

Прикладная биохимия и микробиология, 2020, T. 56, № 5, стр. 452-464
Альтернативный синтез цефазолина с использованием синтетазы цефалоспоринов-кислот
А. В. Скляренко 1, *, И. А. Грошкова 1, А. И. Сидоренко 1, С. В. Яроцкий 1
1 Федеральное государственное бюджетное учреждение “Национальный исследовательский центр
“Курчатовский институт” (НИЦ – “Курчатовский институт”)
123182 Москва, Россия
* E-mail: asklyarenko@yandex.ru
Поступила в редакцию 09.03.2020
После доработки 01.04.2020
Принята к публикации 22.04.2020
Аннотация
Сопоставлены два пути химико-биокаталитического синтеза антибиотика цефазолина (CEZ) из 7-амино-цефалоспорановой кислоты (7-ACA) с использованием в качестве биокатализатора иммобилизованной рекомбинантной синтетазы цефалоспоринов-кислот. Первый путь состоял в химическом замещении 3-ацетокси-группы 7-ACA с помощью 2-меркапто-5-метилтиадиазола с последующим биокаталитическим ацилированием аминогруппы полученной 7-амино-3-[2-метил-1,3,4-тиадиазол-5-ил)-тиометил]-3-цефем-4-карбоновой кислоты (TDA) метиловым эфиром 1(Н)-тетразолилуксусной кислоты. Альтернативой являлось биокаталитическое ацилирование аминогруппы 7-ACA с образованием полупродукта (S-p CEZ), который затем без выделения из реакционной смеси был химически трансформирован в CEZ. Изучение и оптимизация каждого из биокаталитических процессов показало ряд важных преимуществ ацилирования 7-ACA по сравнению с ацилированием TDA в отношении выхода процесса, концентрации продукта в конечной реакционной смеси и толерантности условий протекания процесса по отношению к активности и стабильности фермента. С учетом несомненных экологических преимуществ процесса химической трансформации S-p CEZ в CEZ по сравнению с процессом получения TDA из 7-ACA сделан вывод о предпочтительности химико-биокаталитического синтеза CEZ по второму пути.
Цефазолин (6R,7R)-3-[(5-метил-1,3,4-тиадиазол-2-ил)тиометил]-8-оксо-7-[(1H-тетразол-1-ил)ацетиламино]-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (CEZ) является одним из важнейших представителей класса цефалоспоринов-кислот, включающего более полутора десятков полусинтетических парентеральных β-лактамных антибиотиков [1, 2]. Этот антибиотик входит в перечень основных лекарственных средств, рекомендуемых Всемирной организацией здравоохранения.
В настоящее время мировое производство CEZ составляет около 1000 метрических т в год (в пересчете на свободную кислоту) [3] и базируется на методах химического синтеза. Исходя из 3-(ацетилоксометил)-7-амино-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновой кислоты (7-амино-цефалоспорановая кислота, 7-ACA) получают 7-амино-3-[2-метил-(1,3,4-тиадиазол-5-ил)-тиометил]-3-цефем-4-карбоновую кислоту (TDA) путем замещения 3-ацетокси-группы действием 2-меркапто-5-метилтиадиазола (MMTD) в неводной среде в присутствии сильных кислот [4, 5]. Затем аминогруппу TDA ацилируют активированными производными 1(Н)-тетразолилуксусной кислоты (TzAA) в неводной или водно-органической среде при низкой температуре (–40°С), используя защиту карбоксильной группы [6, 7].
Перспективной альтернативой химическому синтезу β-лактамных антибиотиков, в том числе CEZ и других цефалоспоринов-кислот, является трансформация, катализируемая ферментами в форме гетерогенных биокатализаторов. Использование таких биокаталитических технологий позволяет снизить экологическую нагрузку на окружающую среду, а также повысить чистоту получаемых препаратов [2, 8–13].
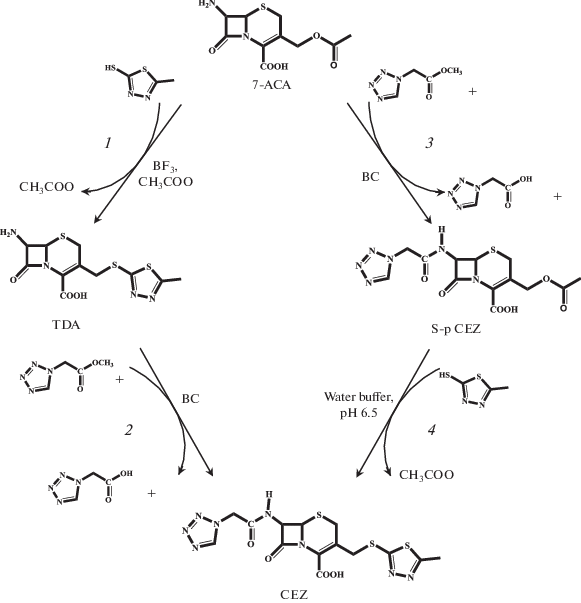
Химико-биокаталитический синтез CEZ из 7-ACA может быть осуществлен двумя путями (рис. 1). В первом случае 7-ACA превращают в TDA традиционным химическим синтезом (рис. 1, трансформация 1), а затем осуществляют биокаталитическое ацилирование аминогруппы TDA с образованием CEZ (рис. 1, трансформация 2). Альтернативным путем является использование биокатализа для ацилирования аминогруппы 7-ACA (рис. 1, трансформация 3) с последующим химическим превращением получаемого полупродукта CEZ (S-p СЕZ) в целевой антибиотик (рис. 1, трансформация 4). В обоих случаях ацилирующим агентом служит активированное производное TzAA, а именно ее метиловый эфир (METzAA), то есть осуществляется процесс синтеза с ацильным переносом (кинетически-контролируемый синтез) [2, 14], где TDA или 7-ACA выступают в качестве исходного β-лактама – ключевой аминокислоты (КА), а METzAA – в качестве ацилирующего агента (АА).
Рис. 1.
Пути химико-биокаталитического синтеза CEZ. Трансформации: 1 – xимический синтез TDA из 7-ACA; 2 ‒ биокаталитический синтез CEZ из TDA; 3 – биокаталитический синтез S-p CEZ из 7-ACA; 4 – xимический синтез CEZ из S-p CEZ.
При проведении кинетически-контролируемого синтеза β-лактамного соединения (CEZ или S-p CEZ) фермент катализирует три конкурирующие реакции [2, 14]:
− синтез целевого продукта путем переноса ацильной группы АА (METzAA) на аминогруппу в положении С7 β-лактамного ядра КА (TDA или 7-ACA), сопровождающийся выделением метилового спирта;
− гидролиз целевого продукта с образованием соответствующей КА и TzAA;
− гидролиз METzAA с образованием TzAA и метилового спирта.
Достигаемый выход целевого продукта определяется соотношением скоростей перечисленных реакций и зависит от условий их проведения и от начальных концентраций исходного β-лактама и АА.
Оптимизация биокаталитического синтеза β‑лактамного антибиотика для разработки экономически выгодного процесса должна быть направлена на достижение высокого выхода продукта трансформации при использовании возможно высокой начальной концентрации КА ($C_{{{\text{KA}}}}^{{\text{o}}}$) и возможно низкого мольного избытка АА над КА $\left( {{{X}^{{\text{o}}}} = \frac{{C_{{{\text{AA}}}}^{{\text{o}}}}}{{C_{{{\text{KA}}}}^{{\text{o}}}}},\,\,{{\text{M}} \mathord{\left/ {\vphantom {{\text{M}} {\text{M}}}} \right. \kern-0em} {\text{M}}}} \right)$ [2]. Это обеспечивает достижение высокой концентрации целевого продукта (CEZ или S-p CEZ) в конечной реакционной смеси при минимизации остаточных концентраций субстратов (TDA или 7-ACA и METzAA) и побочного продукта (TzAA), что необходимо для разработки эффективной процедуры выделения антибиотика.
Одним из ферментов, высокоспецифичных к синтезу CEZ и других цефалоспоринов-кислот, является синтетаза цефалоспоринов-кислот (Cephalosporin-acid synthetase, CASA КФ 3.5.1.11), называемая также цефазолин синтетазой [2]. Фермент синтезируется штаммами Escherichia coli, которые были получены методами химического мутагенеза, такими как штаммы 1787 (Коллекция культур Государственного научного центра по антибиотикам, Россия) [15–17], FU-99-S [18], ВКПМ B-10182 [19, 20] и CGMCC No. 3508 [19]. Путем клонирования гена CASA, выделенного из штамма ВКПМ B-10182, были получены штаммы E. coli ВКПМ B-12206 [20, 21] и ВКПМ B-12316 [3], являющиеся высокоэффективными продуцентами рекомбинантной CASA. Ген CASA был идентифицирован как прямой гомолог гена пенициллин G ацилазы (Рenicillin G acylase, PGA) из штамма E. coli ATCC 9637, несущего множественные мутации, способствующие повышению уровня синтеза фермента и его синтетазной активности [20].
В данной работе в качестве биокатализатора был использован фермент CASA из рекомбинантного штамма E. coli ВКПМ B-12316, иммобилизованный путем ковалентного связывания с эпокси-активированным макропористым носителем [3] (Immobilized Enzyme CASA – IECASA).
В обзоре, посвященном синтезу цефалоспоринов-кислот [2], были суммированы предыдущие исследования по биокаталитическому получению CEZ, катализируемому PGA или CASA из штаммов E. coli [15–18, 22–24]. Высокий выход 98% был достигнут при трансформации 7-ACA в полупродукт S-p СЕZ путем биокаталитического ацилирования с помощью METzAA, но лишь при низкой начальной концентрации 7-ACA ($C_{{{\text{KA}}}}^{{\text{o}}}$ = = 50 мМ) и трехкратном мольном избытке METzAA над 7-ACA ${{X}^{{\text{o}}}}$ = 3 M/M. Биокатализатором служила PGA, иммобилизованная на глиоксилагарозе [23, 24]. При биокаталитической трансформации TDA в CEZ в водной среде с использованием в качестве биокатализатора CASA из мутантного штамма E. coli FU-99-S, иммобилизованной в полиакриламидном геле [2, 18], или CASA из рекомбинантного штамма E. coli ВКПМ B-12206, ковалентно связанной с макропористым носителем [2, 21], был достигнут выход CEZ более 90%, однако лишь при относительно низкой начальной концентрации TDA ($C_{{{\text{KA}}}}^{{\text{o}}}$ = 60–80 мМ) и высоком мольном избытке METzAA над TDA (${{X}^{{\text{o}}}}$ ≥ 4 M/M).
Ранее авторами было проведено исследование влияния рН и исходных концентраций TDA и METzAA на эффективность кинетически-контролируемого синтеза CEZ, учитывающее рН-зависимости растворимости всех компонентов реакционной смеси [3]. На основе полученных данных был разработан способ биокаталитического синтеза CEZ в водной среде при регулируемом ступенчатом градиенте рН, делающий возможным осуществление процесса при высокой исходной концентрации TDA ($C_{{{\text{KA}}}}^{{\text{o}}}$ = 150–200 мМ) и обеспечивающий выход CEZ 92–95% при ${{X}^{{\text{o}}}}$ ≤ 3.5 М/М. Использованный при этом биокатализатор, полученный нами путем иммобилизации рекомбинантной CASA из штамма E. coli ВКПМ B-12316 на макропористом носителе, продемонстрировал хорошую стабильность в ходе операционных испытаний, осуществленных в Сычуаньском индустриальном институте антибиотиков (Китай) [3].
Цель работы – разработка способа получения CEZ, включающего биокаталитический синтез полупродукта из 7-ACA методом ацильного переноса и сопоставление эффективности двух альтернативных методов получения антибиотика при использовании биокатализатора IECASA.
МЕТОДИКА
Материалы. Образцы METzAA (чистота 98%, определена методом высокоэффективной жидкостной хроматографии, ВЭЖХ) и MMTD, (чистота 97%, ВЭЖХ) предоставлены Сычуаньским индустриальным институтом антибиотиков (Китай). Остальные использованные реагенты: 7-ACA (“Anhui BBCA Pharmaceutical”, Китай, чистота 97%, ВЭЖХ); натриевая соль CEZ (“Sigma-Aldrich”, США стандарт, чистота 98%, ВЭЖХ); TzAA (“Sigma-Aldrich” США стандарт, чистота 99.9%, ВЭЖХ). Образец S-p CEZ с ВЭЖХ чистотой 94.8%, использовавшийся в качестве стандарта при анализах методом ВЭЖХ, а также для изучения растворимости, был выделен осаждением при рН 2.0 из реакционной смеси, полученной путем биокаталитического синтеза с последующей очисткой методом перекристаллизации.
Получение биокатализатора IECASA. Использованный в работе образец IECASA был получен путем выделения CASA из биомассы клеток штамма E. coli VKPM B-12316 и иммобилизации фермента на макропористом эпокси-активированном носителе Seplite LX-1000EP (“Sunresin New Materials”, Китай) как это описано в работе [3]. Синтетазная активность IECASA, определенная по синтезу CEZ [3], составила 420 ME/г влажного биокатализатора при содержании сухих веществ 37.2%.
За 1 международную единицу ферментативной (синтетазной) активности образца в реакции синтеза цефазолина (1 МЕ) принимали количество фермента, катализирующее получение 1 мкмоль продукта за 1 мин в растворе, содержащем 60 мМ TDA и 240 мМ METzAA при рН 7.5 и 30°С.
Анализ методом ВЭЖХ. Анализы проб методом ВЭЖХ осуществляли в изократическом режиме с использованием хроматографа фирмы Gilson (США), оснащенного УФ-детектором, на колонке Spherisorb ODS, 250 × 4 мм с размером частиц 7.5 мкм, при температуре 30°С, скорости потока 1.0 мл/мин и детекции при 214 или 254 нм. В качестве мобильной фазы использовали смесь 50 мМ фосфатно-аммонийного буфера с метанолом. Условия анализа различных реакционных смесей методом ВЭЖХ и времена удерживания компонентов (RT) приведены в табл. 1.
Таблица 1.
Условия анализа реакционных смесей методом ВЭЖХ
| Реакционная смесь | Компонент | Мобильная фаза | Детекция, нм | RT, мин | |
|---|---|---|---|---|---|
| pH буфера | Содержание метанола, % (об./об.) | ||||
| Биокаталитический синтез S-p CEZ | 7-ACA | 2.1 | 21 | 254 | 3.2–3.5 |
| S-p CEZ | 5.5–6.5 | ||||
| TzAA | 1 | 214 | 3.0–3.3 | ||
| METzAA | 10–11 | ||||
| Модификация S-p CEZ с образованием CEZ | 7-ACA | 2.1 | 28 | 254 | 2.8–3.0 |
| S-p CEZ | 3.9–4.3 | ||||
| MMTD | 5.3–5.7 | ||||
| CEZ | 9.0–9.5 | ||||
| Биокаталитический гидролиз S-p CEZ | 7-ACA | 2.1 | 21 | 254 | 3.2–3.5 |
| Биокаталитический гидролиз CEZ | TDA | 4.0 | 24 | 254 | 6.8–7.2 |
| Растворимость 7‑ACA | 7-ACA | 2.1 | 21 | 254 | 3.2–3.5 |
| Растворимость S-p CEZ | S-p CEZ | 2.1 | 21 | 254 | 5.5–6.5 |
| Растворимость CEZ | CEZ | 4.0 | 28 | 254 | 6.0–6.5 |
Проведение биокаталитического синтеза S-p CEZ. Синтез S-p CEZ осуществляли в стеклянном реакторе, оснащенном механической лопастной мешалкой и системами контроля и поддержания рН и температуры, при начальном объеме реакционной смеси 75 мл. Исходный раствор субстратов (7-АСА и METzAA) с выбранными начальными концентрациями ($C_{{{\text{KA}}}}^{{\text{o}}}$ и $C_{{{\text{AA}}}}^{{\text{o}}}$) готовили при 30°С и постоянном перемешивании. Аликвоту 7-АСА суспрендировали в 0.3 М фосфатно-натриевом буфере (ФБ) с рН 8.3, суспензию перемешивали до прекращения снижения рН, затем при интенсивном перемешивании небольшими порциями добавляли 2 М NaOH вплоть до полного растворения 7-АСА при рН 6.8–7.2. В полученный раствор вносили аликвоту METzAA, перемешивали до растворения и доводили объем раствора до 75 мл 0.3 М ФБ с рН 7.0. При умеренном перемешивании при 30°С запускали процесс синтеза внесением IECASA в раствор субстратов в количестве 1.8 г влажного биокатилизатора (концентрация фермента в реакционной смеси CE = 10 МЕ/мл). В ходе синтеза рН спонтанно снижался до 6.0, а затем его поддерживали на этом уровне путем добавления 2 М NaOH до завершения процесса. Процесс останавливали, отделяя биокатализатор от реакционной смеси фильтрованием на пористом стеклянном фильтре.
Для изучения динамики синтеза S-p CEZ в пробах, отбираемых из реакционной смеси по ходу процесса, методом ВЭЖХ определяли содержание четырех компонентов: S-p CEZ, 7-АСА, METzAA и TzAA. Процесс синтеза проводили до достижения стабильного плато на зависимости текущей концентрации S-p CEZ от времени. Максимальную степень трансформации 7-АСА в S-p CEZ (максимальный выход S-p CEZ ${{\eta }^{{{\text{max}}}}},$ %) рассчитывали по формуле ${{\eta }^{{{\text{max}}}}} = \frac{{C_{{{\text{prod}}}}^{{\max }}}}{{C_{{{\text{KA}}}}^{{\text{o}}}}} \times 100,$ где $C_{{{\text{prod}}}}^{{\max }}$ – максимальная концентрация целевого продукта (средние данные на плато); $C_{{{\text{KA}}}}^{{\text{o}}}$ – начальная концентрация КА.
Определение бимолекулярных констант реакций гидролиза CEZ и S-p CEZ. Процессы гидролиза CEZ или S-p CEZ, катализируемые IECASA, проводили в конических колбах, помещенных в водяной шейкер-инкубатор, при постоянном перемешивании при 30°С в 0.3 М ФБ, рН 6.5, варьируя начальные концентрации гидролизуемого β‑лактамного соединения (С o, мМ) в пределах от 5 до 30 мМ. Каждая реакционная смесь, объемом 10 мл содержала 30–40 мг влажного биокатализатора с известным содержанием сухих веществ. Текущую концентрацию образующейся аминокислоты (TDA или 7-ACA) в реакционной смеси определяли методом ВЭЖХ, отбирая пробы каждые 3–5 мин. По линейному участку кривой накопления продукта определяли начальную скорость гидролиза CEZ или S-p CEZ в пересчете на 1 г сухого биокатализатора (V, мМ мин–1 г–1). Результаты, согласно методу Лайнувера–Берка [25], представляли в виде зависимости 1/V от 1/C o, полученные прямые обрабатывали в программе Exсel и рассчитывали бимолекулярные константы гидролитического процесса, отнесенные к 1 г сухого биокатализатора (Vmax/Km, мин–1 г–1), где Km (мМ) – константа Михаэлиса, Vmax (мМ мин–1 г–1) – максимальная скорость ферментативной реакции.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Растворимость соединений, вовлеченных в процессы биокаталитического синтеза CEZ и S-p CEZ. В реакционной смеси, получаемой при биокаталитическом синтезе β‑лактамного антибиотика методом ацильного переноса, присутствуют четыре компонента: целевой продукт (CEZ или S-p CEZ), КА (TDA или 7-ACA), АА (METzAA) и побочный продукт (TzAA). Учет их растворимости является важным инструментом оптимизации процессов биокаталитической трансформации и последующего выделения целевого продукта. [3, 26]. Имеющиеся в литературе данные о растворимости указанных выше соединений, а также результаты изучения растворимости 7-ACA, S-p CEZ и CEZ, полученные в настоящей работе, приведены в табл. 2.
Таблица 2.
Кислотно-основные свойства и растворимость соединений, вовлеченных в процессы получения CEZ
| Соединение | Структура | Электрохимическая природа | Константы ионизации | Характеристическая растворимость, мМ* | Условия определения, ссылка | |
|---|---|---|---|---|---|---|
| pK1 | pK2 | |||||
| METzAA | 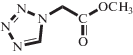 |
Неэлектролит | _ | _ | 402 | 20°C, H2O [15] |
| 1440 | 30°C, 0.3 M ФБ, pH 6.5 [3] | |||||
| TzAA | 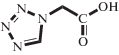 |
Электролит, монокарбоновая кислота | 2.2 | _ | 616 | 20°C, H2O [15] |
| CEZ | 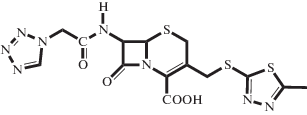 |
Электролит, монокарбоновая кислота | 2.38 | _ | 0.66 | 20°C, H2O [15] |
| 2.33 ± 0.05 | _ | 0.37 ± 0.2 | 30°C, 0.3 M ФБ (настоящая работа) | |||
| S-p CEZ | 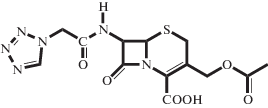 |
Электролит, монокарбоновая кислота | 2.16 ± 0.05 | _ | 32.6 ± 1.6 | 30°C, 0.3 M PhNaB (настоящая работа) |
| TDA | 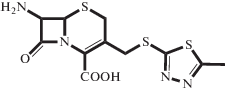 |
Электролит, аминокислота | 2.46 | 5.04 | 0.29 | 20°C, H2O [15] |
| Н/о** | 5.21 | 0.28 | 30°C, 0.3 M ФБ [3] | |||
| 7-ACA | 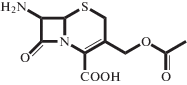 |
Электролит, аминокислота | 2.64 | 4.83 | 2.64 | 20°C, 0.1 М NaCl [28] |
| Н/о** | 4.70 ± 0.05 | 2.79 ± 0.14 | 30°C, 0.3 M ФБ (настоящая работа) | |||
Ранее [3] в условиях, моделирующих процесс синтеза CEZ, были изучены растворимость неэлектролита METzAA, рН-зависимость растворимости аминокислоты TDA и продемонстрирован эффект перенасыщения раствора TDA при снижении рН. Полученные данные позволили разработать эффективный метод биокаталитического синтеза CEZ в ступенчатом градиенте рН с использованием исходной концентрации TDA ($C_{{{\text{KA}}}}^{{\text{o}}}$ = 150–200 мМ), многократно превосходящей ее растворимость при нейтральных рН, оптимальных для функционирования биокатализатора.
В настоящем исследовании были изучены рН-зависимости растворимости таких компонентов реакционной смеси синтеза S-p CEZ, как КА (7-ACA) и целевой продукт S-p CEZ, а также антибиотика CEZ, получаемого в той же реакционной смеси без выделения полупродукта путем замещения в нем 3-ацетокси-группы с помощью MMTD. Влияние pH на растворимость 7-АСА, S-p CEZ и CEZ изучали методом насыщения [26, 27] в условиях, подобранных ранее для биокаталитического синтеза CEZ (0.3 М ФБ, 30°С) [3]. Методика проведения эксперимента описана в работах [3, 26].
Зависимость от рН растворимости (S, М) монокарбоновых кислот S-p CEZ и CEZ, а также TzAA описывается уравнением (1):
(1)
$S = {{S}^{{\text{o}}}} + \frac{{{{S}^{{\text{o}}}}{{K}_{1}}}}{{\left[ {{{{\text{H}}}^{ + }}} \right]}}.$Зависимость от рН растворимости (S, мМ) аминокислот (7-ACA и TDA), имеющих по две ионогенные группы, описывается уравнением (2):
(2)
$S = {{S}^{ \pm }}\frac{{{{{\left[ {{{{\text{H}}}^{ + }}} \right]}}^{{\text{2}}}} + {{K}_{1}}\left[ {{{{\text{H}}}^{ + }}} \right] + {{K}_{1}}{{K}_{2}}}}{{{{K}_{1}}\left[ {{{{\text{H}}}^{ + }}} \right]}}.$В условиях полного протонирования карбоксильной группы аминокислоты, то есть при нейтральных и щелочных значениях рН, применимо уравнение (2.1):
(2.1)
$S = {{S}^{ \pm }} + {{S}^{ \pm }}{{K}_{2}}\frac{{\text{1}}}{{\left[ {{{{\text{H}}}^{ + }}} \right]}}.$[H+] – концентрация ионов водорода при заданном рН, мМ;
K1 – константа ионизации карбоксильной группа монокарбоновой кислоты или аминокислоты, мM;
K2 – константа ионизации аминогруппы аминокислоты, мM;
S o – растворимость индивидуальной незаряженной формы монокарбоновой кислоты (характеристическая растворимость электролита), мM;
S± – растворимость индивидуальной электро-нейтральной цвиттерионной формы аминокислоты (характеристическая растворимость электролита), мM.
Использование приведенных выше уравнений позволяло рассчитывать константы, определяющие растворимость электролитов [26].
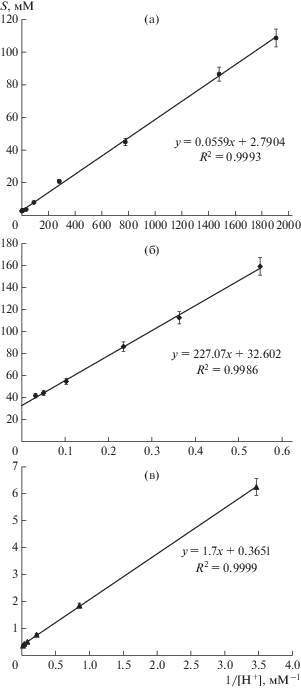
На рис. 2а показана линеаризация в координатах уравнения (2.1) (S vs. 1/[H+]) экспериментальных данных, полученных при изучении влияния рН на растворимость аминокислоты 7-ACA в области рН, близкой к значению pK2, известному из литературных данных (табл. 2) [28]. Линеаризация экспериментальных данных, полученных при изучении влияния рН на растворимость монокарбоновых кислот S-p CEZ и CEZ в кислой области рН, представлена на рис. 2б и 2в соответственно. Рассчитанные значения характеристической растворимости (S± или So) и констант ионизации аминогруппы аминокислоты (pK2) или карбоксильной группы монокарбоновых кислот (pK1) сопоставлены с опубликованными ранее данными в табл. 2. Величины pK2 и S± для 7-ACA, определенные нами в 0.3 М ФБ при 30°С, практически совпадали с константами, определенными в 0.1 М NaCl при 20°С [28]. Сопоставление констант, определенных для 7-ACA и TDA в одинаковых условиях (0.3 М ФБ, 30°С), показало, что замена 3‑ацетокси-группы на MMTD приводило к смещению величины рK2, характеризующей ионизацию аминогруппы в С7 положении β-лактама, на 0.5 единиц в щелочную область и к 10-кратному уменьшению S±. Это влечет за собой существенное снижение растворимости аминокислоты в диапазоне рН 6.0–8.0, используемом для осуществления биокаталитического синтеза. В случае монокарбоновых кислот S-p CEZ и CEZ замена 3-ацетокси-группы на MMTD также снижала характеристическую растворимость в 10 раз. При этом величина рK1, характеризующая ионизацию карбоксильной группы в С4 положении β-лактама, незначительно смещалась в щелочную область (на 0.17).
Рис. 2.
Зависимость растворимости КА и продуктов биокаталитического синтеза от рН (30°C, 0.3 M ФБ): а – 7-ACA – в координатах уравнения (2.1); б – S-p CEZ, в – CEZ в координатах уравнения (1).
Сопоставление констант, определенных для CEZ в воде при 20°С [15] и в 0.3 М ФБ при 30°С, свидетельствовало о том, что увеличение ионной силы среды и повышение температуры не влияло на величину pK1, но почти вдвое снижало характеристическую растворимость S o.
На рис. 3 показаны рассчитанные с использованием констант, представленных в табл. 2, теоретические кривые зависимости растворимости компонентов реакционных смесей, получаемых при биокаталитическом синтезе CEZ и S-p CEZ, от рН.
Рис. 3.
Теоретические кривые зависимости растворимости от рН, рассчитанные с использованием констант в табл. 2: 1 – TDA, 2 – 7-ACA (30°C, 0.3 M ФБ), уравнение (2); 3 – CEZ; 4 – S-p CEZ (30°C, 0,3 M ФБ) уравнение (1); 5 – TzAA (20°C, вода), уравнение (1); 6 – METzAA (30°C, 0.3 M ФБ).
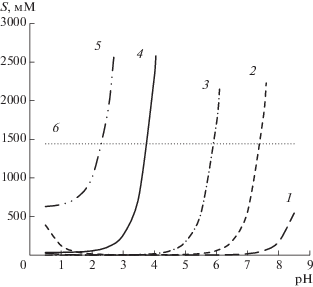
Растворимость METzAA, не являющегося электролитом, не зависела от рН. Ранее было установлено [3], что в 0.3 М ФБ при 30°С она составляла 1440 мМ (табл. 2), что позволяло использовать METzAA в высоких концентрациях как АА при биокаталитическом синтезе. В случае TzAA низкое значение рK1 и высокое значение So (табл. 2) уже при рН 2.7 обеспечивали высокую растворимость S = 2560 мМ (рис. 3, 5). В области рН 6.0–8.0, где протекают реакции, катализируемые IECASA [3], растворимость этого побочного продукта процессов синтеза с ацильным переносом столь высока, что его выпадение в осадок не может осложнять процесс при доступных исходных концентрациях субстратов.
При биокаталитическом синтезе CEZ низкая растворимость TDA в рабочем диапазоне рН 6.0–8.0 (рис. 3, кривая 1) являлась основным осложняющим фактором для приготовления исходной реакционной смеси и осуществления процесса при высокой исходной концентрации КА ($C_{{{\text{KA}}}}^{{\text{o}}}$) без выпадения осадка в ходе синтеза [3]. В диапазоне рН 6.0–8.0 растворимость 7-ACA примерно в 32 раза превосходила растворимость TDA (рис. 3, кривые 1, 2), что обеспечивало существенные преимущества процесса биокаталитического синтеза S-p CEZ по сравнению с синтезом CEZ, как на стадии приготовления исходной реакционной смеси, так и при проведении биокаталитической трансформации.
Кривая, описывающая зависимость растворимости S-p CEZ от рН, сдвинута в сторону низких значений рН по сравнению с соответствующей кривой для CEZ (рис. 3, кривые 3 и 4), а уровень значений S для полупродукта более чем в 130 раз превосходит растворимость CEZ при том же рН. Например, при рН 4.0 растворимость S-p CEZ и CEZ составляла 2300 мМ и 17 мМ соответственно. В рабочем диапазоне рН 6.0–8.0 оба продукта прекрасно растворимы, что обеспечивало их сохранение в растворе в процессе биокаталитической трансформации. Растворимость продуктов в области низких рН важна для разработки метода выделения их из конечной реакционной смеси путем осаждения. Относительно высокая растворимость S-p CEZ при рН около 2.0 была негативным фактором, так как влекла за собой потери продукта при осаждении. В этой связи целесообразна разработка процесса химической трансформации S-p CEZ в CEZ непосредственно в конечной реакционной смеси после биокаталитической трансформации без выделения полупродукта.
Биокаталитический синтез полупродукта CEZ, катализируемый IECASA. Биокаталитическую трансформацию 7-ACA в S-p CEZ осуществляли в 0.3 М ФБ при 30°С, CE = 10 МЕ/мл, варьируя $C_{{{\text{KA}}}}^{{\text{o}}}$ в диапазоне 55–320 мМ и ${{X}^{{\text{o}}}}$ в диапазоне 1.5–3.5 М/М при $C_{{{\text{AA}}}}^{{\text{o}}}$ не более 830 мМ, что не превышало растворимость METzAA в условиях опыта (табл. 2, рис. 3, кривая 6). Высокая растворимость 7-ACA при рН близких к нейтральному (рис. 3, кривая 2), позволила во всем использованном диапазоне $C_{{{\text{KA}}}}^{{\text{o}}}$ растворять ее при рН ~ 7.0. Это являлось существенным преимуществом перед процедурой приготовления раствора КА для синтеза CEZ [3], когда из-за низкой растворимости TDA ее необходимо растворять при высоком рН (вплоть до 8.5), неблагоприятном для стабильности β-лактама, при этом удавалось достичь $C_{{{\text{KA}}}}^{{\text{o}}}$ не более 200 мМ. Начинать синтез CEZ приходилось также при рН 8.2–8.3, неблагоприятном для активности и стабильности IECASA [3].
Синтез S-p CEZ начинали при рН 6.8–7.2 после последовательного внесения METzAA и биокатализатора в раствор 7-ACA и проводили при спонтанно снижающемся рН с 7.0 до 6.0, т.е. в условиях, оптимальных для функционирования и стабильности IECASA. Далее поддерживали рН 6.0 до завершения трансформации. Снижение рН по ходу процесса синтеза β-лактама обусловлено образованием свободной TzAA – продукта побочных гидролитических реакций, в первую очередь METzAA (рис. 1). Снижение рН может сопровождаться выпадением в осадок неацилированной КА, если ее остаточная концентрация, превысит растворимость данного соединения при текущем рН, как это наблюдалось при трансформации слабо растворимой TDA в CEZ в спонтанном градиенте рН от 8.2–8.3 до 6.0 [3]. Критическое значение рН, при котором происходило выпадение КА, зависит от констант ионизации аминокислоты, определяющих рН-зависимость ее растворимости (табл. 2), а также от операционных параметров $C_{{{\text{KA}}}}^{{\text{o}}}$ и ${{X}^{{\text{o}}}},$ определяющих при заданных температуре и CE степень трансформации КА в целевой β-лактам к моменту достижения критического рН. Для осуществления процесса синтеза CEZ был разработан ступенчатый градиент рН, при котором на фоне самопроизвольного снижения рН реакционной смеси от начального значения 8.2–8.3 последовательно проводили его искусственное поддержание на уровнях 6.8 и 6.0, что обеспечивало сохранение TDA в растворе [3].
В настоящем исследовании трансформацию 7‑ACA в S-p CEZ в диапазоне рН от 7.0 до 6.0 осуществляли в спонтанном градиенте, и при этом процесс не был осложнен выпадением КА даже при высоком избытке METzAA (${{X}^{{\text{o}}}}$ = 3.0 М/М), способствующем интенсивному выделению TzAA, в сочетании с $C_{{{\text{KA}}}}^{{\text{o}}}$ вплоть до 275 мМ. Растворимость 7-ACA закономерно падала при снижении рН (рис. 3, кривая 2) до S = 60 мМ при рН 6.0, однако в выбранных операционных условиях ее уровень во всем рабочем диапазоне рН был достаточно высок для сохранения нетрансформированной КА в растворе. Снижение избытка АА до ${{X}^{{\text{o}}}}$ = 2.5 М/М позволяло осуществить синтез S-p CEZ в спонтанном градиенте рН и при более высокой начальной концентрации 7-ACA $C_{{{\text{KA}}}}^{{\text{o}}}$ = 320 мМ.
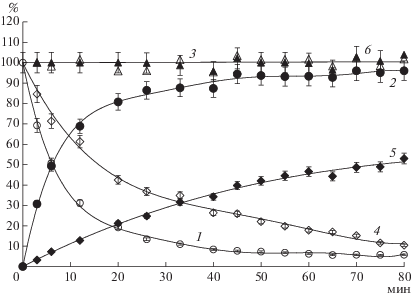
На рис. 4 показана зависимость от времени состава реакционной смеси, образующейся при кинетически-контролируемом синтезе S-p CEZ, катализируемом IECASA, а именно, динамика расхода КА 7-ACA и АА METzAA, а также динамика накопления целевого β-лактама S-p CEZ и побочного продукта TzAA. При этом динамика относительной концентрации S-p CEZ отражает зависимость выхода целевого продукта синтеза от времени по отношению к содержащему β-лактам субстрату (7-ACA). Важной особенностью процесса является наличие продолжительного плато на кривой накопления S-p CEZ, когда достигается максимальный выход продукта (${{\eta }^{{{\text{max}}}}}$). Как и в случае синтеза CEZ, катализируемого IECASA [3], наличие плато объясняется установлением продолжительного кинетического равновесия между процессами синтеза и гидролиза β-лактамного продукта. Сохранение 100%-ного баланса как по β-лактам-, так и по тетразолил-содержащим компонентам (рис. 4) свидетельствовало об отсутствии в системе неконтролируемых побочных процессов, в том числе связанных с разрушением β-лактама.
Рис. 4.
Изменение состава реакционной смеси (относительные концентрации, %) от времени при синтезе S-p CEZ, катализируемом IECASA (30°C, 0.3 M ФБ, CE = 10 МЕ/мл, $C_{{{\text{KA}}}}^{{\text{o}}}$ = 320 мМ, $C_{{{\text{AA}}}}^{{\text{o}}}$ = 800 мМ, ${{X}^{{\text{o}}}}$ = 2.5 М/М). 1 – 7‑ACA; 2 – S-p CEZ, рассчитаны по отношению к $C_{{{\text{KA}}}}^{{\text{o}}};$ 3 – баланс (%) по β-лактаму, сумма относительных концентраций 7‑ACA и S-p CEZ; 4 – METzAA; 5 – TzAA, рассчитаны по отношению к $C_{{{\text{AA}}}}^{{\text{o}}};$ 6 – баланс (%) по TzAA – сумма относительных концентраций S-p CEZ, METzAA и TzAA.
С целью оптимизации процесса синтеза S-p CEZ, катализируемого IECASA, была осуществлена серия экспериментов при различных $C_{{{\text{KA}}}}^{{\text{o}}}$ (55–320 мМ) и ${{X}^{{\text{o}}}}$ (1.5–3.5 М/М) в стандартных условиях, выбранных ранее [3] для синтеза CEZ (30°С, CE = 10–12 МЕ/мл, 0.3 М ФБ). Был использован спонтанный градиент рН от рН 7.0 ± 0.2 до рН 6.0, с последующим поддержанием рН 6.0 ± 0.1 с помощью добавления 2 М раствора NaOH до завершения процесса. Эффективность процесса характеризовали величиной максимального выхода S-p CEZ (${{\eta }^{{{\text{max}}}}},$ %), рассчитанного как среднее значение на плато накопления целевого продукта. Время достижения максимальной концентрации S-p CEZ в реакционной смеси составляло 25–50 мин, в зависимости от операционных условий.
Полученные результаты по синтезу S-p CEZ, катализируемому IECASA, обобщены в виде зависимостей максимального выхода продукта от $C_{{{\text{KA}}}}^{{\text{o}}}$ (рис. 5, кривые 1, 1а) и от ${{X}^{{\text{o}}}}$ (рис. 6, кривая 1), а также сопоставлены с полученными ранее результатами по биокаталитическому синтезу CEZ [3] (рис. 5, кривые 2, 3; рис. 6, кривые 2, 3). Осуществление синтеза CEZ в спонтанном градиенте рН (рис. 5, кривая 3; рис. 6, кривая 3) позволило достичь $\eta _{{{\text{CEZ}}}}^{{{\text{max}}}}$ = (92 ± 2)%, но только при $C_{{{\text{KA}}}}^{{\text{o}}}$ = = 100–140 мМ и ${{X}^{{\text{o}}}}$ = 3.1–3.5 М/М (рис. 5, кривая 3). Повышение исходной концентрации TDA до $C_{{{\text{KA}}}}^{{\text{o}}}$ ≥ 145 мМ приводило к падению выхода CEZ до 85% и ниже (рис. 6, кривая 3) из-за выпадения осадка TDA при снижении рН в ходе процесса. Выпадение TDA в осадок удавалось предотвратить при осуществлении синтеза CEZ в ступенчатом градиенте рН (рис. 5, кривая 2; рис. 6, кривая 2). При этом в диапазоне концентраций TDA $C_{{{\text{KA}}}}^{{\text{o}}}$ = 150–200 мМ при ${{X}^{{\text{o}}}}$ = 3.1–3.5 М/М достигался выход ${{\eta }^{{{\text{max}}}}}$ = (93.5 ± 1.5)%. Эти условия могут быть приняты как оптимальные для синтеза CEZ, катализируемого IECASA.
Рис. 5.
Зависимость максимального выхода β-лактамного продукта (${{\eta }^{{{\text{max}}}}},$ %) от исходной концентрации КА ($C_{{{\text{KA}}}}^{{\text{o}}},$ мМ) при синтезе с ацильным переносом, катализируемом IECASA (30°C, 0.3 M ФБ, CE = = 10–12 МЕ/мл): 1 – синтез S-p CEZ из 7-ACA и METzAA; спонтанный градиент рН в диапазоне от 7.0 ± 0.2 до 6.0, далее поддержание рН 6.0 ± 0.1; ${{X}^{{\text{o}}}}$ = = 2.9–3.1 М/М; 1а – то же при ${{X}^{{\text{o}}}}$ = 2.5 М/М; 2 – синтез CEZ из TDA и METzAA при ${{X}^{{\text{o}}}}$ = 3.1–3.5 М/М, ступенчатый градиент рН от 8.2–8.3 с поддержанием рН 6.8 ± 0.1 и рН 6.0 ± 0.1; 3 – синтез CEZ из TDA и METzAA при ${{X}^{{\text{o}}}}$ = 3.1–3.5 М/М, спонтанный градиент рН от 8.2–8.3 до 6.0, далее поддержание рН 6.0 ± 0.1.
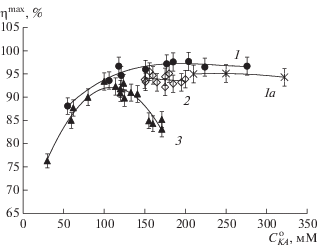
Рис. 6.
Зависимость максимального выхода β-лактамного продукта (${{\eta }^{{{\text{max}}}}},$ %) от исходного мольного избытка АА над КА (${{X}^{{\text{o}}}},$ М/М) при синтезе с ацильным переносом, катализируемом IECASA при 30°C, 0.3 M ФБ, CE = 10 МЕ/мл: 1 – синтез S-p CEZ из 7‑ACA и METzAA при $C_{{{\text{KA}}}}^{{\text{o}}}$ = 180–330 мМ; спонтанный градиент рН в диапазоне от 7.0 ± 0.2 до 6.0, далее поддержание рН 6.0 ± 0.1; 2 – синтез CEZ из TDA и METzAA; $C_{{{\text{KA}}}}^{{\text{o}}}$ = 150–200 мМ; ступенчатый градиент рН от 8.2–8.3 до 6.0 с поддержанием уровней рН 6.8 ± ± 0.1 и рН 6.0 ± 0.1; 3 – то же при $C_{{{\text{KA}}}}^{{\text{o}}}$ = 145–170 мМ; спонтанный градиент рН от 8.2–8.3 до 6.0, далее поддержание рН 6.0 ± 0.1.
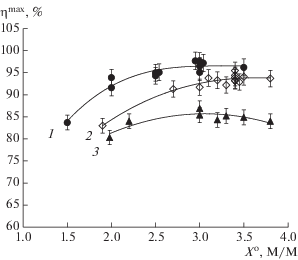
S-p CEZ удавалось синтезировать с более высоким выходом ${{\eta }^{{{\text{max}}}}}$ = (96.5 ± 1.5)% в спонтанном градиенте рН в пределах концентраций 7-ACA $C_{{{\text{KA}}}}^{{\text{o}}}$ = 175–275 мМ при ${{X}^{{\text{o}}}}$ = 3.0 М/М ( рис. 5, кривая 1; рис. 6, кривая 1). Существенно, что при высоких концентрациях 7-ACA кривая зависимости ${{\eta }^{{{\text{max}}}}}$ от ${{X}^{{\text{o}}}}$ (рис. 6, кривая 1) выходила на плато при ${{X}^{{\text{o}}}}$ = 3.0 М/М, а снижение мольного избытка до ${{X}^{{\text{o}}}}$ = 2.5 М/М сопровождалось незначительным падением выхода до ${{\eta }^{{{\text{max}}}}}$ = (95.0 ± 1.5)% (рис. 5, кривая 1а).
Увеличение выхода в процессе кинетически-контролируемого синтеза, катализируемого IECAS-A, при замене TDA на 7-ACA являлось, в частности, результатом меньшей специфичности фермента к побочной реакции гидролиза целевого β-лактама (S-p CEZ по сравнению CEZ). Специфичность IECASA к гидролизу CEZ и S-p CEZ оценивали по бимолекулярным константам соответствующих процессов, определенным по методу Лайнувера–Берка (рис. 7). Бимолекулярные константы гидролитических процессов (Vmax/Km, мин–1 г–1), рассчитанные по представленным прямым, составляли 0.24 ± 0.01 мин–1 г–1 и 0.18 ± 0.01 мин–1 г–1 для CEZ и S-p CEZ соответственно, т.е. целевой продукт кинетически-контролируемого синтеза S-p CEZ гидролизовался IECASA на 30% менее эффективно, чем продукт синтеза CEZ.
Рис. 7.
Определение бимолекулярных констант гидролиза CEZ (1) и S-p CEZ (2), катализируемого IECASA при 30°С и 0.3 М ФБ, методом Лайнувера–Берка.
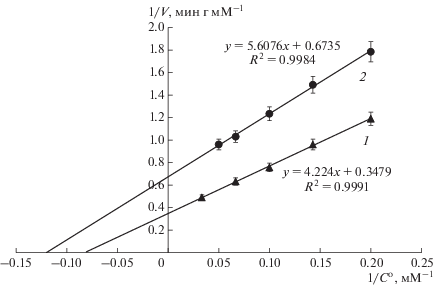
Оптимизация процесса синтеза β-лактама, катализируемого IECASA, была направлена на достижение максимального выхода целевого продукта при максимальной допустимой величине $C_{{{\text{KA}}}}^{{\text{o}}}$ и минимальной допустимой величине ${{X}^{{\text{o}}}},$ что обеспечивало высокую концентрацию целевого продукта в реакционной смеси после трансформации при минимизации содержания примесей (остаточная КА и TzAA – продукт побочных гидролитических процессов). В табл. 3 результаты ряда экспериментов по синтезу CEZ и S-p CEZ в различных условиях сопоставлены по параметрам эффективности процесса, таким как максимальный выход продукта по отношению к β-лактамному субстрату (${{\eta }^{{{\text{max}}}}},$ %) и состав конечной реакционной смеси после трансформации. В конечной реакционной смеси определяли концентрации целевого продукта (CEZ или S-p CEZ) – Сprod (мМ), КА (TDA или 7-ACA) – СКА (мМ) и суммарную концентрацию АА и побочного продукта (METzAA и TzAA) – СME + TzAA (мМ). Последние два компонента учитываются суммарно, так как перед выделением целевого продукта остаточный METzAA легко может быть гидролизован до TzAA.
Таблица 3.
Параметры эффективности процессов синтеза CEZ и S-p CEZ, катализируемых IECASA
| Синтезируемый β-лактам | Условия процесса | $\eta _{{}}^{{{\text{max}}}}{\text{,}}$ % | Конечная реакционная смесь | |||||
|---|---|---|---|---|---|---|---|---|
| Исходный рН | Градиент рН | $C_{{{\text{KA}}}}^{{\text{o}}}{\text{,}}$ мМ | ${{X}^{{\text{o}}}}{\text{,}}$ М/М | Сprod, мМ | СКА, мМ | СME + TzAA, мМ | ||
| CEZ | 8.3 | Ступенчатый | 200 | 3.4 | 93.9 | 190 | 12 | 490 |
| S‑p CEZ | 7.05 | Спонтанный | 204 | 3.0 | 97.7 | 200 | 5 | 415 |
| 7.20 | Спонтанный | 276 | 3.0 | 95.7 | 265 | 12 | 565 | |
| 7.20 | Спонтанный | 322 | 2.5 | 94.5 | 305 | 18 | 500 | |
Осуществление синтеза CEZ в оптимальных условиях с использованием ступенчатого градиента рН позволило при ${{X}^{{\text{o}}}}$ = 3.4 М/М, исходя из максимально допустимой $C_{{{\text{KA}}}}^{{\text{o}}}$ = 200 мМ (табл. 3), осуществить трансформацию с выходом ${{\eta }^{{{\text{max}}}}}$ = 93.9% и достичь в реакционной смеси концентрации антибиотика Сprod = 190 мМ при СКА = 12 мМ и СME + TzAA = 490 мМ. Процесс синтеза S-p CEZ при практически такой же $C_{{{\text{KA}}}}^{{\text{o}}}$ = 204 мМ был осуществлен в более простом спонтанном градиенте рН с существенно более высоким выходом ${{\eta }^{{{\text{max}}}}}$ = 97.7%, несмотря на использование меньшего избытка ацилирующего агента ${{X}^{{\text{o}}}}$ = 3.0 М/М (табл. 3). Это обеспечило повышение концентрации целевого β-лактама в реакционной смеси до Сprod = 200 мМ при соответствующем снижении СКА и СME + TzAA до 5 и 415 мМ соответственно. Использование более высоких исходных концентраций 7-ACA $C_{{{\text{KA}}}}^{{\text{o}}}$ = 276 мМ и $C_{{{\text{KA}}}}^{{\text{o}}}$ = 322 мМ (табл. 3) позволило достичь высокого выхода S-p CEZ около 95% и увеличить концентрацию целевого продукта в реакционной смеси в 1.3–1.5 раза по сравнению с оптимальным процессом синтеза CEZ практически без увеличения остаточного содержания КА. При этом по сравнению с оптимальным процессом синтеза CEZ величина СME + TzAA возросла на 15% при ${{X}^{{\text{o}}}}$ = 3.0 М/М и лишь на 2% при ${{X}^{{\text{o}}}}$ = 2.5 М/М.
S-p CEZ может быть трансформирован в CEZ непосредственно в реакционной смеси, полученной при биокаталитическом синтезе, после отделения биокатализатора. Инкубация с MMTD, взятом в трехкратном мольном избытке по отношению к S-p CEZ, позволяла при 65°С и рН 6.0–6.5 достичь степени трансформации полупродукта в CEZ, равной 96.5%. Оптимизация процесса трансформации будет осуществлена в ходе дальнейших исследований, связанных с разработкой процесса выделения и очистки CEZ.
Сопоставление биокаталитического синтеза CEZ, исходя из TDA, и S-p CEZ, исходя из 7-ACA, (рис. 1, стадии 2 и 3) свидетельствовало о ряде важных преимуществ второго процесса:
− приготовление исходного раствора 7-ACA при нейтральном рН (вместо рН 8.2–8.5 для раствора TDA) с достижением более высокой $C_{{{\text{KA}}}}^{{\text{o}}};$
− осуществление процесса синтеза S-p CEZ в простом спонтанном градиенте рН 7.0–6.0, благоприятном для активности и стабильности фермента, вместо ступенчатого градиента рН от 8.3 до 6.0, используемого при синтезе CEZ;
− повышение выхода целевого β-лактама в оптимизированных для каждого процесса условиях с (93.5 ± 1.5)% до (96.5 ± 1.5)%;
− повышение концентрации целевого β-лактама в конечной реакционной смеси в 1.3–1.5 раза;
− возможность использования продукта биокаталитического ацилирования 7-ACA метиловым эфиром TzAA в качестве полупродукта для получения не только CEZ, но и цефалоспоринового антибиотика цефтезола.
Получение CEZ через трансформации 3 и 4 (рис. 1) может быть осуществлено как единый процесс без выделения S-p CEZ из реакционной смеси. Следует отметить также, что процесс химической трансформации S-p CEZ в CEZ (рис. 1, трансформация 4), протекающий при 30°С в водной среде, с экологической точки зрения предпочтительнее получения TDA из 7-ACA (рис. 1, трансформация 1), осуществляемого при низкой температуре (–40°С) в среде токсичных реагентов (BF3, CH3CN). Можно заключить, что химико-биокаталитический синтез CEZ по пути прямого биокаталитического ацилирования 7-ACA представляется перспективной заменой традиционному пути, использующему биокаталитическое ацилирование TDA.
Работа выполнена в рамках Государственного задания № 593-00003-19 ПР “Фундаментальные и прикладные научные работы в области биотехнологии”.
Список литературы
Ныс П.С., Курочкина В.Б., Скляренко А.В., Вейнберг Г.А. // Антибиотики и химиотерапия. 2000. Т. 45. № 11. С. 36–42.
Sklyarenko A.V., Eldarov M.A., Kurochkina V.B., Yarotsky S.V. // Appl. Biochem. Microbiol. 2015. V. 51. № 6. P. 627–640.
Wang Lu, Sklyarenko A.V., Li Duanhua, Sidorenko A.I., Zhao Chen, Li Jinjun, Yarotsky S.V. // Bioprocess Biosyst. Eng. 2018. V. 41. № 12. P. 1851–1867.
Патент США. 1995. № 5387679.
Saikawa I., Takano S., Momonoi K., Takakura I., Tanaka K., Kutani C. // Chem. Pharm. Bull. 1985. V. 33. № 12. P. 5534–5538.
Durckheimer W., Blumbach J., Lattrel R., Scheunemann K.H. // Angew. Chem. 1985. V. 24. № 3. P. 180–202.
Fernández-Lafuente R., Guisan J.M., Pregnolato M., Terreni M. // Tetrahedron Lett. 1997. V. 38. № 26. P. 4693–4696.
Elander R.P. // Appl. Microbiol. Biotechnol. 2003. V. 61. № 5–6. P. 385–392.
Barber M.S., Giesecke U., Reichert A., Minas W. // Adv. Biochem. Eng. Biotechnol. 2004. V. 88. P. 179–215.
Schmidt F.-R. The Mycota X. Industrial Applications. / Ed.: K. Esser, M. Hofrichter. Berlin, Heidelberg: Springer-Verlag, 2010. V. 5. P. 101–121.
Volpato G., Rodrigues R.C., Fernandez-Lafuente R. // Curr. Med. Chem. 2010. V. 17. № 32. P. 3855–3873.
Rajasekar V.W. // Enz. Eng. 2016. V. 5. № 1. P. 138–139.
Rodriguez-Herrera R., Puc L.E.C., Sobrevilla J.M.V., Luque D., Cardona-Felix C.S., Aguilar-González C.N., Flores-Gallegos A.C. Enzymes in the Pharmaceutical Industry for β-Lactam Antibiotic Production / Ed. M. Kuddus: Acad. Press, 2019. Ch. 36. P. 627–643.
Kurochkina V.B., Sklyarenko A.V. // Enzymatic Synthesis of beta-Lactam Antibiotics / Ed. G.E. Zaikov. New-York: Nova Science Publishers, 2008. Ch. 20. P. 175–204.
Курочкина В.Б., Ныс П.С. // Антибиотики и химиотерапия. 1999. Т. 44. № 5. С. 12–16.
Kurochkina V.B., Nys P.S. // Biocatal. Biotransform. 2002. V. 20. № 1. P. 35–41.
Nys P.S., Kurochkina V.B. // Appl. Biochem. Biotechnol. 2000. V. 88. № 1–3. C. 221–229.
Пaтeнт PФ. 2000. № 2210596.
Пaтeнт PФ. 2011. № 2420581.
Eldarov M.A., Sklyarenko A.V., Mardanov A.V., Beletsky A.V., Zhgun A.A., Dumina M.V., Medvedeva N.V., Satarova D.E., Ravin N.V., Yarockii S.V. // Appl. Biochem. Microbiol. 2015. V. 51. № 5. P. 505–510.
Эльдаров М.А., Скляренко А.В., Думина М.В., Медведева Н.В., Жгун А.А., Сатарова Д.Э., Сидоренко А.И., Епремян А.С., Яроцкий С.В. // Биомедицинская химия. 2015. Т. 61. № 5. С. 646–651.
Park C.B., Lee S.B., Ryu D.D. // J. Mol. Catal. B Enzym. 2000. V. 9. № 4–6. P. 275–281.
Fernández-Lafuente R., Guisan J.M., Pregnolato M., Terreni M. // Tetrahedron Lett. 1997. V. 38. № 26. P. 4693–4696.
Hernandez-Justiz O., Fernandez-Lafuente R., Guisan J.M., Negri P., Pagani G., Pregnolato M. // J. Org. Chem. 1997. V. 62. № 26. P. 9099–9106.
Березин И.В., Клесов А.А. // Практический курс химической и ферментативной кинетики. М.: Издательство МГУ, 1976. С. 79.
Kurochkina V.B., Sklyarenko A.V., Satarova D.E., Yarosky S.V. // Bioprocess Biosyst. Eng. 2011. V. 34. № 9. P. 1103–1117.
McDonald M.A., Bommarius A.S., Rousseau R.W. // Chem. Eng. Sci. 2017. V. 165. P. 81–88.
Булычева М.С., Ныс П.С., Савицкая Е.М. // Антибиотики. 1977. Т. 22. № 12. С. 1073–1076.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология