

Прикладная биохимия и микробиология, 2021, T. 57, № 4, стр. 353-361
Закисление культуральной среды продуктами метаболизма глюкозы препятствует синтезу внеклеточной гетерологичной α-амилазы бактериями Bacillus subtilis 168
А. В. Качан 1, *, А. Н. Евтушенков 1
1 Белорусский государственный университет
220030 Минск, Беларусь
* E-mail: av.kachan@mail.ru
Поступила в редакцию 27.11.2020
После доработки 07.01.2021
Принята к публикации 22.02.2021
Аннотация
Синтез α-амилазы AmyM3 из Bacillus flexus 406 в клетках рекомбинантного штамма B. subtilis 168-28 существенно подавлялся при глубинном культивировании бактерий в питательной среде с добавлением 1% глюкозы. Подавление синтеза α-амилазы глюкозой в клетках Bacillus subtilis происходило на уровне транскрипции при участии белка катаболитного контроля CcpA. Удаление предполагаемого участка cre в гене amyM3 приводило к исчезновению эффекта катаболитной репрессии, однако количество синтезированной внеклеточной α-амилазы в присутствии глюкозы оставалось сниженным. Было показано, что сдвиг рН среды культивирования до значений 5.8–6.0, обусловленный метаболитами, выделяемыми бактериями B. subtilis при утилизации глюкозы, препятствовал синтезу активной внеклеточной α-амилазы, вероятно, оказывая влияние на пост-секреторный фолдинг фермента. Это наблюдение демонстрирует новый аспект обусловленного регулятором CcpA влияния предпочтительных источников углерода на продукцию внеклеточной α-амилазы бактериями B. subtilis.
Микроорганизмы характеризуются избирательной утилизацией содержащихся в среде питательных субстратов, отдавая предпочтение веществам, подвергающимся катаболизму с меньшими энергетическими затратами, например, глюкозе. В присутствии легко метаболизируемого источника углерода происходит подавление экспрессии генов транспорта и утилизации других питательных субстратов. Такой способ регуляции метаболизма получил название катаболитной репрессии [1]. Катаболитный контроль подразумевает также активацию ряда генов, способствующих катаболизму предпочитаемого субстрата. Ключевую роль в катаболитном контроле экспрессии генов хорошо изученной почвенной бактерии B. subtilis играет глобальный транскрипционный регулятор CcpA. Данный белок семейства LacI/GalR активируется при повышении внутриклеточной концентрации фруктозо-1,6-бисфосфата, одного из первичных интермедиатов гликолитического расщепления глюкозы [2]. В присутствии глюкозы происходит активация регулируемой фруктозо-1,6-бисфосфатом протеинкиназы, фосфорилирующей белок HPr, который, после данной модификации, образует комплекс с CcpA. В комплексе с HPr CcpA cвязывается со специфическими участками, называемыми cre (catabolite-responsive element), обнаруженными в более чем 60 регулируемых им оперонах. Под катаболитным контролем в клетках B. subtilis находятся гены ряда метаболических путей, дыхания, споруляции [3].
CcpA преимущественно действует в качестве репрессора транскрипции. В клетках B. subtilis 168 ген amyE, кодирующий α-амилазу (КФ 3.2.1.1), является мишенью репрессирующего действия белка CcpA. Регулятор связывается с последовательностью азотистых оснований 5'-TGT AAG CGC TTA CA-3', расположенной на 4 нуклеотида ниже области – 10 промотора, осуществляя репрессию транскрипции amyE в присутствии глюкозы [4]. Для некоторых оперонов B. subtilis, например, кодирующих ферменты превращения ацетил-КоА в ацетат, CcpA выступает в качестве активатора. Направленный мутагенез участков cre в генах биосинтеза биотехнологически значимых метаболитов позволяет повысить продуктивность бактерий при их выращивании в богатых углеводами питательных средах.
Ранее из хромосомной ДНК Bacillus flexus 406 был изолирован ген amyM3, кодирующий термостабильную α-амилазу AmyM3 [5]. После клонирования данного гена в клетках B. subtilis 168 бактерии эффективно синтезировали гетерологичный фермент.
Цель работы – охарактеризовать влияние катаболитного регулятора CcpA на синтез α-амилазы AmyM3 и получить мутантный вариант кодирующего ее гена, не чувствительный к катаболитной репрессии.
МЕТОДИКА
Объекты исследования. Штамм бактерий Escherichia coli XL1-Blue (endA1 gyrA96(NalR) thi-1 recA1 relA1 lac glnV44 F' [::Tn10 proAB+ lacIq Δ(lacZ)M15] hsdR17($r_{K}^{--}m_{K}^{--}$)) был использован в работе для проведения молекулярного клонирования плазмидных векторов. Штамм бактерий E. coli TG-1 (K-12 glnV44 thi-1 Δ(lac-proAB) Δ(mcrB-hsdSM)5, $\left( {r_{K}^{--}m_{K}^{--}} \right)$ F′ [traD36 proAB+lacIqlacZΔM15]) использовали для получения препаратов плазмидных ДНК перед трансформацией клеток B. subtilis. Штаммы E. coli XL1-Blue и TG-1, а также штамм B. subtilis 168 (trpC2) взяты из коллекции микроорганизмов кафедры молекулярной биологии Белорусского государственного университета (Минск). Штамм B. subtilis СR2 (trpC2 ccpA::ermAM(EmR)) был получен замещением центральной части хромосомного гена ccpA бактерий B. subtilis 168 (trpC2) на маркерный ген устойчивости к эритромицину, для чего в клетки B. subtilis 168 вводили интегративную плазмиду pMTL21c-ccpA-Em. Штамм B. subtilis СR3 (trpC2 ccpA::spc(SpR)) был получен замещением центральной части хромосомного гена ccpA бактерий B. subtilis 168 (trpC2) на маркерный ген устойчивости к спектиномицину.
Штаммы B. subtilis 168-28 (trpC2 amyE::[amyM3 aphA3(KmR)]) и 168-22 (trpC2 amyE::[amyM3-cre1 aphA3(KmR)]) получены путем введения в клетки бактерий B. subtilis 168 интегративных векторов pMTL21c-amyE-amyM3-Km и pMTL21c-amyE-amyM3-cre1-Km соответственно.
Штаммы B. subtilis СR2-28 (trpC2 ccpA::ermAM(EmR) amyE::[amyM3 aphA3(KmR)]) и СR2-22 (trpC2 ccpA::ermAM(EmR) amyE::[amyM3-cre1 aphA3(KmR)]) получены введением в клетки бактерий B. subtilis CR2 плазмид pMTL21c-amyE-amyM3-Km и pMTL21c-amyE-amyM3-cre1-Km соответственно.
Штаммы B. subtilis 168-MZ (trpC2 amyE::[PamyM3-lacZ ermAM(EmR)]) и СR3-MZ (trpC2 ccpA::spc(SpR) amyE::[PamyM3-lacZ ermAM(EmR)]) получены введением в клетки бактерий B. subtilis 168 и CR3 соответственно плазмидной ДНК pMTL21c-amyE-lacZ-amyM3, которая позволяла встроить в состав кодирующей последовательности гена amyE B. subtilis репортерную конструкцию, обеспечивающую экспрессию гена β-галактозидазы (КФ 3.2.1.23) lacZ E. coli под контролем промоторного участка гена amyM3. Штамм B. subtilis 168-СZ (trpC2 amyE:: [PamyM3-сre1-lacZ ermAM(EmR)]) содержал аналогичную инсерцию, в которой в промоторном участке гена amyM3 отсутствовал предполагаемый сайт cre.
В работе также использовался штамм B. subtilis 568 (amyE::ermAM(EmR)), который был создан на основе B. subtilis 168 и содержал ген α-амилазы amyE, инактивированный путем инсерции маркера устойчивости к эритромицину.
Культивирование. Бактерии выращивали при температуре 37°С в полноценной жидкой или на агаризованной пептонно-дрожжевой среде следующего состава (г/л): пептон ферментативный – 10.0, дрожжевой экстракт – 5.0, NaCl – 5.0, pH – 7.0. Для оценки активности ферментов, синтезируемых различными штаммами B. subtilis, культивирование бактерий проводили в жидкой питательной среде для споруляции (ПСС) [6]. При выращивании в жидкой среде для аэрации и перемешивания культур (240 об./мин) использовали орбитальный шейкер IST-4075 (“Jeio Tech”, Республика Корея). При необходимости в питательные среды вносили антибиотики в следующих концентрациях: ампициллин – 100 мкг/мл, канамицин – 50 мкг/мл, спектиномицин – 100 мкг/мл, эритромицин – 300 мкг/мл для E. coli и 3 мкг/мл для B. subtilis, хлорамфеникол – 5 мкг/мл для B. subtilis.
Измерение pH в питательных средах, растворах и отбираемых пробах бактериальных культур осуществляли с помощью рН-метра HI 83141 (“HANNA Instruments”, Германия).
Конструирование интегративных плазмид. Для введения в состав хромосомной ДНК B. subtilis 168 чужеродных генетических конструкции были использованы интегративные плазмиды, полученные на основе вектора pMTC21c, не способного реплицироваться в клетках B. subtilis [7]. При создании плазмиды pMTL21c-ccpA-Em проводили амплификацию фрагмента хромосомной ДНК B. subtilis 168, содержащего ген ccpA, в реакции ПЦР с олигонуклеотидами ссpF (5'-TTT TGC TAG CAA TAT TAC GAT CTA CGA TGT A-3') и ccp-R (5'-TTC TCG AGT TAT GAC TTG GTT GAC TTT CTA AGC T-3'). Для проведения всех описанных в работе реакций ПЦР использовали амплификатор SureCycler 8800 (“Agilent Technologies”, США), высокоточную ДНК-полимеразу Diamant HF и реагенты производства ОДО “Праймтех” (Беларусь). Реакционная смесь для ПЦР объемом 50 мкл содержала около 50 нг тотальной ДНК B. subtilis 168, 0.2 ммоль/л каждого дНТФ, 0.5 мкмоль/л каждого праймера, 0.02 E/мкл ДНК-полимеразы и соответствующий буфер. Продукты амплификации разделяли с помощью электрофореза в агарозном геле. Целевые продукты очищали из геля с помощью набора Agarose Gel Extraction Kit (“Jena Bioscience”, Германия), после чего полученный фрагмент ДНК объединяли в ходе реакции лигирования с плазмидой pMTC21c, предварительно линеаризированной с помощью эндонуклеазы рестрикции SmaI. После клонирования рекомбинантных молекул в клетках E. coli XL1-Blue верификацию получения нужных плазмид осуществляли с помощью рестрикционного анализа. Выделение плазмидной ДНК, электрофорез ДНК в агарозном геле, рестрикционный анализ и лигирование, кальциевую трансформацию клеток E. coli проводили в соответствии с общепринятыми методиками [8]. В работе использовались препараты эндонуклеаз рестрикции и ДНК-лигазы фирм “Thermo Scientific” (Литва) и “New England Biolabs” (Великобритания).
После встраивания в состав вектора pMTL21c участка ДНК с геном ccpA полученную плазмиду обрабатывали эндонуклеазами ClaI и Bsp119I для удаления части кодирующей последовательности гена ccpA. Линейную плазмиду с делецией обрабатывали ДНК-полимеразой фага T4 (“Thermo Scientific”, Литва) для создания ровных концов, после чего лигировали с фрагментом ДНК, содержащим маркер устойчивости к эритромицину, который был получен путем обработки плазмиды pMUTIN-4 [9] ферментами XbaI, Sau3AI и ДНК-полимеразой фага T4. Рекомбинантную плазмиду pMTL21c-ccpA-Em клонировали в клетках E. coli XL1-Blue.
Трансформация бактерий B. subtilis плазмидной ДНК производилась по общепринятой методике [10]. После трансформации проводили отбор колоний, клетки которых проявляли устойчивость к эритромицину (что указывало на присутствие в геноме бактерий вставки pMTL21c-ccpA-Em, интегировавшейся в состав локуса ccpA путем гомологичной рекомбинации), при этом были чувствительными к хлорамфениколу (маркер устойчивости к которому присутствует в плазмиде pMTL21c). Такой фенотип указывал на то, что в клетках происходила двойная рекомбинация, в результате которой участок хромосомного гена ccpA замещался на поврежденную копию ccpA, при этом последовательность плазмиды pMTL21с в хромосому не внедрялась. Наличие в отобранном штамме B. subtilis СR2 данной генетической перестройки подтверждали с помощью ПЦР на матрице геномной ДНК штамма c праймерами ccp-F и ссp-R по образованию единственного продукта реакции длиной 2061 п.н., в то время как размер амплифицируемого участка ДНК B. subtilis 168 в аналогичной реакции составлял 1017 п.н.
Аналогичная стратегия была использована для конструирования других использованных в работе интегративных плазмид и получения с их помощью рекомбинантных штаммов B. subtilis. Плазмиды pMTL21c-amyE-amyM3-Km и pMTL21c-amyE-amyM3-cre1-Km обеспечивали инсерцию в состав локуса amyE маркера устойчивости к канамицину из векторной молекулы pKS1 [11] и гена α-амилазы amyM3 B. flexus 406 [5]. Плазмида pMTL21c-amyE-amyM3-cre1-Km позволяла интегрировать в локус amyE B. subtilis 168 мутантный вариант гена с делецией предполагаемого участка cre. Полученные с использованием данных плазмид штаммы B. subtilis 168-28, 168-22, СR2-28 и СR2-22 содержали ген amyM3 в составе хромосомного локуса amyE. Такая инсерция приводила к инактивации гена amyE, кодирующего собственную α-амилазу B. subtilis 168.
Благодаря полученной в работе плазмиде pMTL21c-amyE-lacZ-amyM3 производилась интеграция в состав кодирующей последовательности гена amyE B. subtilis репортерной конструкции, обеспечивающей экспрессию гена β-галактозидазы lacZ E. coli (изолирован из плазмиды pMUTIN-4) под контролем промоторного участка гена amyM3. Репортерный вектор получали путем встраивания в состав плазмиды pMTL21c-amyE фрагментов из плазмиды pMUTIN-4, содержащих маркер устойчивости к эритромицину и ген lacZ. Затем полученную плазмиду линеаризировали обработкой ферментом SmaI. Промоторный участок amyM3 получали путем амплификации в реакции ПЦР с использованием плазмиды pALTER-1-amyM3 в качестве матричной ДНК и олигонуклеотидов cpm-F (5'-CGA TCG CCT ATT TGG CTT TTC CCC ATT CGC TAC TTC GAA AAA GCA GG TAG C-3') и cpm-R (5'-GAT CCG CGG CGG CCG CCC ATG ATT CGG TGC AAC CGT TGG CAC-3'). Объединение промоторного участка гена amyM3 с линейным интегративным вектором проводили c помощью методики кольцевой полимеразной реакции (CPEC) [12]. Реакционная смесь объемом 60 мкл включала 10 нг/мкл фрагмента гена amyM3, 10 нг/мкл линеаризированного вектора, 0.2 ммоль/л каждого дНТФ и 0.02 E/мкл ДНК-полимеразы. Реакцию осуществляли при следующем температурном режиме: 98°С 30 с; 20 циклов: 98°С 10 с, 70°С 30 с, 72°С 5 мин; 72°С 10 мин. Аналогичным образом в работе была сконструирована плазмида pMTL21c-amyE-lacZ-amyM3-cre, содержащая ген lacZ под контролем промотора amyM3 c мутацией предполагаемого участка cre. Репортерные конструкции вводились в клетки штаммов B. subtilis 168 и CR2, с последующим отбором содержащих инсерции в локус amyE бактерий. Полученные штаммы B. subtilis 168-MZ, B. subtilis 168-CZ и СR3-MZ характеризовались отсутствием амилолитической активности, устойчивостью к эритромицину и чувствительностью к хлорамфениколу.
Направленный мутагенез предполагаемого участка cre гена amyM3 проводили с помощью набора Altered Sites® II in vitro Mutagenesis System (“Promega Corp.”, США) в соответствии с рекомендуемым производителем протоколом. Наличие изменений в последовательности ДНК подтверждали с помощью секвенирования.
Определение концентрации D-глюкозы в бактериальных культурах проводили глюкозооксидазным-пероксидазным методом с использованием набора D-glucose assay kit (GOPOD-Format) (“Megazyme”, Ирландия).
Оценку накопления α-амилазы в культуре бактерий осуществляли, выращивая штаммы до середины экспоненциальной фазы в среде ПСС, после чего культуры разделяли на две части, в одну из которых вносили раствор D-глюкозы до конечной концентрации 1% [6]. В течение дальнейшего культивирования в периодически отбираемых пробах культуры оценивали количество клеток по величине оптической плотности при длине волны 600 нм, а также измеряли активность внеклеточных α‑амилаз с реагентом I2/KI, как описано [6]. Продуктивность клеток выражали в виде отношения рассчитанных значений активности к значению D600 культуры (E/D600).
Оценку активности гена β-галактозидазы lacZ в пробах выращенных аналогичным образом бактериальных культур проводили в соответствии с широко используемой методикой [13].
Статистический анализ данных. Все данные представляют собой средние значения, полученные по результатам по меньшей мере трех независимых экспериментов. Статистическая обработка результатов измерений проводилась в программе Microsoft Office Excel 2003. Погрешность измерений оценивали по величине среднеквадратичного отклонения (σ).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
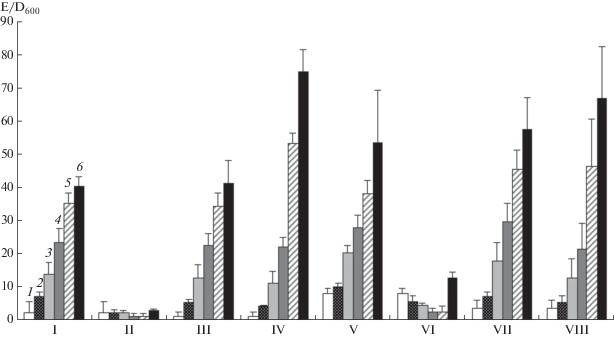
При выращивании в жидкой питательной среде ПСС бактерии штамма B. subtilis 168-28 вступали в стационарную фазу роста на 2–3 ч после начала отбора проб в середине экспоненциальной фазы. Внесение глюкозы до концентрации 1.0% в этой фазе роста культуры вызывало задержку в достижении стационарной фазы. При оценке количества α-амилазы в культуре бактерий B. subtilis 168-28, выращиваемых в данных условиях, было обнаружено, что активность фермента через 3 ч после внесения в культуру глюкозы составляла около 5% от активности фермента в культуре без глюкозы, как показано на рис. 1. Было выдвинуто предположение, что в клетках B. subtilis 168-28 ген amyM3 подвергается катаболитной репрессии по механизму, характерному для гена amyE, т.е. обусловленному транскрипционным регулятором CcpA. Это предположение подтверждалось тем, что в культуре бактерий B. subtilis CR2-28, у которых ген ccpA был инактивирован, активность α‑амилазы продолжала увеличиваться несмотря на присутствие глюкозы в среде культивирования. Через 3 ч культивирования как в среде ПСС с глюкозой, так и без глюкозы в культуре B. subtilis CR2-28 регистрировали сопоставимые уровни амилолитической активности (около 23 E/D600). Через 5 и 7 ч после внесения в культуру глюкозы бактерии продолжали активно синтезировать фермент (активность 53.3 ± 3.2 и 74.9 ± 6.7 E/D600 соответственно), в то время как в культуре, в которую глюкозу не добавляли, через 5 и 7 ч культивирования прирост активности снижался (активность 34.6 ± 3.7 и 41.4 ± 6.7 E/D600 соответственно).
Рис. 1.
Продуктивность (среднее значение E/D600 ± σ) различных штаммов B. subtilis по α-амилазе: I, II – 168-28; III, IV – CR2-28; V, VI – 168‑22; VII, VIII – CR2-22. В культуры I, III, V, VII глюкозу не вносили, в культуры II, IV, VI и VIII вносили глюкозу до концентрации 1.0% в середине экспоненциальной фазы. Активность фермента оценивали непосредственно после внесения в культуры глюкозы (1) или через 1 (2) 2 (3), 3 (4), 5 (5) и 7 (6) ч после этого.
Для поиска участков связывания белка CcрA в нуклеотидной последовательности гена amyM3 с помощью базы данных DBTBS был получен список 44 участков cre, функциональное значение которых в регуляции различных генов и оперонов B. subtilis 168 было подтверждено экспериментально [14]. В программе FIMO [15] было осуществлено сканирование нуклеотидной последовательности гена amyM3 (код доступа в Genbank: JX429073.1) полученной консенсусной последовательностью cre. В результате анализа в последовательности гена amyM3 было обнаружено 14 участков, проявляющих сходство с последовательностью cre (p < 0.001). Результаты поиска представлены на рис. 2. Наибольшее подобие с консенсусом последовательности cre B. subtilis обладал участок 5'-TGG TAA CGT TTA CA-3', который локализовался в положении 588–601 проанализированной последовательности гена amyM3, между областью – 10 предполагаемого промотора гена и последовательностью Шайна-Дальгарно, как показано на рис. 2. Такое расположение соответствует локализации участков cre во многих генах, для которых подтверждена регуляция белком CcpA [3]. Значительное сходство с консенсусом наблюдалось как на прямой, так и на обратной цепи ДНК в положении 588–601, что указывает на выраженную палиндромную структуру этого участка. Менее четким сходством с консенсусной последовательностью cre обладали 5 участков, расположенных внутри кодирующей последовательности гена (координаты 682–695, 703–716, 780–793, 1052–1065 и 1286–1299 на рис. 2). Другие выявленные участки не имели ряда важных структурных особенностей, характерных для областей cre B. subtilis, например, консервативной пары CG, либо располагались на значительном расстоянии от точки старта транскрипции, поэтому их роль в зависимой от белка CcpA репрессии гена amyM3 маловероятна.
Рис. 2.
Результаты поиска областей cre в гене amyM3 c помощью программы FIMO. Подчеркнуты предполагаемые промотор и RBS-сайт, в рамке – кодирующая последовательность гена. Черным фоном выделена наиболее вероятная последовательность cre, серым – менее вероятные.

Чтобы определить, задействован ли обнаруженный участок 5'-TGG TAA CGT TTA CA-3' в регуляции активности гена α-амилазы, осуществляемой CcpA, был проведен его направленный мутагенез, в результате которого вышеуказанная 14-нуклеотидная последовательность была заменена на сайт узнавания эндонуклеазы SpeI 5'-ACT AGT-3'. Мутантный вариант гена amyM3 с удаленным таким образом предполагаемым участком cre внедряли в состав хромосомного локуса amyЕ бактерий B. subtilis, имеющих как функциональный, так и инактивированный ген ccpA. В результате были получены штаммы B. subtilis 168-22 и CR2-22 соответственно.
Результаты оценки активности α-амилазы в культурах бактерий B. subtilis 168-22 и CR2-22 приведены на рис. 1. В культуре B. subtilis CR2-22 динамика накопления внеклеточной α-амилазы существенно не отличалась от характерной для штамма B. subtilis CR2-28. Продуктивность клеток бактерий B. subtilis 168-22 по α-амилазе в присутствии глюкозы была низкой (4.64 ± 0.42 E/D600), хотя в первые 2 ч после внесения глюкозы она была значимо больше, чем в культуре штамма B. subtilis 168-28, имеющего нативную последовательность гена amyM3 (2.28 ± 0.51 E/D600). Был сделан вывод, что подавление синтеза фермента глюкозой обусловлено фактором CcpA, однако регулятор либо действует через связывание с другим участком гена amyM3, либо может оказывать опосредованное влияние через иные факторы.
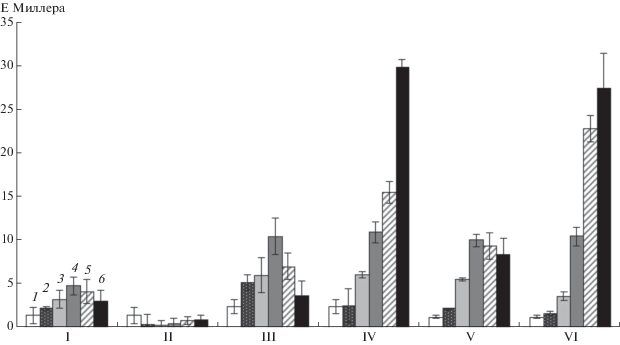
Чтобы детальнее изучить влияние регулятора CcpA на функционирование гена α-амилазы AmyM3, в хромосомы бактерий B. subtilis 168 и CR3 вводилась репортерная конструкция, содержащая кодирующий β-галактозидазу ген lacZ, выше которого был встроен фрагмент ДНК, содержащий промоторный участок гена amyM3 либо этот промотор с мутацией предполагаемого участка cre. Оценка активности β-галактозидазы в клетках полученных штаммов B. subtilis 168-MZ, 168-CZ и CR3-CZ показала, что промотор гена amyM3 наиболее эффективно инициировал транскрипцию в начале стационарной фазы роста культуры, после чего активность гена постепенно снижалась. Как видно на рис. 3, при внесении в культуру глюкозы наблюдалось практически полное подавление активности нативного промотора, однако промотор с мутацией в предполагаемом участке cre обеспечивал повышенный уровень транскрипции гена-репортера при добавлении в среду культивирования глюкозы. Соизмеримо высокая активность гена lacZ, находящегося под контролем нативного промотора amyM3, обнаруживалась и в клетках штамма, имеющего инактивированный ген ccpA. Полученные результаты свидетельствуют о том, что белок CcpA контролирует инициацию транскрипции в промоторе гена amyM3, при наличии глюкозы подавляя экспрессию гена путем контакта с участком cre, имеющим нуклеотидную последовательность 5'-TGG TAA CGT TTA CA-3'.
Рис. 3.
Активность гена β-галактозидазы (среднее значение E Миллера ± σ), находящегося под контролем различных вариантов промотора гена amyM3, в клетках различных штаммов B. subtilis: I, II –168-MZ; III, IV – 168‑CZ; V, VI – CR3-MZ. В культуры I, III, V глюкозу не вносили, в культуры II, IV и VI вносили глюкозу до концентрации 1.0% в середине экспоненциальной фазы. Измерения проводили непосредственно после внесения в культуры глюкозы (1) или через 1 (2), 2 (3), 3 (4), 4 (5) и 5 (6) ч после этого.
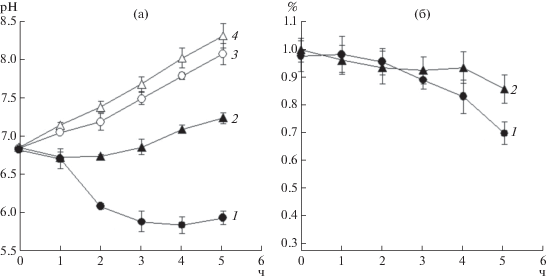
Интересен тот факт, что при внесении в культуру глюкозы, несмотря на сохранение высокой активности промотора amyM3 с делецией участка cre, у штамма B. subtilis 168-22, содержащего ген с данным промотором, синтез внеклеточной α-амилазы существенно подавляется. Известно, что метаболизм глюкозы клетками B. subtilis 168 происходит путем гликолиза с последующим окислительным декарбоксилированием пирувата. Конечный продукт этого процесса, ацетил-КоА, превращается в силу “эффекта избыточного метаболизма (overflow metabolism)” преимущественно в ацетат. Регулятор CcpA выступает активатором соответствующих генов, одновременно блокируя гены метаболизма ацетил-КоА в цикле трикарбоновых кислот. Уксусная кислота, а также пируват, ацетоин и 2,3-бутандиол выделяются клетками во внешнюю среду в качестве основных побочных продуктов утилизации глюкозы [16, 17]. В работе была проведена оценка изменения pH питательной среды в ходе культивирования бактерий B. subtilis 168-28 и СR2-28 в среде ПСС с 1.0% глюкозы. Результаты, представленные на рис. 4, подтверждали известные из литературы факты, что утилизация бактериями глюкозы приводила к интенсивному выделению в среду культивирования кислот, приводя к сдвигу значения pH культуры до 5.8–6.0 через 2 ч после добавления углевода [18]. Закисления среды при культивировании штамма, дефектного по гену ccpA, практически не наблюдалось, что может объясняться нарушением регуляции экспрессии ряда генов углеводного и азотного обмена, находящихся под контролем CcpA [19]. Как показано на рис. 4, за 5 ч культивирования количество глюкозы в культурах штаммов B. subtilis 168-28 и CR2-28 снижается до 0.7 и 0.86% соответственно.
Рис. 4.
Значение pH (среднее значение ± σ) среды ПСС (а) и остаточная концентрация глюкозы (среднее значение % ± σ, б) при культивировании бактерий B. subtilis 168-28 (1, 3) и CR2-28 (2, 4) в присутствии 1% глюкозы (1, 2) или без ее добавления (3, 4). Измерения проводили каждый час, начиная с момента внесения в культуры глюкозы.
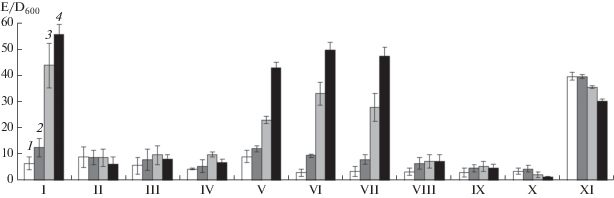
Полученные результаты позволяют предположить, что низкие значения внеклеточной амилолитической активности при культивировании бактерий B. subtilis 168-22 в среде с глюкозой обусловлены фактором CcpA, однако не прямой репрессией гена α-амилазы, лишенного участка cre (транскрипция гена в клетках данного штамма в среде с глюкозой сохранялась на высоком уровне), а опосредовано, через снижение pH среды культивирования продуктами утилизации углевода. Чтобы подтвердить наличие такого влияния, бактерии B. subtilis 168-22 и B. subtilis CR2-22 культивировали в питательной среде ПСС, периодически внося в нее раствор, содержащий 0.5 М ацетата или 1 М HCl в таких количествах, чтобы обеспечить снижение рН среды культивирования до значений 5.8–6.0, соответствующих наблюдаемым в содержащей глюкозу среде. Для оценки стабильности α-амилазы AmyM3 в такой среде в культуру бактерий B. subtilis 568, не способных синтезировать α-амилазу, фермент вносили до концентрации 40 E/мл. Представленные на рис. 5 результаты показывают, что подавление синтеза α-амилазы бактериями B. subtilis 168-22 происходило как при утилизации глюкозы, в ходе чего происходит снижение pH внеклеточной среды до 5.8–6.0 (рис. 4), так и при внесении в культуру уксусной или соляной кислоты. В культурах штамма B. subtilis CR2-22, клетки которого лишены регулятора СcpA, необходимого для выделения кислот, образующихся при метаболизме глюкозы, синтез внеклеточной амилазы также мог быть блокирован доведением рН до 5.8–6.0. С другой стороны, смещение pH до значения 7.5, производимое путем внесения 2 М раствора гидроксида аммония, при выращивании B. subtilis 168-22 в среде ПСС с глюкозой позволяло восстановить накопление амилолитической активности в среде культивирования, что служило доказательством влияния рН среды культивирования на синтез внеклеточной α-амилазы.
Рис. 5.
Продуктивность (среднее значение E/D600 ± σ) штаммов B. subtilis 168-22 (I–V), СR2-22 (VI–X) по α-амилазе в условиях сдвига pH. В культуры II, V, VII и X вносили глюкозу до концентрации 1.0% в середине экспоненциальной фазы. В культуру V также вносили 2 М NH4OH для поддержания значения рН 7.5. Для поддержания значения рН 5.9 в культуры III и VIII вносили 0.5 М уксусную кислоту, в культуры IV, IX, X – 1 М НСl. XI – культура B. subtilis 568 c добавлением 40 E/мл AmyM3, в которую вносили 1 М НСl для поддержания значения рН 5.9. Активность амилаз измерена непосредственно после внесения в культуры глюкозы (1) или через 1 (2), 3 (3) и 5 (4) ч после этого.
Следует отметить, что инактивация внесенного в культуру B. subtilis 568 фермента в анализируемых условиях была незначительной, рассчитанное время полуинактивации α-амилазы в такой культуре составляло около 10 ч. Оптимум активности данного фермента ранее был определен в диапазоне значений рН от 6.0 до 7.0.
На основании полученных результатов можно сделать вывод о том, что катаболизм глюкозы до кислотных продуктов, контролируемый при участии транскрипционного фактора CcpA, и закисление ими культуральной жидкости препятствует синтезу внеклеточной α-амилазы AmyM3 бактериями B. subtilis. Это объясняет, почему бактерии, имеющие ген amyM3 с удаленным участком cre, выделяли лишь незначительные количества α-амилазы в культуре с добавлением глюкозы.
Анализ приведенных наблюдений позволяет предположить, что чувствительность α-амилазы AmyM3, синтезируемой клетками B. subtilis 168, к кислому рН среды проявлялся на стадии транслокации через мембрану либо пост-транслокационного фолдинга. Известно, что многие гетерологичные α-амилазы, синтезируемые в клетках B. subtilis 168, как и ряд других внеклеточных ферментов, секретируясь в развернутом виде, подвергаются внеклеточному фолдингу с участием липопротеина PrsA [20]. Затруднения на этапе принятия секретируемыми белками нативной третичной структуры, обычно связанные с их сверхпродукцией или отсутствием необходимых для фолдинга факторов, приводят к накоплению несвернутых полипептидов в просвете между мембраной и клеточной стенкой. В клетках B. subtilis это инициирует секреторный стресс, сопровождающийся повышенной экспрессией генов мембранных протеаз HtrA и HtrB, которые обеспечивают деградацию дефектных белков [21]. В исследовании Манабэ с соавт. было показано, что одним из факторов, влияющих на внеклеточный фолдинг щелочной α-амилазы AmyK38, является значение внеклеточного рН – смещение рН среды культивирования бактерий с 6.8 до 8.4 приводило с существенному повышению уровня внеклеточной активности α-амилазы и снижению экспрессии маркеров секреторного стресса [22]. Вероятно, фолдинг α-амилазы AmyM3, выделяющейся из клеток B. subtilis, также нарушается при снижении рН до значений 6.0.
Обнаруженное влияние низких значений рН среды культивирования на способность бактерий B. subtilis выделять функциональную α-амилазу является еще одним аспектом действия регулятора CcpA, активирующегося при утилизации клетками легко метаболизируемых углеводов, на продукцию внеклеточной α-амилазы бактериями В. subtilis. Данное влияние следует учитывать в процессах ферментации, при использовании богатых углеводами питательных сред для выращивания штаммов-продуцентов внеклеточных белков.
Дальнейшие исследования позволят детальнее понять, что происходит с α-амилазой AmyM3 после секреции и какие генетические изменения клеток B. subtilis могут уменьшить негативный эффект внеклеточного рН на синтез этого фермента.
Работа выполнялась в рамках задания государственной программы научных исследований “Биотехнологии” за 2018–2020 гг.
Список литературы
Deutscher J. // Curr. Opin. Microbiol. 2008. V. 11. № 2. P. 87–93.
Fujita Y. // Biosci. Biotechnol. Biochem. 2009. V. 73. № 2. P. 245–259.
Lorca G.L., Chung Y.J., Barabote R.D., Weyler W., Schilling C.H., Saier Jr. M.H. // J. Bacteriol. 2005. V. 187. № 22. P. 7826–7839.
Weickert M.J., Chambliss G.H. // Proc. Natl. Acad. Sci. USA. 1990. V. 87. № 16. P. 6238–6242.
Kachan A.V., Evtushenkov A.N. // Cent. Eur. J. Biol. 2013. V. 8. № 4. P. 346–356.
Nicholson W.L., Chambliss G.H. // J. Bacteriol. 1985. V. 161. № 3. P. 875–881.
Chambers S.P., Prior S.E., Barstow D.A., Minton N.P. // Gene. 1988. V. 68. № 1. P. 139–149.
Маниатис Т., Фрич Э., Сэмбрук Дж. Методы генетической инженерии. Молекулярное клонирование / Ред. А.А. Баев, К.Г. Скрябин. М.: Мир, 1984. 480 с.
Vagner V., Dervyn E., Ehrlich S.D. // Microbiology. 1998. V. 144. № 11. P. 3097–3104.
Spizizen J., Anagnostopoulos C. // J. Bacteriol. 1961. V. 81. № 5. P. 741–746.
Shatalin K., Neyfakh A. // FEMS Microbiol Lett. 2005. V. 245. № 2. P. 315–319.
Кулик Е.В., Селезнева Ю.В., Качан А.В., Русь О.Б., Евтушенков А.Н. // Изв. НАН Беларуси. Сер. биол. наук. 2020. Т. 65. № 3. С. 319–327.
Ferrari E., Howard S.M., Hoch J.A. // J. Bacteriol. 1986. V. 166. № 1. P. 173–179.
Sierro N., Makita Y., de Hoon M., Nakai K. // Nucl. A-cids Res. 2008. V. 36. P. D93–D96.
Grant C.E., Bailey T.L., Noble W.S. // Bioinformatics. 2011. V. 27. № 7. P. 1017–1018.
Speck E.L., Freese E. // J. Gen. Microbiol. 1973. V. 78. № 2. P. 261–275.
Blencke H.-M., Homuth G., Ludwig H., Mäder U., Hecker M., Stülke J. // Metab. Eng. 2003. V.5. № 2. P. 133–149.
Turinsky A.J., Moir-Blais T.R., Grundy F.J., Henkin T.M. // J. Bacteriol. 2000. V. 182. № 19. P. 5611–5614.
Sonenshein A.L. // Nat. Rev. Microbiol. 2007. V. 5. № 12. P. 917–927.
Vitikainen M., Hyyryläinen H.-L., Kivimäki A., Kontinen V.P., Sarvas M. // J. Appl. Microbiol. 2005. V. 99. № 2. P. 363–375.
Hyyryläinen H.-L., Bolhuis A., Darmon E., Muukkonen L., Koski P., Vitikainen M., Sarvas M., Prágai Z., Bron S., van Dijl J.M., Kontinen V.P. // Mol. Microbiol. 2001. V. 41. № 5. P. 1159–1172.
Manabe K., Kageyama Y., Tohata M., Ara K., Ozaki K., Ogasawara N. // Microb. Cell Fact. 2012. V. 11.https://doi.org/74-10.1186/1475-2859-11-74
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология