

Прикладная биохимия и микробиология, 2020, T. 56, № 5, стр. 436-445
Влияние комплексов железа с оксидом азота на реакционную способность цистеинов гемоглобина
О. В. Космачевская 1, *, Э. И. Насыбуллина 1, К. Б. Шумаев 1, Н. Н. Новикова 2, А. Ф. Топунов 1
1 Институт биохимии им. А.Н. Баха, Федеральный исследовательский центр
“Фундаментальные основы биотехнологии” Российской академии наук
119071 Москва, Россия
2 Национальный исследовательский центр “Курчатовский институт”
123182 Москва, Россия
* E-mail: rizobium@yandex.ru
Поступила в редакцию 12.03.2020
После доработки 17.04.2020
Принята к публикации 22.04.2020
Аннотация
Эритроцитарный гемоглобин (Hb) человека имеет два реакционноспособных остатка цистеина, расположенных на поверхности β-субъединиц. Эти остатки цистеина играют важную роль в настройке функций Hb. Известно, что они участвуют в транспорте внутриклеточного оксида азота (NO), в редокс-сигнализации, а также в регуляция равновесия димерной и тетрамерной форм Hb. В работе показано, что включение Cys-93β как лигандов в состав комплексов железа с NO является еще одним способом регуляции реакционной способности SH-групп. Такие комплексы стабилизируют SH-группы в виде тиолят-аниона (R–S–), реакционная способность которого значительно выше, чем у протонированной формы тиола (Cys-SH). Благодаря этому включенные в комплексы тиолы проявляют повышенную реакционную способность по отношению к электрофильным агентам, таким как ThioGlo1. С другой стороны, в составе комплексов тиолы оказываются защищенными от окисления гидропероксидом трет-бутила. Включение SH-групп в состав комплексов железа с NO можно рассматривать как защиту тиолов от необратимого окисления в условиях окислительного стресса.
Значение цистеинов для физиологии клетки огромно, поскольку эти аминокислоты подвержены широкому спектру посттрансляционных модификаций, многие из которых обратимы. Тиоловые группы обладают такими свойствами, как нуклеофильность, способность к обратимому окислению-восстановлению и связыванию металлов, благодаря которым они чрезвычайно чувствительны к кислотно-основному, окислительно-восстановительному и электрофильно-нуклеофильному равновесию [1–5]. Даже незначительное изменение концентрации окислителей или электрофилов отражается на состоянии белковых SH-групп. Именно поэтому цистеины играют ключевую роль в передаче редокс-сигналов в живых системах.
Критическими для функционирования белков являются SH-группы, входящие в активный центр, а также локализованные на поверхности белковой молекулы. Поэтому такие цистеины расположены в эволюционно консервативных доменах молекул [6]. Взаимодействовать с белковыми SH-группами могут различные соединения, обладающие электрофильными свойствами, к которым относятся активные формы кислорода и азота, активные карбонильные соединения, а также ионы металлов – железа, цинка, меди, кальция и др. [7]. Наиболее распространенные посттрансляционные модификации сульфгидрильных групп: окисление до дисульфидов (R–S–S–R) и цистеинсульфеновой кислоты (R–SOH), нитрозилирование (R–SNO), сульфгидрирование (R–S–SH), образование аддуктов Михаэля и гемитиоацеталей [4, 8–10]. Перечисленные посттрансляционные модификации изменяют физико-химические свойства белков, их реакционную способность и способность к агрегации.
Показано, что сульфгидрильные группы играют важную роль в системе проведения сигнала внутри клетки. Часто именно через модификацию SH-групп ферментов, рецепторов, транскрипционных факторов и других белков-регуляторов происходит интеграция различных информационных потоков для выработки оптимального ответа клетки на внешнее воздействие [4, 9–11]. Упомянутые посттрансляционные модификации, в отличие от фосфорилирования, как правило являются неферментативными. Еще одной модификацией SH-групп цистеинов является формирование динитрозильных комплексов железа (ДНКЖ) – комплексов, содержащих ион железа (Fe2+) и две молекулы оксида азота (NO). ДНКЖ, связанные с цистеинами белков, являются стабильной формой депонирования NO в организме [12, 13], однако нельзя исключить, что эти комплексы функционируют и в качестве регуляторов активности белковой молекулы.
Гемоглобины (Hb) многих позвоночных имеют реакционноспособные остатки цистеина, локализованные на поверхности белковой молекулы [14]. Hb человека содержит два таких остатка в β-субъединицах тетрамера (Cys-93β). Показано, что с участием этих цистеинов и формируются ДНКЖ [15, 16] (рис. 1). В качестве второго лиганда, связывающего железо в ДНКЖ (обозначен на рис. 1 как L), обычно выступает другая аминокислота в белке. Мы предполагем, что чаще всего таким лигандом является остаток тирозина.
Рис. 1.
Схематичное изображение ДНКЖ, связанных с SH-группой β-субъединицы гемоглобина. L – второй лиганд, связывающий железо в ДНКЖ.
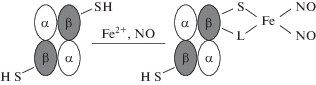
Ранее была показана способность ДНКЖ, связанных с Cys-93β, защищать Hb от окислительной модификации, вызванной гидроперекисями [15–17], сформулирована гипотеза о сайтспецифическом антиоксидантном действии ДНКЖ по отношению к белкам. В модельных системах с низкомолекулярными ДНКЖ, содержащими тиольные лиганды, было показано, что в основе антиоксидантных свойств комплексов лежат следующие эффекты: связывание ионов железа и NO, перехватывание активных форм кислорода и азота, кооперативное взаимодействие с другими антиоксидантами, способность восстанавливать оксофферильную форму гема [15, 16, 18, 19]. Мы предположили, что ДНКЖ, связанные с Hb, могут быть и модуляторами реакционной активности Cys-93β.
Цель работы – изучение влияния ДНКЖ на реакционную способность SH-групп Hb при действии на него гидропероксида трет-бутила. Последний является водорастворимым аналогом гидропероксидов липидов и широко используется для моделирования процессов перекисного окисления в живых системах [19, 20].
МЕТОДИКА
Использованные реактивы. В работе были использованы следующие реактивы: метгемоглобин (окисленный Hb) человека, гидропероксид трет-бутила (t-BOOH), метилглиоксаль, ПААГ, ДДС-Na, Трис, глицин, NaN3, дитиотреитол (ДТТ), диметилсульфоксид (ДМСО), 4-гидрокси-(2,2,6,6-тетраметилпиперидин-1-ил)оксил (4-гидрокси-TEMPO), кумасси бриллиантовый синий R-250 – “Sigma-Aldrich” (США); 3H-нафтол[2,1-b]пиран-s-карбоновая кислота (ThioGlo1) – “Calbiochem” (США); остальные реактивы – “Химмед” (Россия).
ДНКЖ синтезировали, как было описано в работах [15–17, 21]. ДНКЖ с глутатионовыми лигандами (GS-ДНКЖ) получали, смешивая растворы FeSO4 и восстановленный глутатион (GSH) в молярном соотношении 1 : 2 в сосуде Тунберга в атмосфере NO. ДНКЖ с фосфатными лигандами синтезировали аналогичным образом, пропуская в сосуде Тунберга газообразный NO через раствор FeSO4 в 100 мМ фосфатном буфере, рН 6.8.
Связанные с Hb ДНКЖ получали добавлением фосфатных ДНКЖ (ДНКЖ-${\text{PO}}_{{\text{4}}}^{--}$) к раствору гемоглобина в 20 мМ К-фосфатном буфере рН 6.8, в молярном соотношении Hb : ДНКЖ = 1 : 3. Концентрацию ДНКЖ рассчитывали по интегральной интенсивности сигнала электронного парамагнитного резонанса (ЭПР) этих комплексов, используя в качестве внешнего стандарта спиновую метку 4-гидрокси-TEMPO. Препараты ДНКЖ хранили при –70°С.
Получение metHb-N3 и его обработка гидропероксидом трет-бутила. Для исключения аллостерических переходов в гемоглобине к 0.15 мМ раствору Hb в 20 мМ К-фосфатном буфере, рН 6.8, добавляли NaN3 до конечной концентрации 12 мМ, при этом раствор приобретал темно-вишневый цвет, что свидетельствовало об образовании низкоспиновой формы HbIII-N3. В дальнейшем при упоминании гемоглобина имется в виду именно этот комплекс.
Для приготовления реакционной смеси к раствору HbIII-N3 добавляли t-BOOH в молярном соотношении 1 : 10. При изучении зависимости реакционных эффектов от концентрации окислителя t-BOOH добавляли до конечных концентраций: 0.6, 1.6, 2.6, 6.8, 11.0 мМ и инкубировали в течение 5 мин при комнатной температуре, после чего проводили измерения.
Определение свободных SH-групп. Количественную оценку сульфгидрильных групп проводили с помощью тиол-специфичной флуоресцентной метки ThioGlo1. При добавлении к раствору белка ThioGlo1 образуется тиоловый аддукт с максимумом испускания флуоресценции при 500 нм при длине волны возбуждения 379 нм [22].
Образцы для анализа готовили следующим образом: к 5 мкл реакционной смеси, содержащей 0.15 мМ Hb (0.15 мМ ), добавляли 5 мкл 2.5 мМ ThioGlo1 в ДМСО и инкубировали 3 мин. Полученный раствор (10 мкл) вносили в кварцевую спектрофлуориметрическую кювету, содержащую 490 мкл 20 мМ К-фосфатного буфера, рН 6.8. Для деоксигенации Hb кювету с реакционной смесью продували аргоном в течение 10 мин.
При измерении содержания SH-групп в образцах глутатиона и глутатионовых ДНКЖ в реакции с t-BOOH реакционная смесь содержала 0.08 мМ GS-ДНКЖ (по Fe2+) или эквивалентное количество GSH в 10 мМ К-фосфатном буфере, рН 7.4, и 0.2 мМ t-BOOH (GSH/GS-ДНКЖ : : t‑BOOH = 1 : 2.5).
Измерение автофлуоресценции триптофана и дитирозина. Состояние остатков триптофана и тирозина в молекуле Hb изучали с помощью флуоресцентной спектроскопии. Образцы белков были приготовлены, как описано выше для получения metHb-N3. В кювету к 490 мкл 20 мМ К-фосфатного буфера, рН 6.8, добавляли 10 мкл раствора белка. Для селективного возбуждения автофлуоресценции триптофана использовали длину волны 295 нм. Флуоресценцию регистрировали при 330 нм. Ширина щели возбуждающего света составляла 5 нм, испускаемого – 10 нм.
Спектрофотометрическую детекцию образования дитирозинов осуществляли при длине волны возбуждения 325 нм и излучения 400 нм (330–600 нм) [23]. Ширина щели возбуждающего и испускаемого света составляла 5 нм.
Образцы Hb перед измерением разбавляли в 25 раз при измерении флуоресценции тритпофана и в 12.5 раз при измерении флуоресценции дитирозинов.
Регистрацию флуоресценции во всех случаях проводили в кварцевых кюветах с длиной оптического пути 1 см на спектрофлуориметре RF-5301 PC (“Shimadzu”, Япония) при высокой чувствительности – “high” и средней скорости сканирования – “medium” (в соответствии с обозначениями на приборе).
ДДС-электрофорез в ПААГ. Электрофорез проводили в блоках 12%-ного ПААГ размером 15 × × 15 × 1 мл по методу Лэммли [24], используя прибор для вертикального электрофореза серии VE (“Хеликон”, Россия). К образцам Hb, приготовленным как описано выше, добавляли буфер для образцов в соотношении 1 : 1, прогревали в течение 5 мин при 95°С и затем наносили по 10 мкл на гель. Буфер для образцов был приготовлен на основе 0.1 М Трис-HCl буфера, рН 6.8, и содержал 4% ДДС-Na, 0.2% бромфенолового синего и 20% глицерина. Для создания восстановительных условий в буфер добавляли раствор 3%-ного ДТТ. В качестве электродного буфера использовали 0.2 М Трис-глициновый буфер, рН 8.3, содержащий 0.1% ДДС-Na .
Электрофорез проводили при 4°С и I = 50 мА, U = 150 B. Необходимые условия электрофореза обеспечивались источником питания Эльф-4 (“НПО ДНК-Технология”, Россия). После окончания процесса разделения белков гель фиксировали и окрашивали раствором Кумасси бриллиантового синего R-250.
Для каждого образца Hb измерения проводили не менее чем в трех повторностях. Статистическую обработку полученных данных проводили на основе расчета средних арифметических значений и их ошибок. Различия показателей по сравнению с контролем определялись методом вариационной статистики с использованием критерия Стьюдента. Данные представлены как среднее ± стандартное отклонение.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Как было отмечено выше, в интактном Hb человека имеется только два доступных реакционноспособных цистеина (Cys-93β) [25], расположенных вблизи гистидина (His-92), координирующего ион железа, что делает их чувствительными к конформационным превращениям белка, вызванным присоединением лиганда к гемовому железу [26, 27]. Реакционная способность SH-групп зависит как от природы гемового лиганда, так и от степени насыщения лигандом гемовых групп.
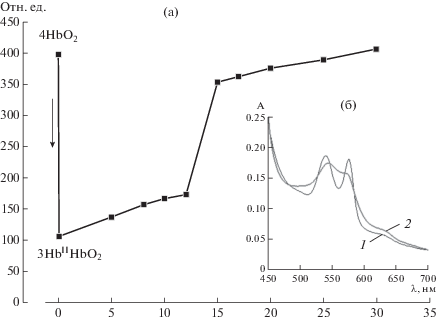
Для оценки реакционной способности сульфгидрильных групп использовали высокоспецифичный флуоресцентный реагент на восстановленные тиолы – ThioGlo1, образующий ковалентные аддукты с восстановленными SH-группами [22]. Поскольку на флуоресцентные характеристики тиоловых аддуктов значительное влияние может оказывать электронное состояние гема, было проведено предварительное исследование взаимосвязи между реакционной способностью SH-групп и конформационным состоянием молекулы белка. Показано, что интенсивность флуоресценции тиолового аддукта с ThioGlo1 меняется в зависимости от степени оксигенации Hb (конформационный R–T-переход) (рис. 2). Этот факт подтвердил наличие аллостерического взаимодействия между гемом и Cys-93β, что позволило использовать данный методический подход для изучения модификаций SH-групп в Hb. Полученные в эксперименте данные согласуются с результатами, полученными в работе [26], в которой было показано, что добавление CO или O2 к deoxyHb приводило к 8-кратному увеличению скорости реакции N-этилмалеимида с SH-группами. Чтобы избежать влияния аллостерии на состояние Cys-93β, к раствору metHb добавляли азид натрия (NaN3) для получения стабильного низкоспинового комплекса HbIII-N3.
Рис. 2.
Влияние степени оксигенации Hb на реакционную способность SH-групп, измеренную с помощью ThioGlo1: а – снижение интенсивности флуоресценции (отн. ед.) ThioGlo1 при деоксигенации Hb (стрелкой показана продувка реакционной смеси аргоном); б – спектры поглощения Hb с разной степенью оксигенации: 1 – оксигенированы все четыре субъединицы (4HbO2); 2 – оксигенирована одна субъединица (3HbIIHbO2).
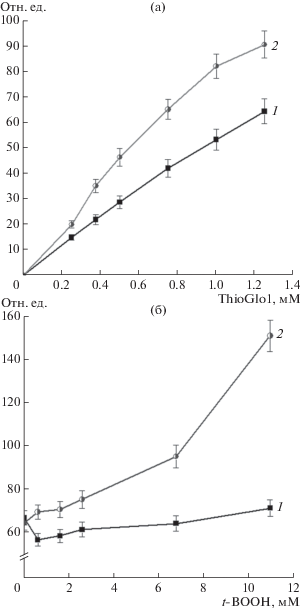
Изменение реакционной способности SH-групп после их включения в состав ДНКЖ изучали в зависимости от концентрации ThioGlo1 (система 1) и от концентрации окислителя – t-BOOH (система 2) (рис. 3). В первой системе флуоресценция тиолового аддукта линейно возрастала при увеличении концентрации ThioGlo1 в реакционной смеси (рис. 3а). В экспериментах с гемоглобиновыми ДНКЖ (Hb-ДНКЖ) интенсивность флуоресценции была выше по сравнению с Hb. В системе 2 интенсивность флуоресценции тиолового аддукта Hb-ДНКЖ также превосходила интенсивность флуоресценции Hb (рис. 3б), причем с увеличением концентрации окислителя возрастала и эта разница. Вероятно, включение SH-групп в состав ДНКЖ продлевали время их нахождения в форме тиолят-аниона (R–S–), характеризующейся выраженными нуклеофильными свойствами, что увеличивало ее реакционную способность в отношении ThioGlo1.
Рис. 3.
Флуоресценция (отн. ед.) аддуктов цистеина с ThioGlo1 в зависимости от концентрации ThioGlo1 (а) и t-BOOH (б): 1 – Hb, 2 – Hb-ДНКЖ.
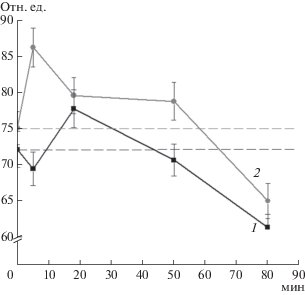
На следующем этапе было изучено изменение во времени флуоресценции тиоловых аддуктов с ThioGlo1 после добавления t-BOOH к раствору Hb в молярном соотношении 10 : 1. t-BOOH в обоих вариантах – Hb и Hb-ДНКЖ сначала вызывал незначительное (~7%) увеличение интенсивности флуоресценции, а затем снижение, которое свидетельствовало об окислении SH-групп (рис. 4). К 80 мин интенсивность флуоресценции падала в обоих вариантах на ~15%. Наблюдаемый вначале скачок флуоресценции, по-видимому, связан с образованием активированных форм тиола: тиолят-аниона (R–S–) и сульфеновой кислоты (R–SOH). У этих форм константа связывания ThioGlo1 отличалась от R–SH. Известно, что образование активированных тиолов происходит в условиях мягкого окисления остатков цистеина [4, 9], что соответствует условиям эксперимента. Следует отметить, что в случае Hb-ДНКЖ время нахождения тиолов в активированном состоянии было дольше, чем для Hb.
Рис. 4.
Изменение во времени флуоресценции (отн. ед.) аддуктов цистеина с ThioClo1 гемоглобина, обработанного пероксидом трет-бутила (Hb : t-BOOH = = 1 мМ : 10 мМ): 1 – Hb, 2 – Hb-ДНКЖ. Пунктирная линия – исходный уровень флуоресценции образца.
Чтобы проверить, действительно ли наблюдаемое повышение флуоресценции тиоловых аддуктов связано с включением SH-групп в состав комплексов, было исследовано взаимодействие t-BOOH с низкомолекулярным тиолом – восстановленным глутатионом (GSH). Добавление к раствору глутатиона t-BOOH в молярном соотношении 1.0 : 2.5 показало, что тиоловые группы GSH окисляются и к концу измерений (20 мин) их количество составляло только половину от исходного уровня (рис. 5, кривая 1).
Рис. 5.
Изменение во времени флуоресценции (отн. ед.) тиоловых аддуктов глутатиона, обработанного пероксидом t-BOOH (GSH/GS-ДНКЖ : t-BOOH = = 1.0 : 2.5 мМ): 1 – GSH, 2 – GS-ДНКЖ. Пунктирная линия – исходный уровень флуоресценции образца.
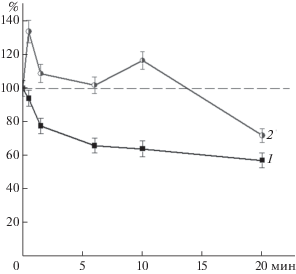
Следует отметить, что как в случае низкомолекулярных глутатионовых ДНКЖ (GS-ДНКЖ), так и в случае белковых (Hb-S-ДНКЖ) в начале происходило увеличение флуоресценции и только затем ее снижение (рис. 5, кривая 2). При этом к 20 мин окислилось только 30% тиолов. Ранее сходные результаты были нами получены при окислении GSH и GS-ДНКЖ хлорноватистой кислотой [19].
Связанные с белком глутатионовые ДНКЖ слабо реагируют с органическими гидропероксидами. Однако разрушение ДНКЖ может быть спровоцировано оксоферрильной формой Hb (Hb–FeIV=O), образующейся в реакции metHb с t-BOOH [16]. Возможный молекулярный механизм антиоксидантного действия ДНКЖ в этом случае связан с восстановлением радикалов Hb–FeIV=O [17, 28, 29]. Антиоксидантное действие также может быть обусловлено восстановлением оксоферрильной формы гема и нитрозилированием гемовой группы оксидом азота, высвобождающимся при распаде ДНКЖ [28, 29]. Оксоферрильная форма гема является очень сильным окислителем, модифицирующим белковую цепь гемопротеидов [30, 31]. При этом в результате одноэлектронного окисления остатков тирозина формируются феноксильные радикалы, которые могут окислять остатки цистеина с образованием тиильных радикалов [32].
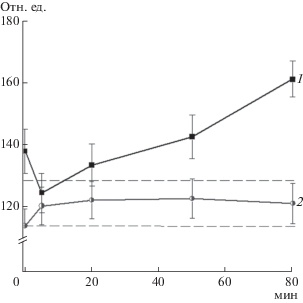
Известно, что в Hb окислению подвергаются остатки Tyr42 и Tyr24. В связи с этим наряду со спектрофлуориметрическими измерениями состояния тиолов было проведено определение состояния тирозиновых остатков. Их флуоресценция возрастала при образовании дитирозинов в результате взаимодействия двух тирозиновых радикалов. В экспериментах с Hb происходило сначала снижение флуоресценции, а затем ее возрастание, которое к концу измерения составляло 15% от исходного уровня (рис. 6). Возрастание флуоресценции тирозинов по времени совпадало со снижением флуоресценции тиоловых аддуктов с ThioGlo1 (рис. 4), что указывало на одновременность окисления тиолов и тирозинов в Hb. Полученные результаты согласуются с результатами работы [32], в которой было показано, что окислению тиолов и образованию тиильного радикала предшествовало появление тирозиновых радикалов. В эксперименте с Hb-ДНКЖ флуоресценция в первые минуты увеличивалась на 6% и далее уже не менялась (рис. 6). Таким образом, наличие ДНКЖ в составе белка защищало остатки тирозина от окисления. Этот факт можно объяснить образованием 3-нитротирозина в реакции NO (источником которого были ДНКЖ) с тирозиновыми радикалами [33].
Рис. 6.
Изменение во времени автофлуоресценции тирозина гемоглобина, обработанного пероксидом t-BOOH (Hb : t-BOOH = 1 мМ : 10 мМ): 1 – Hb, 2 – Hb-ДНКЖ. Пунктирная линия показывает исходный уровень флуоресценции образца.
Поскольку наблюдаемые различия в свойствах тиолов могут быть вызваны конформационными изменениями белка под действием ДНКЖ, была исследована собственная флуоресценция Hb, обусловленная свечением остатков триптофана. Этот метод широко применяется для оценки конформационного состояния белков. Полученные спектры испускания флуоресценции Hb и Hb-ДНКЖ представлены на рис. 7а. Анализ спектров показал, что формирование Hb-ДНКЖ не приводило к сдвигу максимума спектра флуоресценции триптофана, положение которого зависит от перестройки ближайшего окружения данной аминокислоты. Это говорит об отсутствии влияния ДНКЖ на конформационное состояние молекулы Hb. Поэтому наблюдаемые различия в реакционных свойствах SH-групп в обоих вариантах скорее всего обусловлены не конформационными изменениями белка, а разным состоянием тиолов.
Рис. 7.
Автофлуоресценция остатков триптофана в гемоглобине: а – спектр автофлуоресценции раствора Hb, б – зависимость интенсивности флуоресценции триптофана в зоне максимума (327 нм) от концентрации t-BOOH: 1 – Hb, 2 – Hb-ДНКЖ. Пунктирная линия показывает исходный уровень флуоресценции образца.
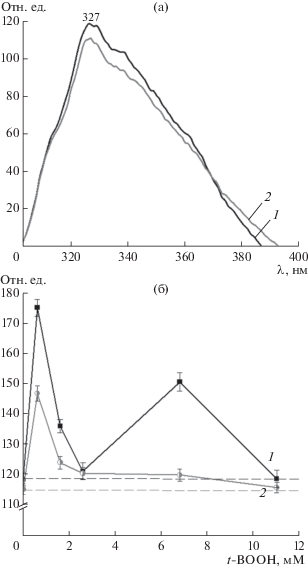
Известно, что в Hb мишенью для атаки гидропероксидами помимо цистеина является триптофан [34, 35], поэтому была изучена зависимость интенсивности испускания флуоресценции триптофана при 327 нм от концентрации t‑BOOH (рис. 7б). Эта зависимость имела волнообразный характер. На кривых, полученных для Hb, присутствуют два экстремума: при концентрациях окислителя 0.6 и 6.8 мМ, в случае Hb-ДНКЖ – явно выражен только первый экстремум. Для обоих вариантов белка можно отметить концентрации t-BOOH, при которых флуоресценция практически не отличается от исходной: без t-BOOH – это 2.6 мМ и 11 мМ. Появление экстремумов флуоресценции триптофана можно объяснить локальными конформационными перестройками молекулы Hb, происходящими в результате окислительной модификации. Конформационные перестройки скорее всего носят неденатурационный характер, причем в случае Hb-ДНКЖ они гораздо менее выражены. Интенсивность флуоресценции во всем диапазоне концентраций t-BOOH не опускалась ниже исходного уровня, что указывало на отсутствие окисления триптофана.
Агрегация молекул Hb была исследована с помощью ДДС-электрофореза в ПААГ с дитиотреитолом, восстанавливающим дисульфидные связи. Анализ представленных на электрофореграмме данных показывает, что в присутствии t-BOOH в обоих вариантах белка образовывались ковалентно-сшитые димеры (рис. 8), но в Hb количество высокомолекулярных форм было больше, чем в Hb-ДНКЖ. Поскольку образцы белка содержали ДТТ, можно заключить, что димеризация Hb была обусловлена не дисульфидными связями. Скорее всего в образовании межбелковых сшивок участвовали тирозиновые радикалы. Поскольку в образце Hb-ДНКЖ окисление тирозинов было менее выражено (рис. 6), чем в Hb, то и формирование сшитых белков происходило менее интенсивно. Полученные данные согласуются с ранее полученными результатами, демонстрирующими способность ДНКЖ предотвращать окислительную модификацию связанного с ними белка [15–17]. Эта способность ДНКЖ объясняется тем, что они выступают одновременно в роли хелатора и антиоксиданта. Однако в случае Hb-ДНКЖ при концентрациях t-BOOH 6.8 и 11 мМ возникали крупные белковые агрегаты, которые не входили в разделяющий гель. Это может быть связано с тем, что при высокой концентрации окислителя динитрозильные комплексы разрушаются, высвобождая ионы Fe2+, реагирующие с пероксидами с образованием свободных радикалов, провоцирующих дальнейшее окисление белка [36]. Двойственная роль ДНКЖ как антиоксиданта при низких концентрациях, окислителя и прооксиданта при высоких неоднократно подчеркивалась в работах [15–18, 21, 37, 38].
Рис. 8.
Электрофорез в 12%-ном ПААГ с ДТТ и ДДС-Na : Hb (a) и Hb-ДНКЖ (б). Концентрация t-BOOH (мМ): 1 – 0, 2 – 0.6, 3 – 1.6, 4 – 2.6, 5 – 6.8, 6 – 11.0. (16 кДа – мономер Hb, 32 кДа – димер).

Можно дать и дополнительное объяснение наблюдаемым фактам. Расположенные на поверхности белка тиолы являются легкодоступными мишенями для активных форм кислорода (АФК). Вступая в реакцию с окислителем, они принимают на себя удар и тем самым защищают другие аминокислоты от необратимой окислительной модификации. Если тиолы заблокированы и не могут прореагировать с АФК, под удар попадают тирозин и триптофан, что приводит к множественным межмолекулярным сшивкам и накоплению крупных белковых агрегатов. Роль Cys-93β в защите Hb от денатурации и полимеризации в реакции с H2O2 была показана в работах [39, 40].
Роль Cys-93β в функционировании Hb и эритроцитов до сих пор до конца не понятна. На важное биологическое значение этих тиолов указывают высокая консервативность поверхностного остатка цистеина у Hb позвоночных [14], аллостерическая регуляция его реакционной способности [26, 27] и высокая внутриклеточная концентрация (~10 мМ, исходя из концентрации Hb в эритроцитах человека при ~40%-ном гематокрите). Активно обсуждается гипотеза об участии Cys-93β в метаболизме NO и в его экспорте [10, 41–45]. Поскольку реакционная способность Cys-93β зависит от степени насыщения Hb кислородом, эти тиолы могут участвовать во внутриклеточной редокс-сигнализации, которая осуществляется через взаимодействие Hb с компонентами мембраны эритроцита [45]. Тиоловые группы могут участвовать в регуляции равновесия димерной и тетрамерной форм Hb, от чего зависит образование ${\text{O}}_{{\text{2}}}^{{ \bullet --}}$ и соответственно H2O2 внутри клетки [46]. Имеются доказательства участия Cys93β в регуляции окисления Hb. Так, этот тиол может реагировать с супероксидом, образующимся в гемовом кармане β-цепи при автоокислении, с образованием тиильного радикала [47]. Показано, что Cys93β замедляет зависимое от H2O2 окисление оксигенированного Hb примерно в два раза [48]. Предложена гипотеза, что Cys-93β является аллостерически регулируемым антиоксидантом в эритроцтах [48]. В работе [39] обсуждается роль Cys93β в обеспечении безопасного пути для передачи электронов, генерируемых в гемовом кармане. Образующийся при этом цистеиновый радикал может быть восстановлен глутатионом, содержание которого в эритроцитах велико.
Полученные на Hb результаты показывают, что ДНКЖ могут быть регуляторами реакционной способности белковых SH-групп, при этом стабильность ДНКЖ определяется редокс-условиями внутри клетки. В нормальных условиях включение SH-групп в ДНКЖ является обратимой посттрансляционной модификацией, которая снижает реакционную способность тиолов, но при этом не исключает участия в редокс-превращениях. В условиях окислительного стресса комплексы распадаются, высвобождая тиолят-анион, который находится в состоянии повышенной готовности к реакции с АФК и электрофильными соединениями, поскольку не нуждается в предварительном депротонировании.
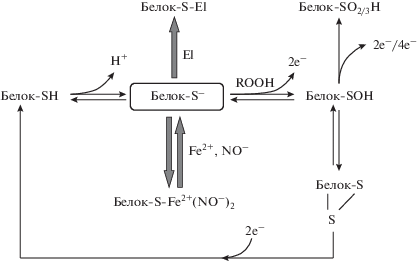
Образование аддуктов с электрофильными соединениями приводит к формированию стабильной модификации белка. Иными словами, ДНКЖ являются своеобразной меткой на реакционноспособные цистеины, предрасполагая их к стабильным модификациям, и обеспечивая тем самым селективность этой модификации. Регуляторное действие ДНКЖ связано еще и с тем, что они влияют на равновесие между тиолом и тиолятом в белке, смещая его в сторону тиолята. Стабилизируя тиолятные анионы, эти комплексы повышают энергетический барьер реакции с пероксидами. Таким образом, ДНКЖ настраивают реакционную способность цистеина по отношению к действию пероксидов и электрофилов, что проиллюстрировано схемой (рис. 9).
Рис. 9.
Предполагаемый механизм регуляции активности тиолов в белках посредством формирования ДНКЖ. El – электрофильные соединения, ROOH – органический гидропероксид.
Настройку реакционной способности цистеинов с помощью ДНКЖ можно отнести к механизмам регуляции структуры и функции белка посредством связывания ионов металлов или их комплексов [1, 7]. Известно, что тиолятная группа является хорошим лигандом для ионов железа, меди и цинка [1, 7, 49, 50]. При этом связывание металлов цистеиновыми лигандами не только изменяет структуру белка, но также оказывает комплексное защитное действие благодаря одновременному снижению реакционной способности тиола и металла. Это позволяет отсрочить образование стабильных модификаций белковых тиолов в условиях незначительного окислительного стресса и, наоборот, ускорить их при высокой степени стресса.
При проведении исследований использовали оборудование Центра коллективного пользования “Промышленные биотехнологии” Федерального исследовательского центра “Фундаментальные основы биотехнологии” Российской академии наук.
Работа выполнена при финансовой поддержке Российского Фонда Фундаментальных Исследований (грант № 19-29-12052) и Министерства науки и высшего образования Российской Федерации.
Список литературы
Giles N.M., Watts A.B., Giles G.I., Fry F.H., Littlechild J.A., Jacob C. // Chem. Biol. 2003. V. 10. № 8. P. 677–693.
Paulsen C.E., Carroll K.S. // ACS Chem. Biol. 2010. V. 5. № 1. P. 47–62.
Paulsen C.E., Carroll K.S. // Chemical Reviews. 2013. V. 113. № 7. P. 4633–4679.
Go Y.M., Chandler J.D., Jones D.P. // Free Radic. Biol. Med. 2015. V. 84. P. 227–245.
Klomsiri C., Karplus P.A., Poole L.B. // Antioxid. Redox Signal. 2010. V. 14. № 6. P. 1065–1077.
Gupta V., Carroll K.S. // Biochim. Biophys. Acta. 2013. V. 1840. № 2. P. 847–875.
Novikova N.N., Kovalchuk M.V., Yurieva E.A., Konovalov O.V., Stepina N.D., Rogachev A.V., Yalovega G.E., Kosmachevskaya O.V., Topunov A.F., Yakunin S.N. // J. Phys. Chem. B. 2019. V. 123. № 40. P. 8370–8377.
Lo Conte M., Carroll K.S. // J. Biol. Chem. 2013. V. 288. № 37. P. 26480–26488.
Wible R.S., Sutter T.R. // Chem. Res. Toxicol. 2017. V. 30. № 3. P. 729–762.
Kosmachevskaya O.V., Shumaev K.B., Topunov A.F. // Biochemistry (Moscow). 2019. V. 84. Suppl. 1. P. S206–S224.
Foyer C.H., Wilson M.H., Wright M.H. // Free Radic. Biol. Med. 2018. V. 122. P. 137–149.
Vanin A.F. // Nitric Oxide. 2016. V. 54. P. 15–29. https://doi.org/10.1016/j.niox.2016.01.006
Vanin A.F. Dinitrosyl Iron Complexes as a “Working Form” of Nitric Oxide in Living Organisms. Cambrighe, UK: Cambridge Scholars Publishing, 2019. 265 p.
Reischl E., Dafre A.L., Franco J.L., Wilhelm Filho D. // Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2007. V. 146. № 1–2. P. 22–53.
Shumaev K.B., Gubkin A.A., Serezhenkov V.A., Lobysheva I.I., Kosmachevskaya O.V., Ruuge E.K., Lankin V.Z., Topunov A.F., Vanin A.F. // Nitric Oxide. 2008. V. 18. № 1. P. 37–46.
Shumaev K.B., Kosmachevskaya O.V., Timoshin A.A., Vanin A.F., Topunov A.F. // Methods Enzymol. 2008. V. 436. P. 445–461.
Shumaev K.B., Petrova N.E., Zabbarova I.V., Vanin A.F., Topunov A.F., Lankin V.Z., Ruuge E.K. // Biochemistry (Moscow). 2004. V. 69. № 5. P. 569–574.
Shumaev K.B., Dudylina A.L., Ivanova M.V., Pugachenko I.S., Ruuge E.K. // Biofactors. 2018. V. 44. № 3. P. 237–244.
Shumaev K.B., Gorudko I.V., Kosmachevskaya O.V., Grigoryeva D.V., Panasenkoe O.M., Vanin A.F., Topunov A.F., Terekhova M.S., Sokolov A.V., Cherenkevich S.N., Ruuge E.K. // Oxid. Med. Cell. Longev. 2019. V. 2019. Article ID 2798154. https://doi.org/10.1155/2019/2798154
Domanski A.V., Lapshina E.A., Zavodnik I.B. // Biochemistry (Moscow). 2005. V. 70. № 7. P. 761–769.
Шумаев К.Б., Губкин А.А., Губкина С.А., Гудков Л.Л., Лакомкин В.Л., Топунов А.Ф., Ванин А.Ф., Рууге Э.К. // Биофизика. 2007. Т. 52. № 3. С. 534–538.
Hoff S., Larsen F.H Andersen M.L., Lund M.N. // Analyst. 2013. V. 138. № 7. P. 2096–2103.
Davies K.J., Delsignore M.E. // J. Biol. Chem. 1987. V. 262. № 20. P. 9908–9913.
Laemmli U.K. // Nature. 1970. V. 227. № 5259. P. 680–685.
Blacken G.R., Wang Y., Lopez J.A., Fu X. // Blood. 2009. V. 114. № 22. P. 4040–4040.
Riggs A. // J. Biol. Chem. 1961. V. 236. № 7. P. 1948–1954.
Benesch R.E., Benesch R. // Biochemistry. 1962. V. 1. № 5. P. 735–738.
Gorbunov N.V., Osipov A.N., Day B.W., Zayas-Rivera B., Kagan V.E., Elsayed N.M. // Biochemistry. 1995. V. 34. № 20. P. 6689–6699.
Gorbunov N.V., Yalowich J.C., Gaddam A., Thampatty P., Ritov V.B., Kisin E.R., Elsayed N.M., Kagan V.E. // J. Biol. Chem. 1997. V. 272. № 19. P. 12 328–12 341.
Reeder B.J., Grey M., Silaghi-Dumitrescu R.-L., Svistunenko D.A., Bülow L., Cooper C.E., Wilson M.T. // J. Biol. Chem. 2008. V. 283. № 45. P. 30 780–30 787.
Vlasova I.I. // Molecules. 2018. V. 23. № 10. e2561. https://doi.org/10.3390/molecules23102561
Bhattacharjee S., Deterding L.J., Jiang J., Bonini M.G., Tomer K.B., Ramirez D.C., Mason R.P. // J. Am. Chem. Soc. 2007. V. 129. № 44. P. 13 493–13 501.
Gunther M.R., Sturgeon B.E., Mason R.P. // Toxicology. 2002. V. 177. № 1. P. 1–9.
Steffek R.P., Thomas M.J. // Free Radic. Res. Commun. 1991. V. 12–13. № 2. P. 489–497.
Jia Y., Buehler P.W., Boykins R.A., Venable R.M., Alayash A.I. // J. Biol. Chem. 2007. V. 282. № 7. P. 4894–4907.
Laguerre M., Bily A., Roller M., Birtic S. // Annu. Rev. Food Sci. Technol. 2017. V. 8. P. 391–411.
Шумаев К.Б., Губкин А.А., Губкина С.А., Гудков Л.Л., Свиряева И.В., Тимошин А.А., Топунов А.Ф., Ванин А.Ф., Рууге Э.К. // Биофизика. 2006. Т. 51. №. 3. С. 472–477.
Шумаев К.Б., Губкина С.А., Ванин А.Ф., Бурбаев Д.Ш., Мох В.П., Топунов А.Ф., Рууге Э.К. // Биофизика. 2013. Т. 58. № 2. С. 239–245.
Winterbourn C.C., Carrell R.W. // Biochem. J. 1977. V. 165. № 1. P. 141–148.
Pimenova T., Pereira C.P., Gehrig P., Buehler P.W., Schaer D.J., Zenobi R. // J. Proteome Res. 2010. V. 9. № 8. P. 4061–4070.
Stamler J.S., Singel D.J., Piantadosi C.A. // Nat. Med. 2008. V. 14. № 10. P. 1008–1009.
Jensen F.B. // J. Exp. Biol. 2009. V. 212. № 21. P. 3387–3393.
Gaston B., May W.J., Sullivan S., Yemen S., Marozkina N.V., Palmer L.A., Bates J.N., Lewis S.J. // J. Appl. Physiol. 2014. V. 116. № 10. P. 1290–1299.
Zhao Y., Wang X., Noviana M., Hou M. // Acta Biochim. Biophys. Sin. 2018. V. 50. № 7. P. 621–634.
Kosmachevskaya O.V., Nasybullina E.I., Blindar V.N., Topunov A.F. // Appl. Biochem. Microbiol. 2019. V. 55. № 2. P. 83–98.
O’Neill J.S., Reddy A.B. // Nature. 2011. V. 469. № 7331. P. 498–503.
Balagopalakrishna C., Abugo O.O., Horsky J., Manoharan P.T., Nagababu E., Rifkind J.M. // Biochemistry. 1998. V. 37. № 38. P. 13 194–13 202.
Vitturi D.A, Sun C.W., Harper V.M., Thrash-Williams B., Cantu-Medellin N., Chacko B.K., Peng N., Dai Y., Wyss J.M., Townes T., Patel R.P. // Free Radic. Biol. Med. 2013. V. 55. P. 119–129.
Jakob U., Eser M., Bardwell J.C.A. // J. Biol. Chem. 2001. V. 275. № 49. P. 38 302–38 310.
Kaim W., Schwederski B. // Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life / Ed. G.N.A. Meyer. Chichester, UK: John Wiley & Sons, 1991. P. 330–350.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология