Trujillo M., Bartesaghi S., Radi R., Wardman P. // Arch. Biochem. Biophys. 2011. V. 506. P. 242–249.
Kirsch M., Lehnig M., Korth H.G., Sustmann R., de Groot H. // Chem. Eur. J. 2001. V. 7. № 15. P. 3313–3320.
Kalita A., Kumar P., Mondal B. // Inorg. Chem. 2013. V. 52. № 19. P. 10897–10903.
Tran N.G., Kalyvas H., Skodje K.M., Hayashi T., Moënne-Loccoz P., Callan P.E., Shearer J., Kirschenbaum L.J., Kim E. // J. Am. Chem. Soc. 2011. V. 133. № 5. P. 1184–1187.
Mondal B., Saha S., Borah D., Mazumdar R., Mondal B. // Inorg. Chem. 2019. V. 58. № 2. P. 1234–1240.
Shumaev K.B., Kosmachevskaya O.V., Chumikina, L.V., Topunov A.F. // Natural Product Communications. 2016. V. 11. № 8. P. 1189–1192.
Nappi A.J., Vass E. // J. Biol. Chem. 2001. V. 276. № 14. P. 11214–11222.
Pietraforte D., Minetti M. // Biochem. J. 1997. V. 321. P. 734–750.
Jia Y., Buehler P.W., Boykins R.A., Venable R.M., Alayash A.I. // J. Biol. Chem. 2007. V. 282. P. 4894–4907.
Alvarez B., Rubbo H., Kirk M., Barnes S., Freeman B.A., Radi R. // Chem. Res. Toxicol. 1996. V. 9. № 2. P. 390–396.
Nuriel T., Hansler A., Gross S.S. // J. Proteomics. 2011. V. 74. № 11. P. 2300–2312.
Quijano R., Alvarez B., Gatti R.M., Augusto O., Radi R. // Biochem. J. 1997. V. 322. P. 167–173.
Balazy M., Kaminski P.M., Mao K., Tan J., Wolin M.S. // J. Biol. Chem. 1998. V. 273. № 48. P. 32009–32015.
Karoui H., Hogg N., Kalyanaraman B. // Arch. Biochem. Biophys. 1996. V. 330. P. 115–124.
Denicola A., Souza J.M., Radi R. // Proc. Natl. Acad. Sci. USA. 1998. V. 95. P. 3566–3571.
Kosmachevskaya O.V., Nasybullina E.I., Shumaev K.B., Novikova N.N., Topunov A.F. // Appl. Biochem. Microbiol. 2020. V. 56. № 5. P. 512–520.
Lobysheva I.I., Serezhenkov V.A., Vanin A.F. // Biochemistry (Moscow). 1999. V. 64. № 2. P. 153–158.
Kapelko V.I., Lakomkin V.L., Abramov A.A., Lukoshkova E.V., Undrovinas N.A., Khapchaev A.Y., Shirinsky V.P. // Oxid. Med. Cell. Longev. 2017. V. 2017. e9456163. https://doi.org/10.1155/2017/9456163
Pisarenko O., Studneva I., Timoshin A., Veselova O. // Pflügers Arch. 2019. V. 471. P. 583–593.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология