

Прикладная биохимия и микробиология, 2019, T. 55, № 5, стр. 451-459
Конъюгат полиаспарагиновой кислоты с гистидином: получение и характеристика как аналога антиоксидантных ферментов
М. И. Камалов 1, Г. Р. Садриева 1, А. М. Павлюк 1, Д. В. Салахиева 1, Н. В. Петрова 1, Т. И. Абдуллин 1, *
1 Институт фундаментальной медицины и биологии, Казанский (Приволжский) федеральный университет
420008 Казань, Россия
* E-mail: tabdulli@gmail.com
Поступила в редакцию 31.10.2018
После доработки 04.03.2019
Принята к публикации 22.04.2019
Аннотация
Полиаминокислоты − перспективная основа для создания биоактивных препаратов и переносчиков лекарственных средств. Методом жидкофазного синтеза получены и охарактеризованы в качестве потенциальных антиоксидантов полиаспарагиновые кислоты, а также их амидные конъюгаты с L-гистидином. Полиаспарагиновые (pAsp) кислоты не обладали радикал-связывающей и каталазной активностью в широком диапазоне концентраций. С использованием редокс-реакции генерации супероксид-аниона системой НАДН/метосульфат феназин установлено, что конъюгаты с L‑гистидином в концентрации ≥100 мкг/мл обладали супероксиддисмутазной (СОД) активностью, сходной с действием марганец-зависимой СОД-2. Показано, что L-гистидин в комбинации с ионами марганца и меди ускорял дисмутацию супероксид-аниона пропорционально концентрации аминокислоты, а конъюгаты с L-гистидином обладали СОД-подобной активностью без добавления кофактора. Конъюгаты с L-гистидином оказывали цитотоксическое действие по отношению к опухолевым клеткам линии PC-3 (IC50 ≈ 0.8 мг/мл, 72 ч) и модулировали жизнеспособность фибробластов кожи человека. Полученные результаты представляют интерес для создания терапевтических препаратов, имитирующих действие супероксиддисмутаз, а также синтетических аналогов других природных ферментов на основе производных pAsp.
Полиаминокислоты (pAA) представляют интерес для создания биоактивных препаратов, систем доставки лекарств и биоматериалов. Они являются (со)полимерами, состоящими из одного или нескольких аминокислотных остатков и имеющими, как правило, нерегулярную (статистическую) первичную структуру [1]. Такие pAA, как поли(γ-глутаминовая кислота) и поли(ε-лизин), могут секретироваться бактериями [2], однако, для получения их различных вариантов более широкое применение получили методы, основанные на химической полимеризации остатков аминокислот и их производных [3].
Важным преимуществом pAA по сравнению с химическими полимерными системами являются повышенная биосовместимость из-за биодеградируемости, малой токсичности (в том числе, продуктов деградации) и низкого уровня накопления в тканях [4]. Иммуногенные свойства гомополипептидов и сополимеров, содержащих менее 3 различных остатков аминокислот, выражены слабо, в отличие от полипептидов, состоящих из 4 и более различных мономерных единиц [5]. С учетом этой особенности был создан препарат глатирамера ацетат (Копаксон, “Teva”, Израиль), представляющий собой синтетический сополимер глутаминовой кислоты, лизина, аланина и тирозина (~6.4 кДа), близкий по составу к основному белку миелина и предназначенный для лечения рассеянного склероза (за счет конкурентного связывания с аутоантителами к этому белку) [6].
Работы в области pAA посвящены созданию на их основе переносчиков низкомолекулярных лекарств и биопрепаратов in vitro и in vivo [1]. Разработаны системы доставки терапевтических биомакромолекул на основе плазмидной ДНК, малой интерферирующей РНК/микроРНК [7], а также белковых препаратов [5] с использованием катионных полипептидов, состоящих из остатков протеиногенных и непротеиногенных аминокислот. Примеры катионных pAA с доказанной способностью повышать биодоступность и терапевтическую эффективность биомакромолекул, включают полилизин и полиорнитин [8], полигистидин [9] и полиаргинин [10]. При этом полиаргинин проявляет активность, характерную для проникающих (в клетки) пептидов [10].
Катионные pAA электростатически взаимодействуют с анионными компонентами поверхности клеток бактерий и млекопитающих. Подобные взаимодействия могут быть использованы в антибактериальной терапии для направленного повреждения клеточной стенки бактерий [11], а также для повышения адгезии таких специализированных клеток млекопитающих, как нейроны, к поверхности биоматериалов и посуды для выращивания культур [12].
Наряду с катионными pAA, их такие анионные аналоги, как полиглутаминовые и полиаспарагиновые кислоты (pAsp), имеют различное биомедицинское применения. К синтезу и химической модификации последних используются разнообразные подходы, кроме того их получение относительно просто [4], что делает их весьма перспективной системой для создания функциональных полипептидов, с помощью которых можно решать медицинские и (био)технологические задачи. В частности, для получения эффективных трансфекционных агентов pAsp можно контролируемо катионизировать путем введения в полипептидный остов различных аминов [13]. Включение в состав pAsp дополнительных (био)химических групп позволяет модулировать их физико-химические свойства [14], а также придавать им биоадгезивную способность [15].
На основе pAsp разработаны лекарственные формы, содержащие ковалентно-связанные или инкапсулированные противоопухолевые антибиотики, обладающие улучшенными фармакокинетическими характеристиками [16, 17], а также стимул-чувствительные материалы в форме наноструктур и гидрогелей с рН-, термо- и редокс-чувствительными свойствами для пролонгированной доставки и контролируемого высвобождения лекарственных средств [15, 18]. В результате модификации pAsp γ-аминомасляной кислотой получен биоактивный конъюгат, формирующий наночастицы и стимулирующий пролиферацию неонатальных фибробластов человека [19].
Перспективным и малоизученным подходом является создание на основе pAsp каталитически активных систем. Имеются свидетельства, что полипептиды на основе остатков алифатических аминокислот (лейцина, аланина, валина и др.) катализируют асимметрическое эпоксидирование хальконоподобных соединений (реакция Жулиа-Колонна), что, предположительно, является одной из форм пребиотического катализа [20]. Модификация поли(L-лейцина) имидазользными группами позволила существенно повысить эффективность эпоксидирования α,β-ненасыщенных кетонов [21].
Наряду с остатком гистидина, остаток аспарагиновой кислоты наиболее часто встречается в составе активных центров ферментов. Можно предположить, что конъюгация pAsp с остатками аминокислот может служить основой создания каталитически активных систем, имитирующих свойства природных ферментов.
Цель работы – определение супероксиддисмутазной активности конъюгата pAsp с остатком гистидина и роль молекулярного окружения остатка гистидина в проявлении этой активности.
МЕТОДИКА
В работе использовали L-аспарагиновую кислоту, L-гистидин, Fmoc-His(Trt)-OH, гексофторфосфат N,N,N',N'-тетраметил-O-(1H-бензотриазол-1-ил)урония (HBTU), триметилхлорсилан (TMSCl), дибутиламин (DBA), трифторуксусную и о-фосфорную кислоты (“Acros Organics”, США), триизопропилсилан (“NB TCI”', Япония) и смолу Ринка (“Biotage”, Швеция), а также метосульфат феназина (PMS) и никотинамидадениндинуклеотид (НАДН) (“Acros Organis”, США), 3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2H-тетразолиум, MTS) (“Promega”, США), 3-(4,5-диметилтиазол-2-ил)-2,5-дифенил-тетразолия бромид (МТТ), 2,2-дифенил-1-пикрилгидразил (DPPH), бычьи супероксиддисмутазу 2 (СОД-2, КФ 1.15.1.1, 100 ед./мл) и каталазу печени (“Sigma-Aldrich”, США), молибдат аммония и пероксид водорода (“Реахим”, Россия), органические растворители (“Татхимпродукт”, Россия), реагенты и материалы для культивирования клеток (“ПанЭко”, Россия).
Полисукцинимиды синтезировали путем термической поликонденсации L-аспарагиновой кислоты (L-Asp) в присутствии о-фосфорной кислоты (2 : 1) в инертной атмосфере (N2) при 185°С. Реакцию проводили в течение 0.75, 1.5, 3 и 5 ч для получения полисукцинимидов (pSI) с различной молекулярной массой (ММ). Продукты (pSI) растворяли в диметилформамид (ДМФА), осаждали добавлением воды, сушили и измельчали. Гидролизовали pSI в водном растворе с добавлением 1 М NaOH при перемешивании с последующим осаждением pAsp (натриевой соли) в метаноле. Выход pAsp составил 60–75%.
Среднечисловую ММ различных pSI определяли методом вискозиметрии [13] с использованием их растворов (10 мг/мл) в ДМФА с 0.1 М LiCl. Динамическую вязкость определяли на вискозиметре Штабингера SVM-3000 (“Anton Paar”, Австрия) с катящимся шариком при угле наклона 45°. Диаметры капилляра и шарика составляли 1.6 и 1.5 мм соответственно.
Метиловый эфир L-His синтезировали по стандартной методике [22]. В круглодонной колбе суспензировали L-His (10 ммоль) в метаноле и добавляли по дозам 20 ммоль TMSCl, смесь перемешивали в течение 24 ч. Продукт – дигидрохлорид метилового эфира L-His (His-OMe) – осаждали и промывали диэтиловым эфиром [22].
Структуру полученных веществ определяли методом ИК-Фурье спектроскопии в режиме неполного внутреннего отражения на спектрометре Frontier (“PerkinElmer”, США) в диапазоне 400–4000 см–1 (разрешение 1 см–1, число сканов 10), а также 1H-ЯМР спектроскопии (400 МГц) на спектрометре AVANCE III 400 (“Bruker BioSpin”, США).
Для получения конъюгатов pAsp с L-His смешивали pSI (0.9 ммоль по сукцинимидным единицам) с His-OMe (0.09–0.54 ммоль) в ДМСО. Реакцию проводили в присутствии DBA в течение 48 ч при комнатной температуре (25°C), затем осаждали продукты этилацетатом, промывали метанолом и высушивали на ротационном испарителе. Далее продукты гидролизовали в щелочных условиях с образованием конъюгатов pAsp–His, которые выделяли аналогично pAsp. Выход pAsp–His составлял 50–76%.
Содержание L-His в конъюгатах определяли колориметрически с помощью реакции Паули [23]. Обрабатывали pAsp–His в течение 30 мин последовательно сульфаниловой кислотой (10 мг/мл), нитритом натрия (50 мг/мл), карбонатом натрия (75 мг/мл) и этанолом (20%). Оптическое поглощение раствора определяли на спектрофотометре Lambda 35 (“PerkinElmer”, США) при 498 нм. Степень модификации конъюгата pAsp–His рассчитывали по молярному соотношению мономерных единиц pAsp к присоединенным остаткам L-His.
Хроматографический анализ pAsp и pAsp–His проводили гель-фильтрацией на колонке Acclaim SEC-1000 (7 мкм, 1000 Å, 7.8 × 300 мм) на хроматографе Dionex Ultimate 3000 (“Thermo Scientific”, США). Вещества элюировали 0.1 M KH2PO4/0.1 M Na2HPO4 буфером, рН 7.0, в изократическом режиме при скорости потока 0.5 мл/мин и детектировали при 220 нм. Данные обрабатывали с использованием программного обеспечения Chromeleon 6.80.
Трипептид L-His синтезировали методом твердофазного синтеза на микроволновом пептидном синтезаторе Initiator SP Wave (“Biotage”, США) согласно методике [24]. Использовали смолу Ринка и Fmoc-защищенный L-His. Для присоединения остатка аминокислоты использовали 2.0 эквивалента Fmoc-His(Trt)-OH (по отношению к смоле), 1.95 эквивалента активатора HBTU и 3 эквивалента диизопропилэтиламина в ДМФА. Депротекцию проводили в 20%-ном растворе пиперидина в ДМФА. Синтезированный трипептид в форме С-амида отщепляли от смолы в смеси 95% трифторуксусной кислоты, 2.5% воды и 2.5% триизопропилсилана в течение 2 ч и осаждали охлажденным диэтиловым эфиром. Осадок растворяли в воде и лиофильно высушивали.
В экспериментах in vitro использовали клетки аденокарциномы простаты человека (линия PC-3, ATCC), а также фибробласты кожи человека (HSF), выделенные как описано ранее [25]. Клетки культивировали в асептических условиях при 37°С в увлажненной воздушной атмосфере с 5% CO2 в питательной среде DMEM, содержавшей эмбриональную бычью сыворотку (10%), L-глутамин (2 мМ), пенициллин (100 ед./мл) и стрептомицин (100 мкг/мл). Для промывки и открепления клеток использовали сбалансированный солевой раствор Хэнкса и 0.25%-ный раствор трипсина с этилендиаминтетрауксусной кислотой соответственно.
Влияние pAsp–His на жизнеспособность клеток оценивали с помощью МТТ-теста [25]. Конъюгат добавляли к клеткам двукратно на 1 и на 2 сут (с заменой питательной среды) в диапазоне концентраций от 1.0 до 2.0 мг/мл, общее время культивирования составило 72 ч. Жизнеспособные клетки детектировали с использованием 0.5 мг/мл индикатора МТТ. Продукт восстановления МТТ клетками растворяли в ДМСО и регистрировали на микропланшетном анализаторе Infinite M200 Pro (“TECAN”, Швейцария) при 555 нм. Процент жизнеспособных клеток рассчитывали относительно контроля (необработанные клетки).
Радикал-связывающую активность pAsp и pAsp–His оценивали с помощью DPPH-теста, основанного на связывании хромогенного радикала DPPH, как было детализировано ранее [24]. Разложение пероксида водорода (H2O2) детектировали с помощью колориметрической реакции, основанной на образовании комплекса H2O2 с молибдатом аммония [24]. Концентрацию H2O2 определяли спектрофотометрически при 240 нм (ε = 43.6 М–1 см–1). Вещества инкубировали с H2O2 (20 мМ) в фосфатно-солевом буфере (ФСБ) и добавляли молибдат аммония (16.2 мМ). Оптическое поглощение раствора при 374 нм, пропорциональное содержанию H2O2, детектировали на спектрофотометре Lambda 35 (“PerkinElmer”, США). В качестве положительного контроля использовали каталазу (1.64 ед./мл).
Супероксид-анион (СОА) генерировали в ФСБ, содержавшем 0.5 мМ НАДН и 0.17 мкМ PMS, при 37°C. В качестве индикатора реакции использовали 2 мг/мл реагента MTS, восстанавливающегося с образованием окрашенного формазана, который детектировали при 490 нм. Оптический сигнал регистрировали в условиях максимальной скорости восстановления MTS. Анализ проводили в 96-луночном планшете на анализаторе Infinite M200 Pro (“TECAN”, Швейцария). Для определения вклада СОА в восстановление индикатора в раствор вносили СОД-2 (10–100 ед./мл).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез и характеристика полипептидов. Для получения pAsp проводили термическую поликонденсацию L-Asp в атмосфере азота в присутствии кислотного катализатора (H3PO4). Образующийся в реакции промежуточный продукт полисукцинимид (pSI) далее подвергали щелочному гидролизу с образованием pAsp (рис. 1а). Полученная с помощью этого метода pAsp (натриевая соль) представляет собой статистический сополимер, состоящий из остатков различных изомеров Asp (L, D, α и β) [3].
Рис. 1.
Схемы получения полисукцинимида (pSI) и натриевой соли полиаспарагиновой кислоты (а), метилового эфира L-гистидина по реакции с триметилхлорсиланом (б) и конъюгата полиаспарагиновой кислоты с L-гистидином (в).
По данным ИК-Фурье спектроскопии (рис. 2а) спектр pSI содержал характерную полосу при 1704 см–1, соответствующую С=О имидной группе, исчезающую после щелочного гидролиза. Спектр pAsp характеризовался наличием типичных для полипептидов полос при 1600 см–1 (валентные колебания С=О амидной связи, амид I) и 1520 см–1 (совместные деформационные колебания N–H и валентные колебания С–N, амид II) [14].
Рис. 2.
Репрезентативные ИК-Фурье спектры (а) pSI (1) и pAsp (2) и эксклюзионные хроматограммы (б) конъюгата pAsp–His (2) и эквивалентной pAsp (10.6 кДа, 1). (Элюент 0.1 M KH2PO4/NaH2PO4 буфер, pH 7.0, скорость потока 0.5 мл/мин).

Варьирование условий и времени полимеризации позволяло получить pSI и pAsp с различной ММ. ММ полимеров оценивали методом вискозиметрии по характеристической вязкости pSI в ДМСО с использованием уравнения: $\left| n \right| = K{{M}^{\alpha }},$ где K = 1.32 × 10–2, α = 0.76 для полисукцинимидов [3]. Для различных pSI со временем полимеризации 0.75, 1.5, 3 и 5 ч среднемассовая ММ составила 3, 4.1, 7.6 и 8.4 кДа (степень полимеризации ~30, 41, 77, 85) соответственно. Ожидаемые ММ различных pAsp составили 4.2, 5.7, 10.6 и 11.8 кДа. ИК-Фурье спектры изученных pSI (как и pAsp) были идентичны между собой (рис. 2а). Таким образом, повышение времени реакции не приводило к побочной модификации полимеров. Для дальнейшего изучения были выбраны pSI и pAsp с ММ 7.6 и 10.6 кДа соответственно, для которых степень полимеризации в условиях реакции выходила на предельные значения.
Для получения конъюгата pAsp с L-гистидином (L-His) аминокислоту предварительно модифицировали по стандартной реакции этерификации с триметилхлорсиланом (рис. 1б), характеризующейся мягкими условиями и незначительной рацемизацией [22]. Сшивку полученного продукта, метилового эфира L-His (His-OMe), с pSI проводили в ДМСО в присутствии DBA (рис. 1в). В результате аминолиза pSI происходило раскрытие сукцинимидных циклов с образованием амидной связи между боковой карбоксильной группой Asp и аминогруппой His-OMe. Для повышения степени модификации полисукцинимидного остова варьировали молярное отношение His-OMe и мономерных единиц pSI от 0.1 до 0.6. Для получения конъюгатов pAsp с гистидином (pAsp–His) выделенные конъюгаты pSI–His-OMe подвергали щелочному гидролизу для раскрытия не прореагировавших сукцинимидных циклов и удаления метильной группы с присоединенных остатков His-OMe.
Содержание L-His в конъюгатах определяли с помощью колориметрической реакции Паули c сульфаниловой кислотой, селективной к остаткам гистидина/тирозина в полипептидах [23], по калибровочному графику для гистидина. Обнаружено, что полученные конъюгаты pAsp–His характеризовались сходной степенью модификации 1.4 ± 0.5%, которая почти не зависела от исходной концентрации His-OMe в реакционной смеси. Известно, что алифатические амины с первичными аминогруппами реагируют эквимолярно с сукцинимидными единицами, образуя полностью замещенные полиаспартамиды [13]. В работе [19] эффективность модификации pAsp остатками алифатических аминокислот уменьшалась с 65 до 15% в ряду глицин, β-аланин, γ-аминомасляная кислота, α-аминокислоты, что отражало эффект пространственной доступности аминогруппы и размера молекулы. Можно предположить, что пониженная доступность α-аминогруппы L-His и больший размер этого аминокислотного остатка обусловливали низкую эффективность его присоединения к полисукцинимидному остову.
По результатам эксклюзионной ВЭЖХ в водном растворе конъюгат pAsp–His обладал значительно большим временем удерживания (t), чем эквивалентная pAsp (рис. 2б). На основании зависимостей t и ММ различных pAsp установлено, что ММ pAsp–His составляла ∼1.1 кДа, что свидетельствовало о частичном расщеплении полипептида в процессе модификации. Обнаружено, что, как pAsp–His, так и pAsp, подвергались постепенному гидролизу при инкубации в водных растворах (данные не показаны), поэтому для тестирования использовали свежеприготовленные растворы полимеров. Можно предположить, что расщепление синтезированных полипептидов, происходило вследствие автокаталитической реакции в водном растворе, механизм которой предполагается установить впоследствии.
Антиоксидантная активность конъюгата pAsp–His и комплексов гистидина. Принимая во внимание, что L-His и его производные входят в состав олигопептидов, выполняющих антиоксидантную функцию [26], было проведено сравнительное изучение способности конъюгата pAsp–His и трипептида глутатиона инактивировать стабильный радикал DPPH и в качестве каталазы разлагать пероксид водорода (H2O2). Соответствующие тесты основаны на колориметрическом определении остаточной концентрации окрашенных индикаторов – DPPH-радикала [24] и молибденового комплекса с H2O2 [24]. Установлено, что конъюгат pAsp–His, также как и не модифицированная pAsp не вызывали обесцвечивания раствора DPPH-радикала и не ускоряли разложение H2O2 в широком (до 2 мг/мл) диапазоне концентраций (данные не показаны). Следовательно, изучаемые полипептиды в отличие от глутатиона (EC50 ≈ 250 µM [24]) не проявляли выраженной радикал-связывающей способности, а также не обладали каталазной активностью.
Дополнительно изучено влияние конъюгата pAsp–His на образование радикала СОА. Для генерации и детекции СОА была оптимизирована редокс-реакция с участием НАДН, PMS и соли тетразолия, MTS. В этой реакции НАДН выступает донором электронов, которые передаются с помощью медиатора PMS на MTS с образованием окрашенного продукта его восстановления, формазана. Подобная реакция лежит в основе тестов на цитотоксичность с использованием тетразолиевых индикаторов, выявляющих восстановленные динуклеотиды НАД(Ф), содержание которых пропорционально количеству и метаболической активности клеток [25].
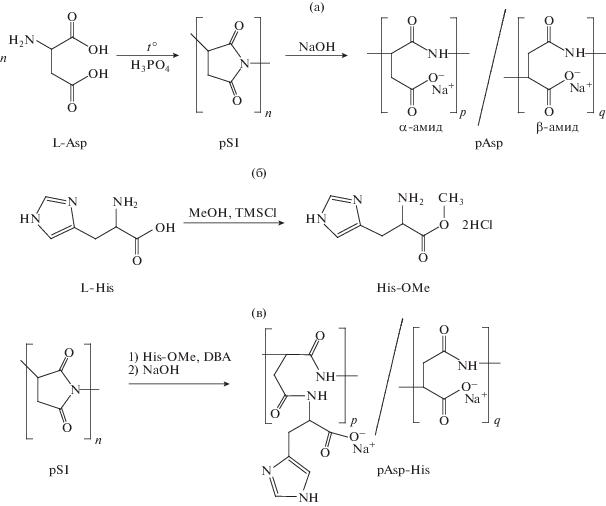
Как показано ранее [27], редокс-реакция ингибируется СОД, поскольку растворенный O2 выступает конкурентным акцептором электронов. Образующийся в результате его одноэлектронного восстановления СОА, в свою очередь, реагирует с MTS или претерпевает дисмутацию с образованием H2O2 и O2. На рис. 3 показана типичная кинетическая кривая реакции восстановления MTS, которая частично ингибируется СОД-2 (пропорционально активности фермента в растворе).
Рис. 3.
Кинетика реакции восстановления MTS редокс-системой НАДН/PMS, сопровождающейся генерацией супероксид-аниона, без ингибиторов (1) и в присутствии СОД-2 (100 ед./мл, 2).
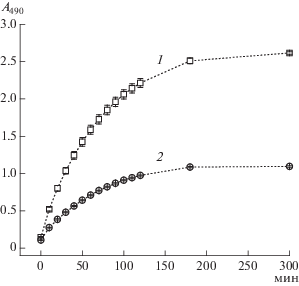
Обнаружено, что конъюгат pAsp–His в концентрации ≥100 мкг/мл проявлял выраженную способность ингибировать восстановление MTS системой НАДН/PMS, сопоставимую с действием 100 ед./мл СОД-2 (~20 мкг/мл) (табл. 1). Исходные компоненты конъюгата (pAsp, L-His и их композиции) не влияли на протекание этой реакции даже в значительно более высоких концентрациях. В совокупности, полученные результаты свидетельствовали о том, что синтезированный конъюгат pAsp–His катализировал разложение СОА вследствие СОД-подобной активности.
Таблица 1.
Влияние pAsp, pAsp–His и их комплексов с ионами металлов (50 мкМ) на сигнал восстановления MTS в условиях максимальной скорости реакции
| № | Пептид | Ион металла | А490 |
|---|---|---|---|
| 1 | pAsp, 200 мкг/мл | – | 2.53 ± 0.01 |
| 2 | pAsp, 200 мкг/мл | Мn+2 | 2.02 ± 0.04 |
| 3 | pAsp, 200 мкг/мл | Сu+2 | 2.36 ± 0.01 |
| 4 | pAsp–His, 100 мкг/мл | – | 0.67 ± 0.01 |
| 5 | pAsp–His, 100 мкг/мл | Мn+2 | 0.38 ± 0.01 |
| 6 | pAsp–His, 100 мкг/мл | Сu+2 | 0.46 ± 0.01 |
| 7 | Контроль (без эффекторов) | 2.38 ± 0.02 | |
| 8 | Контроль (СОД-2) | 0.73 ± 0.01 | |
Следует отметить, что компоненты активных центров супероксиддисмутаз могут обладать супероксиддисмутазной активностью. Способность катализировать дисмутацию СОА была, в частности, обнаружена в отсутствие пептидного компонента у солей марганца, а также комплексов гистидин-содержащих олигопептидов с медью и никелем [28]. Функция аминокислотных остатков активного центра СОД заключается, по-видимому, в усилении и регуляции каталитических свойств металлического кофактора (марганца, меди, никеля и железа) путем образования координационных связей c апоферментом [28].
В связи с этим было дополнительно изучено действие кофакторов Mn2+ и Cu2+ на активность конъюгата pAsp–His и его компонентов (табл. 1 и 2). Эти ионы металлов ингибировали реакцию восстановления MTS пропорционально концентрации. Для обнаружения действия пептидных соединений была подобрана концентрация иона металла, вызывающая незначительное ингибирование (50 мкМ). Установлено, что добавление L-His, но не L-Asp, усиливало ингибирующий эффект Mn2+ пропорционально концентрации аминокислоты в миллимолярном диапазоне (табл. 2). Фактор ингибирования композиций Mn2+ с L-His (1 и 10 мМ) составил 1.3 и 5.8 раз соответственно.
Таблица 2.
Влияние гистидина, трипептида гистидина и их комплексов с ионами металлов (50 мкМ) на сигнал восстановления MTS в условиях максимальной скорости реакции
| № | Пептид | Ион металла | А490 |
|---|---|---|---|
| 1 | L-His, 10 мM | – | 2.39 ± 0.03 |
| 2 | ННН, 200 мкг/мл | – | 2.35 ± 0.02 |
| 3 | L-His, 1 мМ | Мn+2 | 1.81 ± 0.06 |
| 4 | L-His, 10 мМ | Мn+2 | 0.41 ± 0.03 |
| 5 | ННН, 200 мкг/мл | Мn+2 | 0.15 ± 0.04 |
| 6 | L-His, 10 мM | Сu+2 | 2.26 ± 0.02 |
| 7 | ННН, 200 мкг/мл | Сu+2 | 1.85 ± 0.38 |
| 8 | Контроль (без эффекторов) | 2.38 ± 0.02 | |
| 9 | Контроль (СОД-2) | 0.73 ± 0.01 | |
Таким образом, полученные результаты показали, что входящий в активный центр многих ферментов остаток L-His оказался каталитически активным в супероксиддисмутазной реакции, усиливающим действие кофактора. Повышенные его концентрации, по-видимому, способствовали образованию координационного соединения с Mn2+, который имитировал активный центр СОД-2. При этом синтетический трипептид L-His (HHH) в композиции с Mn2+ проявлял еще более высокую ингибирующую активность (15.9 раз) при концентрации 200 мкг/мл (~0.5 мМ), что можно объяснить эффектом пространственного сближения аминокислотных остатков в пептиде. Ионы меди заметно ингибировали изучаемую реакцию в 1.3 раза только в композиции с HHH (табл. 2).
Полученные результаты позволили раскрыть некоторые важные особенности проявления супероксиддисмутазной активности у соединений, содержащих остатки L-His. В отличие от L-His и его трипептида, действие конъюгата pAsp–His незначительно усиливалось ионами металлов (табл. 1), то есть для проявления активности pAsp–His не требовался ион металла в качестве кофактора. Можно предположить, что синтетическая pAsp создавала молекулярное окружение, способствующее в отсутствие редокс-активных металлов дисмутации СОА, катализируемой L-His. На возможность ускорения подобной реакции только пептидным компонентом указывает сохранение остаточной супероксиддисмутазной активности у апофермента после удаления кофактора [28].
Влияние конъюгата pAsp–His на жизнеспособность клеток. Для выявления биоактивных свойств у конъюгата pAsp–His методом пролиферативного МТТ-теста изучено его влияние на жизнеспособность фибробластов кожи человека HSF и опухолевых клеток линии PC-3 (рис. 4). Обнаружено, что pAsp–His проявлял цитотоксический эффект по отношению к клеткам PC-3 (IC50 = 782 ± 23 мкг/мл, 72 ч). При этом во всем изученном диапазоне концентраций конъюгат ингибировал жизнеспособность HSF сходным образом на 10–20% (рис. 4), что указывало на его модулирующее, а не цитотоксическое действие.
Рис. 4.
Зависимость жизнеспособности клеток (% от необработанных клеток, принятых за 100%) аденокарциномы простаты PC-3 (1) и фибробластов кожи человека (2) от концентрации конъюгата pAsp–His по данным МТТ-теста (72 ч).
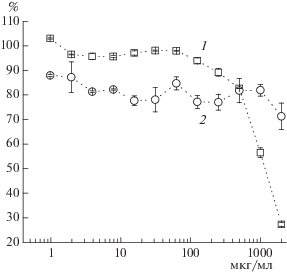
Известно, что опухолевые клетки обладают измененным метаболизмом, обусловливающим повышенное содержание АФК и чувствительность к веществам анти- и прооксидантного действия [29]. Можно предположить, что конъюгат pAsp–His смещал баланс в сторону образования из СОА более реакционноспособных АФК (H2O2 и гидроксил-радикала), и поэтому оказывал более выраженное воздействие на опухолевые клетки, чем на условно нормальные HSF.
Полученные в работе результаты показали новые подходы к созданию каталитически активных производных pAsp и L-His, аналогов ферментов. Благодаря выявленной активности, конъюгат pAsp–His можно рассматривать в качестве прототипа макромолекулярного аналога СОД и возможной альтернативы рекомбинантным СОД [30] и их низкомолекулярным пептидомиметикам [31]. Эти препараты тестируются в качестве компонентов ряда терапевтических средств, обладающих цитопротекторным и противоопухолевым действием [30]. Преимущество предложенных аналогов СОД состоит в относительной простоте получения и модификации структуры, а также возможности их применения в отсутствие потенциально токсичных металлов переходной группы, участвующих в реакциях фентоновского окисления [24].
Таким образом, в работе получен и охарактеризован конъюгат полиаспарагиновой кислоты с L-гистидином. С использованием оптимизированного теста на супероксид-анион установлено, что разработанный конъюгат обладал активностью СОД, не требующей металлического кофактора. Показана роль молекулярного окружения остатка гистидина в проявлении этой активности. Выявлена повышенная цитотоксичность конъюгата к опухолевым клеткам по сравнению с нормальными фибробластами. Эти результаты могут быть использованы для создания синтетических пептидных препаратов – аналогов природных супероксиддисмутаз.
Работа выполнена с использованиием оборудования Междисциплинарного центра коллективного пользования КФУ.
Авторы благодарят сотрудника НИЛ “Реологические и термохимические исследования” КФУ И.Т. Ракипова за проведение вискозиметрических измерений.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (проект №18-34-00766), в рамках программы повышения конкурентоспособности Казанского (Приволжского) федерального университета среди ведущих мировых научно-образовательных центров на 2013–2020 гг.
Список литературы
Lalatsa A., Schätzlein A.G., Mazza M., Le T.B.H., Uchegbu I.F. // J. Control. Release. 2012. V. 161. № 2. P. 523–536.
Kunioka M. // Macromol. Biosci. 2004. V. 4. № 3. P. 324–329.
Thombre S.M., Sarwade B.D. // J. Macromol. Sci. A. 2005. V. 42. № 9. P. 1299–1315.
Itaka K., Ishii T., Hasegawa Y., Kataoka K. // Biomaterials. 2010. V. 31. № 13. P. 3707–3714.
Akagi T., Baba M., Akashi M. Polymers in Nanomedicine / Ed. S. Kunugi, T. Yamaoka. Berlin, Heidelberg: Springer Berlin Heidelberg. 2012. P. 31–64.
Lalive P.H., Neuhaus O., Benkhoucha M., Burger D., Hohlfeld R., Zamvil S.S., Weber M.S. // CNS Drugs. 2011. V. 25. № 5. P. 401–414.
Hoyer J., Neundorf I. // Acc. Chem. Res. 2012. V. 45. № 7. P. 1048–1056.
Ramsay E., Gumbleton M. // J. Drug. Target. 2002. V. 10. № 1. P. 1–9.
Midoux P., Pichon C., Yaouanc J.J., Jaffres P.A. // Brit. J. Pharmacol. 2009. V. 157. № 2. P. 166–178.
Zhao Z.X., Gao S.Y., Wang J.C., Chen C.J., Zhao E.Y., Hou W.J., Feng Q., Gao L.Y., Liu X.Y., Zhang L.R. et al. // Biomaterials. 2012. V. 33. № 28. P. 6793–6807.
Carvajal-Rondanelli P., Arostica M., Alvarez C.A., Ojeda C., Albericio F., Aguilar L.F., Marshall S.H., Guzman F. // Amino Acids. 2018. V. 50. № 5. P. 557–568.
Якунина Л.Д., Курбанов Р.А., Бондарь О.В., Абдуллин Т.И. // Клеточная трансплантология и тканевая инженерия (КТТИ). 2012. Т. VII. № 3. С. 173–176.
Salakhieva D., Shevchenko V., Németh C., Gyarmati B., Szilágyi A., Abdullin T. // Int. J. Pharm. 2017. V. 517. № 1. P. 234–246.
Németh C., Szabó D., Gyarmati B., Gerasimov A., Varfolomeev M., Abdullin T., László K., Szilágyi A. // Eur. Polym. J. 2017. V. 93. P. 805–814.
Lim S., Nguyen M.P., Choi Y., Kim J., Kim D. // Biomacromoleciles. 2017. V. 18. № 8. P. 2402–2409.
Rivera-Rodríguez G.R., Alonso M.J., Torres D. // Eur. J. Pharm. Biopharm. 2013. V. 85. № 3. P. 481–487.
Lee M., Jeong J., Kim D. // Biomacromolecules. 2015. V. 16. № 1. P. 136–144.
Jang J., Cha C. // Biomacromolecules. 2018. V. 19. № 2. P. 691–700.
Kim J.-H., Son C.M., Jeon Y.S., Choe W.-S. // J. Polym. Res. 2011. V. 18. № 5. P. 881–890.
Carrea G., Colonna S., Kelly D.R., Lazcano A., Ottolina G., Roberts S.M. // Trends Biotechnol. 2005. V. 23. № 10. P. 507–513.
Qiu W., He L., Chen Q., Luo W., Yu Z., Yang F., Tang J. // Tetrahedron Lett. 2009. V. 50. № 37. P. 5225–5227.
Li J., Sha Y. // Molecules. 2008. V. 13. № 5. P. 1111–1119.
B Laboratory Protocols in Applied Life Science / P.S. Bisen. Boca Raton, London, New York: CRC Press, Taylor & Francis Group, 2014. 1826 p.
Akhmadishina R.A., Garifullin R., Petrova N.V., Kamalov M.I., Abdullin T.I. // Front. Pharmacol. 2018. V. 9. P. 115.
Tsepaeva O.V., Nemtarev A.V., Abdullin T.I., Grigor’eva L.R., Kuznetsova E.V., Akhmadishina R.A., Ziganshina L.E., Cong H.H., Mironov V.F. // J. Nat. Prod. 2017. V. 80. № 8. P. 2232–2239.
Guiotto A, Calderan A, Ruzza P, Borin G. // Curr. Med. Chem. 2005. V. 12. № 20. P. 2293–2315.
Berridge M.V., Herst P.M., Tan A.S. // Biotechnol. Annu. Rev. 2005. V. 11. P. 127–152.
Barnese K., Gralla E.B., Valentine J.S., Cabelli D.E. // Proc. Natl. Acad. Sci. USA. 2012. V. 109. № 18. P. 6892–6897.
Liou G.-Y., Storz P. // Free Radical Res. 2010. V. 44. № 5. P. 479–496.
Carillon J., Rouanet J.M., Cristol J.P., Brion R. // Pharm. Res. 2013. V. 30. № 11. P. 2718–2728.
Salvemini D., Cuzzocrea S. // Crit. Care Med. 2003. V. 31. № 1. P. S29–S38.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология