

Журнал неорганической химии, 2022, T. 67, № 2, стр. 193-202
Апатитные фосфаты кальция: жидкофазное формирование, термические превращения, терминология и идентификация
И. Е. Глазов a, *, В. К. Крутько a, О. Н. Мусская a, А. И. Кулак a
a Институт общей и неорганической химии НАН Беларуси
220072 Минск, ул. Сурганова, 9/1, Беларусь
* E-mail: che.glazov@mail.com
Поступила в редакцию 19.07.2021
После доработки 10.09.2021
Принята к публикации 15.09.2021
- EDN: BOKEBU
- DOI: 10.31857/S0044457X22020040
Аннотация
Синтезированы апатитные фосфаты кальция при pH 9–11, Ca/P = 1.50–1.67 в варьируемых условиях выделения. Разработана комплексная методика идентификации включений аморфного фосфата кальция при осаждении гидроксиапатита в неравновесных условиях, включающая методы РФА/ИКС/ДТА. Максимальная стабилизация включений аморфной фазы в структуре гидроксиапатита происходит при pH осаждения 9, быстром взаимодействии реагентов, отсутствии стадии созревания осадка под маточным раствором и тщательной дегидратации этанолом с последующим прогревом при 400°С. С формированием аморфных включений связаны несоответствия в описании физико-химических свойств апатитных фосфатов кальция. Предложена схема жидкофазного формирования и термических превращений апатитных фосфатов кальция в условиях различной степени равновесности и образования: однофазного стехиометрического гидроксиапатита, бифазных композитов на основе кальцийдефицитных гидроксиапатитов, аморфного фосфата кальция.
ВВЕДЕНИЕ
Используемые для замещения поврежденной костной ткани синтетические биоматериалы на основе апатитных фосфатов кальция (ФК), таких как гидроксиапатит (ГА) и трикальцийфосфат (ТКФ), вызывают большой интерес [1, 2], обусловленный их сродством к минеральной составляющей костной ткани – биогенному апатиту [3]. Биогенные апатиты костной ткани имеют мольное отношение Ca/P = 1.37–1.77 [4] за счет инкорпорации примесных ионов, в отличие от стехиометрического ГА Ca10(PO4)6(OH)2 с мольным отношением Ca/P = 1.67 и апатитного ТКФ (apatitic TCP) Ca9HPO4(PO4)5OH [5] с Ca/P = 1.50. Подобные отклонения от стехиометрии характерны не только для апатитов природного происхождения, но и для синтетических апатитных ФК. По данным теоретических расчетов [6], беспримесный и бездефектный стехиометрический ГА с отношением Ca/P = 1.6(6) имеет моноклинную решетку P21/b. Однако у природных и синтетических апатитов сингония кристаллической решетки относится к гексагональному типу (P63/m) [7] вследствие образования дефектов и/или инкорпорации примесных ионов, что позволяет отнести их к нестехиометрическим ГА [6]. В отсутствие примесных ионов сингония апатитных ФК сохраняется при отношении 1.33 < Ca/P ≤ 1.67, что соответствует формуле Ca10 –x(HPO4)x(PO4)6 –x(OH)2 –x, где 0 ≤ x < 2 [8]. Устойчивость гексагональной сингонии ГА связана с гибкостью кристаллической решетки, способной дополнительно вмещать анионы F–, Cl–, ${\text{NO}}_{3}^{ - },$ ${\text{CO}}_{3}^{{2 - }},$ ${\text{SiO}}_{4}^{{4 - }}$ и катионы от ${\text{NH}}_{4}^{ + }$ до La3+ и As5+ [3, 6, 9–12]. Именно гибкость решетки и многообразие возможных составов апатитных ФК обусловливают вариабельность параметров кристалличности, термической устойчивости, морфологии и растворимости материалов на их основе [6]. Известно, что у апатитного ТКФ отношение постоянных решетки a/c превышает этот показатель у стехиометрического ГА на 0.22% [13]. Подобное искажение решетки является достаточным для увеличения растворимости от стехиометрического ГА с KПР ~ 155 [14] до апатитного ТКФ с KПР ~ 114. Кроме того, термодинамическая устойчивость апатитного ТКФ (∆Hf = –12708 кДж/моль) меньше, чем стехиометрического ГА (∆H f = –13477 кДж/моль) [15], что соответствует повышенной реакционной способности и биоактивности апатитного ТКФ.
Одним из подходов к управлению составом биоматериалов на основе апатитных ФК является варьирование условий их осаждения и выделения [16]. Зависимость состава апатитов от величины pH, ионной силы, температуры, диэлектрической проницаемости маточного раствора и времени созревания [17] определяют физико-химические характеристики аморфного ФК (АФК) CaxHy(PO4)z · nH2O, n = 3.0–4.5 [2], который в первую очередь образуется при жидкофазном синтезе ФК. В отсутствие примесных ионов мольное отношение Ca/P для АФК составляет 1.00–1.50 и зависит от величины pH и диэлектрической проницаемости среды осаждения. Метастабильная природа АФК обусловливает его склонность к гидролизу с образованием устойчивых фаз – от дикальцийфосфата дигидрата CaHPO4 · 2H2O при pH 6 до стехиометрического ГА при pH 11. Апатитные ФК образуются при pH ≥ 9 из АФК с отношением Ca/P ≈ 1.50 (аморфный ТКФ), структура которого представлена классическими кластерами Познера [17] Ca9(PO4)6 · nH2O, n = 3.0–4.5.
Нарушение условий получения апатитных ФК способствует отклонению их состава от стехиометрического и образованию сопутствующих фаз, таких как CaO и кристаллический ТКФ Ca3(PO4)2, что сопровождается изменением свойств материалов на их основе. Низкотемпературные апатиты являются рентгеноаморфными, поэтому фазовый состав и отношение Ca/P апатитных ФК оценивают по их превращениям после 800°С [7]. Высокая чувствительность фазового состава апатитов к условиям получения, а также их рентгеноаморфная структура обусловливают необходимость разработки единой схемы взаимных превращений АФК и апатитных ФК во избежание разночтений и несоответствий терминологии при их описании.
В настоящей работе исследованы фазовые переходы апатитных ФК в различных условиях жидкофазного синтеза, выделения и термообработки с целью разработки комбинированной идентификации и приведения к единообразию терминологии, встречающейся при их описании: однофазный/стехиометрический ГА, нестехиометрический/кальцийдефицитный ГА, многофазный ФК, аморфный/апатитный/кристаллический ТКФ. Для достижения поставленной цели апатитные ФК синтезировали в условиях индуцированных отклонений (скорость взаимодействия реагентов, время выдерживания осадков под маточным раствором, дегидратация) при сохранении величины pH осаждения 9 либо 11 и отношения Ca/P = 1.50 или 1.67.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез ФК осуществляли взаимодействием растворимых солей Ca2+ и ${\text{HPO}}_{4}^{{2 - }}{\text{:}}$ CaCl2 · 2H2O (Sigma Aldrich) либо Ca(NO3)2 · 4H2O (Carl Roth), (NH4)2HPO4 (Carl Roth); величину pH регулировали добавлением раствора аммиака (0.907 г/см3, ч. д. а., База № 1 химреактивов, РФ).
Серия 1. Апатитные ФК получали в соответствии с широко используемыми методиками синтеза ГА [18–20] и ТКФ [21]. Осаждение ГА-1 осуществляли медленным смешиванием реагентов при pH 11 и мольном отношении Ca/P = 1.67, после чего осадок выдерживали под маточным раствором в течение 4 сут. Синтез ТКФ-1 проводили быстрым смешиванием реагентов при pH 9 и мольном отношении Ca/P = 1.50, осадок выдерживали под маточным раствором в течение 30 мин и отделяли на фильтре.
Серия 2. Синтез апатитных ФК проводили в условиях отклонения взаимодействия реагентов от вышеприведенных методик (серия 1) без изменения величины pH и отношения Ca/P. Синтез ГА-2 осуществляли быстрым смешиванием реагентов с последующим выдерживанием осадков под маточным раствором в течение 30 мин и фильтрованием. Осаждение ТКФ-2 проводили при медленном смешивании реагентов и выдерживании осадка под маточным раствором в течение 4 сут.
Серия 3. Образцы АФК синтезировали согласно известной методике [22] при варьировании величины pH осаждения и отношения Ca/P. Осадки получали быстрым смешиванием реагентов без выдерживания под маточным раствором и быстро отделяли на фильтре. Образцы АФК-1 и АФК-2 осаждали при pH 9 и Ca/P = 1.50, а образец АФК-3 – при pH 11 и Ca/P = 1.67. Осадки апатитных ФК и АФК промывали дистиллированной водой до pH 7.0–7.5, а образцы АФК-1 и АФК-3 дополнительно обезвоживали этанолом для предотвращения гидролиза. Образцы идентифицировали после высушивания при 60°С до ксерогелей с постоянной массой и последовательной термообработки на воздухе при 400, 600, 700, 800, 1000 и 1200°С в течение 2–5 ч.
Рентгенофазовый анализ (РФА) проводили на дифрактометре Advance D8 (Bruker, Германия) с СuKα-излучением, λ = 1.5405 Å. Фазовый состав образцов и размер кристаллитов определяли с использованием программного обеспечения DIFFRAC.EVA. Дифрактограммы ФК сравнивали со стандартами ICDD PDF-2 и литературными данными: АФК [13], α-ТКФ #01-070-0364 [23], β-ТКФ #01-070-2065 [5], ГА #01-084-1998 [18, 24]. ИК-спектроскопию (ИКС) образцов осуществляли на ИК-Фурье-спектрометре Tensor-27 (Bruker, Германия) в диапазоне 400–4000 cм–1 с использованием таблеток бромида калия (2 мг образца на 800 мг бромида калия). Положение полос в ИК-спектрах образцов сравнивали с литературными данными: АФК [22], α-ТКФ [23], β-ТКФ [5, 25], ГА [5, 18, 24], пирофосфат-ион ${{{\text{P}}}_{{\text{2}}}}{\text{O}}_{7}^{{4 - }}$ [5, 26]. Совмещенный термический анализатор STA 409 PC LUXX (Netzsch, Германия) применяли для проведения дифференциального термического анализа (ДТА) образцов на воздухе при скорости нагрева 10 град/мин с использованием для сравнения литературных данных: АФК [13], ГА [5, 24, 27].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Основные различия между методиками получения образцов ГА-1 [18–20], ТКФ-1 [21] и АФК-1 [22] заключались в варьировании степени равновесности условий получения, которая увеличивалась при снижении скорости взаимодействия реагентов и увеличении времени выдерживания осадков под маточным раствором и наоборот. Синтез ГА-1 осуществляли в относительно равновесных условиях медленного взаимодействия реагентов (скорость введения ${\text{HPO}}_{4}^{{2 - }}$ ~ 10–5 моль/с) и длительного созревания осадка (в течение 4 сут), что позволило получить стехиометрический ГА – наиболее термодинамически устойчивый апатитный ФК [15]. Образец ТКФ-1 осаждали в неравновесных условиях быстрого взаимодействия ${\text{HPO}}_{4}^{{2 - }}$ (~0.1 моль/c) и выдерживали под маточным раствором в течение 30 мин, что способствовало образованию метастабильных фаз ФК.
По данным РФА (рис. 1а), основной фазой ГА-1 вплоть до 700°С является рентгеноаморфный апатит с размером кристаллитов 13.6–25.0 нм (табл. 1), который на дифрактограммах представлен широкими рефлексами при 2θ = 25.8°, 31.8°, 32.8°, 34.0°, 39.8°. Подобное уширение рефлексов обусловлено нарушением дальнего порядка [28] в структуре низкотемпературных апатитов, для описания которых подходит термин “аморфизированный”, в отличие от “аморфных” соединений, которые на дифрактограммах представлены рентгеноаморфным гало вследствие полного отсутствия в структуре дальнего порядка. Дальнейшее нагревание до 1200°С сопровождается уменьшением ширины рефлексов ГА-1, что указывает на повышение кристалличности (табл. 1) гексагонального ГА в соответствии со схемой превращений (рис. 2, I).
Рис. 1.
Дифрактограммы ГА-1 (а), ТКФ-1 (б), АФК-1 (в) и АФК-2 (г, 1), АФК-3 (г, 2), ТКФ-2 (г, 3), ГА-2 (г, 4) после прогрева при различных температурах. * – ГА, a – α-ТКФ, b – β-ТКФ.
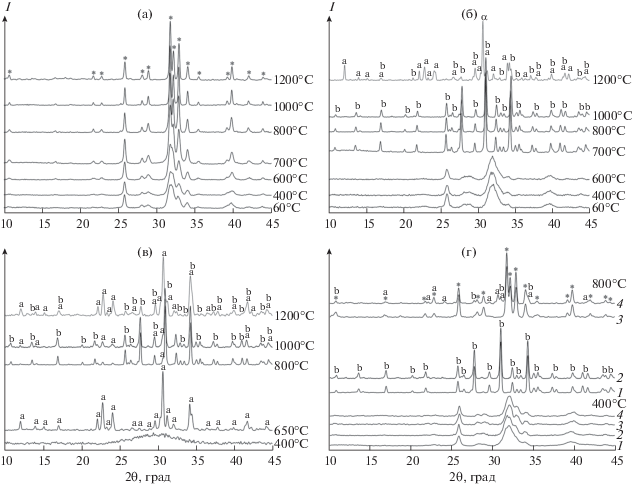
Таблица 1.
Размер кристаллитов основной фазы ФК при различных температурах
| Параметр | Температура прогрева, °С | ||||||
|---|---|---|---|---|---|---|---|
| 60 | 400 | 600 | 700 | 800 | 1000 | 1200 | |
| Образец ГА-1 | |||||||
| Размер кристаллитов, нм | 13.6 | 14.4 | 17.6 | 25.0 | 39.6 | 58.8 | 75.5 |
| Фазовый состав | Сa10(PO4)6(OH)2 | ||||||
| Образец ГА-2 | |||||||
| Размер кристаллитов, нм | 12.7 | 13.2 | 17.1 | 22.1 | 51.3 | – | 75.7 |
| Фазовый состав | Сa10–x(HPO4)x(PO4)6 –x(OH)2 –x Ca9(PO4)6 |
Сa10(PO4)6(OH)2 α,β-Ca3(PO4)2 |
|||||
| Образец ТКФ-1 | |||||||
| Размер кристаллитов, нм | 8.5 | 8.5 | 8.6 | 61.0 | 61.0 | 62.0 | 45.0 |
| Фазовый состав | Сa9HPO4(PO4)5OH Ca9(PO4)6 |
β-Ca3(PO4)2 | α-Ca3(PO4)2 β-Ca3(PO4)2 |
||||
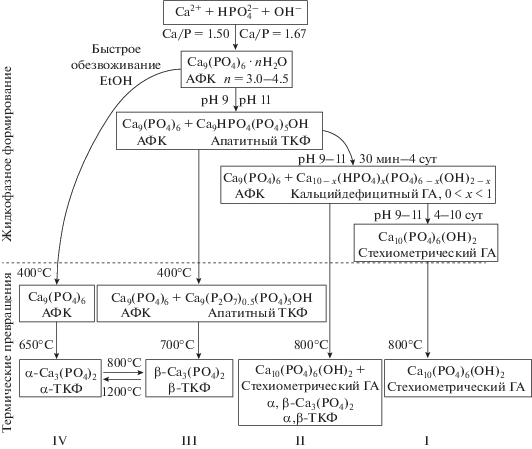
Дифрактограммы ксерогеля ТКФ-1 после 60–600°С (рис. 1б) представлены уширенными рефлексами при 2θ = 25.9°, 32.0°, 34.0°, 39.7° аморфизированного апатита с расчетным размером кристаллитов ~8.5 нм, который не кристаллизуется до 600°С (табл. 1). Причем апатит в составе ТКФ-1 характеризуется повышенной аморфизацией, т.е. пониженной кристалличностью в сравнении с однофазным ГА (рис. 1а), что может быть связано с присутствием аморфной фазы АФК (рис. 2, III), которая образуется в неравновесных условиях получения ТКФ-1 и является причиной устойчивости образца к термической кристаллизации. Смесь апатита и АФК в составе ТКФ-1 после 700–1000°C превращается в кристаллический β-ТКФ, а после 1200°С – в α-ТКФ (табл. 1).
Термообработка при 800–1200°С апатита с Ca/P = 1.50 является распространенным способом получения индивидуальных α- и β-ТКФ, которые не могут быть получены жидкофазным осаждением напрямую (рис. 2, III). Термические превращения апатитных ФК при 800°С описывают [5, 7] уравнением реакции (1):
(1)
$\begin{gathered} {\text{C}}{{{\text{a}}}_{{10--x}}}{{({\text{HP}}{{{\text{O}}}_{{\text{4}}}})}_{x}}{{({\text{P}}{{{\text{O}}}_{4}})}_{{6--}}}_{x}{{({\text{OH}})}_{{2--}}}_{x} \to \\ \to (1--x){\text{C}}{{{\text{a}}}_{{10}}}{{({\text{P}}{{{\text{O}}}_{4}})}_{6}}{{({\text{OH}})}_{2}} + 3x{{\beta - C}}{{{\text{a}}}_{3}}{{({\text{P}}{{{\text{O}}}_{4}})}_{2}}, \\ \end{gathered} $Наименее устойчивой метастабильной фазой ФК является АФК, который получают в очень неравновесных условиях, задаваемых быстрым введением ${\text{HPO}}_{4}^{{2 - }}$ (~0.1 моль/c), дегидратацией этанолом и прогревом при 60–400°С для максимального подавления гидролиза. Аморфная фаза в составе АФК-1 при pH 9 не превращается в апатит в течение 30 мин и на дифрактограммах после 400°С (рис. 1в) характеризуется широким рентгеноаморфным гало при 2θ = 25°–35°. Положение гало АФК обусловливает возможность его перекрывания с наиболее интенсивными апатитными рефлексами (рис. 1а, 1б), что подтверждает предположение об аморфизации апатита в составе ТКФ-1 (рис. 1б) за счет присутствия включений АФК. После 650°С АФК кристаллизуется в α-ТКФ (∆Hf = –4113 кДж/моль [30]), который является термодинамически менее устойчивым по сравнению с β-ТКФ (∆Hf = –4121 кДж/моль), что подчиняется правилу ступеней Оствальда: продуктом фазового перехода является состояние с энергией, максимально близкой к исходной, а не наиболее устойчивое состояние [17, 23]. После 800–1000°С α-ТКФ превращается в β-ТКФ, который после 1200°С снова переходит в α-модификацию (рис. 2, IV).
Полученные физико-химические характеристики образцов ГА-1, ТКФ-1, АФК-1 использовали в качестве стандартов сравнения при идентификации образцов АФК-2, АФК-3, ГА-2, ТКФ-2, осаждаемых в условиях отклонения от известных методик. Отклонения условий получения образцов АФК-2 и АФК-3 от АФК-1 (pH 9, Ca/P = 1.50) создавали отсутствием обработки этанолом для АФК-2 (pH 9, Ca/P = 1.50) либо увеличением pH осаждения до 11 и отношения Ca/P до 1.67 для АФК-3, что сопровождалось частичным превращением аморфной фазы в аморфизированный апатит (рис. 1г, 400°С, кривые 1, 2). Образование апатита в составе АФК-2 при pH 9 подтверждает необходимость дегидратации АФК этанолом. В случае АФК-3 при pH 11 аморфная фаза частично превращается в апатит до обработки этанолом, что указывает на повышенную скорость данного превращения при pH 11 по сравнению с pH 9. Предполагается, что дегидратация этанолом способствует увеличению содержания включений аморфной фазы в образце АФК-3. В таком случае его повышенная кристалличность (рис. 1г, 400°С, кривая 2) в сравнении с АФК-2 (рис. 1г, 400°С, кривая 1) может быть связана с большим размером кристаллитов АФК-3, осажденного при pH 11, Ca/P = 1.67. Образцы АФК-2 и АФК-3 после 800°С представлены однофазным β-ТКФ (рис. 1г, 800°С, кривые 1, 2), что соответствует превращению апатитного ТКФ согласно уравнению реакции (1).
Образец ТКФ-2 осаждали при pH 9, Ca/P = = 1.50 в условиях медленного взаимодействия реагентов и длительного созревания, что способствовало образованию однофазного аморфизированного апатита (рис. 1г, 400°С, кривая 3). После 800°С образец ТКФ-2 представлен однофазным ГА (рис. 1г, 800°С, кривая 3), что указывает на неустойчивость апатитного ТКФ при его длительном выдерживании под маточным раствором (рис. 2, I). Превращение апатитного ТКФ в стехиометрический ГА происходит путем растворения-осаждения [6] с выделением ионов ${\text{HPO}}_{4}^{{2 - }},$ причем движущей силой превращения является увеличение термодинамической устойчивости апатита при росте его отношения Ca/P.
Синтез ГА-2 проводили быстрым взаимодействием реагентов при pH 11, Ca/P = 1.67 и выдерживанием под маточным раствором в течение 30 мин. Полученный апатит (рис. 1г, 400°С, кривая 4) при нагревании меньше склонен к кристаллизации, чем однофазный ГА (табл. 1), за счет присутствия включений АФК. Прогрев при 800°С способствовал превращению апатита в составе ГА-2 в смесь 75 мас. % ГА, 18 мас. % α-ТКФ и 7 мас. % β-ТКФ (рис. 1г, 800°С, кривая 4). Согласно уравнению реакции (1), такая смесь ГА и β-ТКФ формируется при разложении кд-ГА с x ≈ 0.1, что указывает на неполноту созревания апатита в условиях получения ГА-2 (рис. 2, II). Образование α-ТКФ в составе ГА после 800°С наблюдалось ранее при синтезе ГА жидкофазным способом [31–33] и было связано с кристаллизацией ~19 мас. % включений АФК, стабилизированных в матрице основной фазы кд-ГА. Соответственно, образец ГА-2 после 400°С представлен бифазным композитом кд-ГА/АФК с Ca/P ≈ 1.62, который после 800°С превращается в многофазный композит ГА/α, β-ТКФ. В отличие от ГА-2, дополнительная фаза α-ТКФ не образуется после прогрева при 800°С бифазных композитов (апатитный ТКФ)/АФК в составе ТКФ-1 (рис. 1б) и АФК-2, АФК-3 (рис. 1г, 800°С, кривые 1, 2). Данный факт может свидетельствовать о дестабилизирующем влиянии основной фазы β-ТКФ на аллотропное превращение α-ТКФ, в отличие от стабилизирующего влияния фазы ГА в составе ГА-2.
По данным РФА образцов АФК-3, ГА-2, ГА-1 (pH 11, Ca/P = 1.67), увеличение степени равновесности условий получения ГА способствует увеличению отношения Ca/P конечного продукта от 1.50 до 1.67 (рис. 2, I).
Метод РФА позволяет идентифицировать системы кд-ГА/АФК, характерным признаком которых является уширение апатитных рефлексов по сравнению с однофазным стехиометрическим ГА, а также образование сопутствующей фазы α-ТКФ в апатитах с pH осаждения 11 после 800–1200°С. Уширение рефлексов является ненадежным признаком присутствия аморфных включений, а образование фазы α-ТКФ не наблюдалось в апатитах с pH осаждения 9, что указывает на необходимость привлечения дополнительных методов анализа включений АФК в структуре апатитных ФК.
Метод ИКС позволяет выявлять функционально-групповые особенности апатитов и идентифицировать включения АФК в составе апатитных ФК. Символьные обозначения колебаний различных функциональных групп для удобства отмечали порядковыми индексами: 1O–P–O, 2O–H апатита, 3P–O(H), 4P–O–P, 5O–C–O, 6H–O–H. ИК-спектры всех типов образцов (рис. 3) содержат полосы валентных асимметричных $\nu _{{as}}^{1},$ симметричных $\nu _{s}^{1}$, деформационных $\delta _{a}^{1}$ и $\delta _{b}^{1}$ колебаний O–P–O иона ${\text{PO}}_{4}^{{3 - }},$ а также полосы валентных ν6 (3200–3600 см–1) и деформационных δ6 (1660 см–1) колебаний H–O–H адсорбированной и структурной воды, удаление которой происходит после 400°C.
Рис. 3.
ИК-спектры ГА-1 (а), ТКФ-1 (б), АКФ-1 (в) и АФК-2 (г, 1), АФК-3 (г, 2), ТКФ-2 (г, 3), ГА-2 (г, 4) после прогрева при различных температурах. O–P–O ($\nu _{{as}}^{1},$ $\nu _{s}^{1},$ $\delta _{a}^{1},$ $\delta _{b}^{1},$), O–HГА (ν2, L2), P–O(H) (ν3), P–O–P $\left( {\nu _{s}^{4}} \right),$ O–C–O ($\nu _{{as}}^{5},$ δ5), H–O–H (ν6, δ6).
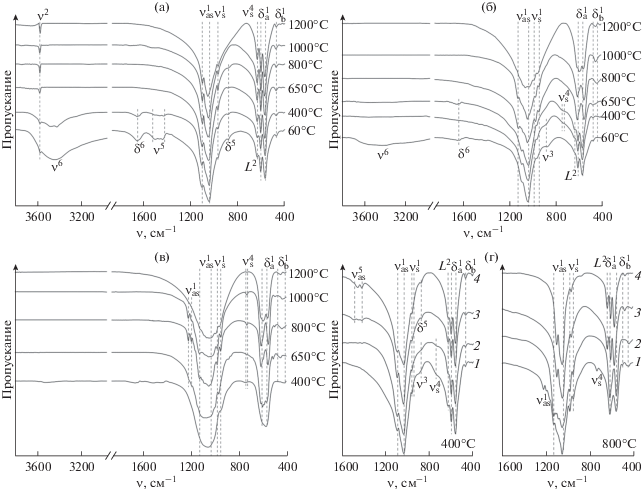
Стехиометрический ГА на ИК-спектрах ГА-1 (рис. 3а) представлен характеристическими полосами апатита $\nu _{{as}}^{1}$ (1090, 1040 см–1), $\nu _{s}^{1}$ (956 см–1), $\delta _{a}^{1}$ (603, 566 см–1), $\delta _{b}^{1}$ (472 см–1), валентных ν2 (3570 см–1) и либрационных L2 (633 см–1) колебаний O–H. Повышение температуры прогрева способствует кристаллизации апатита и уменьшению ширины полос колебаний O–P–O и O–H [34]. Полосы валентных асимметричных $\nu _{{as}}^{5}$ (1485, 1448, 1417 см–1) и деформационных δ5 (876 см–1) колебаний O–C–O свидетельствуют о внедрении в структуру апатита ионов ${\text{CO}}_{3}^{{2 - }},$ источником которых является CO2, растворенный в сильнощелочной среде при получении ГА-1. Карбонатное замещение в структуре ГА-1 относится к Б-типу [35] и сопровождается образованием катионных вакансий, что указывает на кальцийдефицитность ГА-1 по внедренным ионам ${\text{CO}}_{3}^{{2 - }}$ и подтверждает условный характер стехиометричности полученного ГА. Удаление ионов ${\text{CO}}_{3}^{{2 - }}$ из структуры апатита наблюдается после 650°C.
Образец ТКФ-1 при 60–650°С на ИК-спектрах (рис. 3б) представлен апатитными полосами $\nu _{{as}}^{1}$ (1092, 1036 см–1), $\nu _{s}^{1}$ (961 см–1), L2 (633 см–1), δ2 (603, 565 см–1), $\delta _{b}^{1}$ (472 см–1) и плечом ν3 при 876 см–1 валентного колебания P–O(H) иона ${\text{HPO}}_{4}^{{2 - }},$ которые являются уширенными за счет присутствия включений АФК. Присутствие плеча ν3 колебаний P–O(H) позволяет определить наличие отклонения отношения Ca/P апатитов от стехиометрического без термообработки образцов, что является одним из преимуществ метода ИКС по сравнению с РФА в идентификации апатитных ФК. После 400–650°С происходит термолиз ионов ${\text{HPO}}_{4}^{{2 - }},$ на что указывает появление плеча при 723 см–1 валентных симметричных колебаний P–O–P $\left( {\nu _{s}^{4}} \right)$ иона ${{{\text{P}}}_{{\text{2}}}}{\text{O}}_{7}^{{4 - }}$ (рис. 2, III). Апатитный ТКФ после 800–1000°С полностью превращается в β-ТКФ, о чем свидетельствует отсутствие на ИК-спектрах полос колебаний O–H, P–O(H), P–O–P (рис. 3б). После 1200°С полосы β-ТКФ $\nu _{{as}}^{1}$ (1120, 1080, 1044 см–1), $\nu _{s}^{1}$ (973, 943 см–1), $\delta _{a}^{1}$ (607, 552 см–1), $\delta _{b}^{1}$ (496, 432, 414 см–1) сменяются полосами α-ТКФ $\nu _{{as}}^{1}$ (1084, 1064, 990 см–1), $\nu _{s}^{1}$ (960 см–1), $\delta _{a}^{1}$ (611, 597, 584, 563 см–1), $\delta _{b}^{1}$ (447, 409 см–1). По данным ИКС, термические превращения ТКФ в диапазоне 60–1200°C протекают в несколько стадий согласно схеме (рис. 2, III).
На ИК-спектрах АФК-1 после 400°С (рис. 3в) наблюдаются характеристические полосы АФК: $\nu _{{as}}^{1}$ (1049 см–1), $\nu _{s}^{1}$ (937 см–1), $\delta _{a}^{1}$ (570 см–1), $\delta _{b}^{1}$ (410 см–1) колебаний O–P–O. Плечо $\nu _{s}^{4}$ (736 см–1) иона ${{{\text{P}}}_{{\text{2}}}}{\text{O}}_{7}^{{4 - }}$ указывает на внедрение ионов ${\text{HPO}}_{4}^{{2 - }}$ в структуру АФК-1, что связано с неполным взаимодействием реагентов в неравновесных условиях получения. После 650 и 1200°C на ИК-спектрах АФК-1 идентифицируются полосы α-ТКФ, а после 800–1000°C – полосы β-ТКФ. Дополнительно на ИК-спектрах АФК-1 после 650–1000°С присутствуют полосы $\nu _{{as}}^{1}$ (1207, 1186, 1142 см–1) и $\nu _{s}^{4}$ (726 см–1) иона ${{{\text{P}}}_{{\text{2}}}}{\text{O}}_{7}^{{4 - }}$ в составе β-ТКФ, которые практически не наблюдаются у α-ТКФ.
Образцы АФК-2 и АФК-3 после 400°C (рис. 3г, кривые 1, 2) представлены уширенными полосами апатитного ТКФ, а после 800°С – полосами β-ТКФ. В случае АФК-2 на ИК-спектрах после 800°С (рис. 3г, кривая 1) присутствуют полосы ионов ${{{\text{P}}}_{{\text{2}}}}{\text{O}}_{7}^{{4 - }},$ что указывает на стабилизацию дополнительных ионов ${\text{HPO}}_{4}^{{2 - }}$ в образце до прогрева за счет неполного взаимодействия реагентов при pH 9. Отсутствие указанных полос на ИК-спектрах АФК-3 с pH осаждения 11 связано с тем, что основной ионной формой фосфорной кислоты при pH 11 является ион ${\text{PO}}_{4}^{{3 - }},$ а при pH 9 – ион ${\text{HPO}}_{4}^{{2 - }}$ [36].
Составы ТКФ-2 и ГА-2 (рис. 3г, кривые 3, 4) после 400°С соответствуют карбонатзамещенному ГА с Б-типом замещения, ионы ${\text{CO}}_{3}^{{2 - }}$ из структуры которого удаляются после 800°С. Уширение апатитных полос на ИК-спектрах АФК-2, АФК-3, ГА-2 в сравнении с однофазным ГА (рис. 3а) обусловлено присутствием включений АФК. Отсутствие полос иона ${{{\text{P}}}_{{\text{2}}}}{\text{O}}_{7}^{{4 - }}$ на ИК-спектрах АФК-3, ГА-2 (рис. 3г, кривые 2, 4) и ТКФ-1 (рис. 3б) после 800°С указывает на отсутствие дополнительных ионов ${\text{HPO}}_{4}^{{2 - }}$ до прогрева. Без ионов ${\text{HPO}}_{4}^{{2 - }}$ состав включений АФК эквивалентен аморфному ТКФ с Ca/P = 1.50 (рис. 2, III).
Данные ИКС позволяют регистрировать присутствие включений АФК по уширению апатитных полос до прогрева, особенно полосы L2 колебаний O–H. Комбинированный анализ методами РФА и ИКС с использованием однофазного стехиометрического ГА в качестве образца сравнения можно считать достаточным для идентификации систем ГА/АФК, но существует потребность в надежном параметре, свидетельствующем о присутствии фазы АФК.
Поэтому образцы апатитов дополнительно исследовали методом ДТА, который позволяет регистрировать присутствие АФК в составе многокомпонентных смесей [13]. Кривые ДТА образцов ФК (рис. 4) группировали по условиям выделения: 1) ГА-1 и ТКФ-2 (рис. 4, кривые 1, 2) получали в равновесных условиях образования однофазного стехиометрического ГА; 2) ТКФ-1 и ГА-2 (рис. 4, кривые 3, 4) синтезировали в неравновесных условиях получения бифазных смесей кд-ГА/АФК; 3) АФК-3, АФК-2 и АФК-1 (рис. 4, кривые 5–7) осаждали в очень неравновесных условиях с дополнительной дегидратацией, которые обусловливали образование АФК либо смеси апатитный ТКФ/АФК.
Рис. 4.
Кривые ДТА образцов: 1 – ГА-1; 2 – ТКФ-2; 3 – ТКФ-1; 4 – ГА-2; 5 – АФК-3; 6 – АФК-2; 7 – АФК-1.
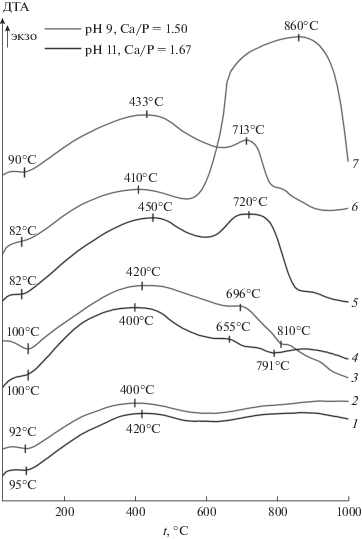
Кривые ДТА стехиометрических однофазных апатитов первой группы (рис. 4, кривые 1, 2) характеризуются присутствием эндотермического пика дегидратации при 92–95°С и широкого экзотермического эффекта при 400–420°С структурной релаксации [28, 37], обусловленной уменьшением концентрации дефектов в структуре аморфизированных материалов при повышении температуры. Дальнейший нагрев ГА до 1000°С сопровождается его кристаллизацией без выраженных термических эффектов, что указывает на термическую устойчивость ГА-1 и ТКФ-2.
На кривых ДТА композитов кд-ГА/АФК в составе образцов второй группы (рис. 4, кривые 3, 4) пик дегидратации сдвинут до 100°С в сравнении с однофазными ГА (рис. 4, кривые 1, 2), это может быть связано с уплотнением апатитов второй группы в процессе фильтрования, что затрудняет удаление воды. При 400 и 420°С наблюдаются эффекты структурной релаксации, которые у апатитов второй группы (рис. 4, кривые 3, 4) обладают повышенной интенсивностью по сравнению с апатитами первой группы (рис. 4, кривые 1, 2) за счет неравновесных условий выделения, способствующих образованию большего количества структурных дефектов. Экзоэффекты у образцов второй группы при 655 и 696°С свидетельствуют о кристаллизации включений АФК, причем на ДТА-кривой ТКФ-1 данный эффект характеризуется повышенной интенсивностью из-за повышенной стабильности АФК в условиях осаждения ТКФ-1 (pH 9, Ca/P = 1.50) по сравнению с ГА-2 (pH 11, Ca/P = 1.67). Последующие эндоэффекты при 791 и 827°С обусловливает преимущественно твердофазное превращение кд-ГА по уравнению реакции (1).
Кривые ДТА образцов третьей группы (рис. 4, кривые 5–7) характеризуются эндоэффектом дегидратации, сдвинутым до 82–90°С, что обусловлено предварительной дегидратацией. Экзоэффекты при 433, 450 и 410°С свидетельствуют о структурной релаксации, а последующие широкие экзоэффекты при 713, 720 и 860°С – о преимущественной кристаллизации АФК. Экзоэффекты фазовых превращений на кривых ДТА третьей группы более интенсивные, а их максимумы сдвинуты в высокотемпературную область по сравнению с бифазными образцами второй группы (рис. 4, кривые 3, 4); это связано с повышенной стабилизацией АФК в матрице ГА в очень неравновесных условиях выделения с дополнительным обезвоживанием. Образец АФК-3 (рис. 4, кривая 5) характеризуется большим содержанием включений аморфной фазы в сравнении с АФК-2 (рис. 4, кривая 6) за счет дополнительной дегидратации этанолом.
Соответственно, присутствие на кривых ДТА экзотермических эффектов при 655–720°С позволяет надежно регистрировать включения АФК в составе апатитных ФК, полученных в неравновесных условиях. Причем увеличение количества аморфной фазы сопровождается увеличением интенсивности характеристического экзоэффекта и сдвигом его максимума в высокотемпературную область, что позволяет сравнивать количество фазы АФК в различных апатитах.
ЗАКЛЮЧЕНИЕ
Синтезированы апатитные ФК при pH 9–11, отношении Ca/P = 1.50–1.67, скорости взаимодействия ${\text{HPO}}_{4}^{{2 - }}$ 10–5–0.1 моль/c и выдерживании осадков под маточным раствором до 4 сут. Синтез в относительно равновесных условиях медленного взаимодействия реагентов HPO42– (~10–5 моль/с) и созревания осадков в течение 4 сут способствовал образованию однофазного стехиометрического ГА с Ca/P ≈ 1.67. В более неравновесных условиях получения комбинированный анализ методами РФА/ИКС/ДТА позволил установить факт формирования бифазных композитов на основе кд-ГА с Ca/P < 1.67 и АФК, которые после 800°С превращались в однофазный β-ТКФ либо смесь ГА/α, β-ТКФ. Формирование апатитного ТКФ с максимальным содержанием АФК наблюдалось при быстром введении ${\text{HPO}}_{4}^{{2 - }}$ (~0.1 моль/c), отсутствии стадии созревания осадка под маточным раствором и дополнительной дегидратации этанолом с последующим прогревом при 400°С. В данных условиях уменьшение pH осаждения до 9 способствовало образованию однофазного АФК, содержащего ионы ${\text{HPO}}_{4}^{{2 - }}.$ На основании зависимости фазового и функционально-группового состава апатитных ФК от условий их осаждения, выделения и термообработки разработана единая схема их превращений при pH 9–11, Ca/P = = 1.50–1.67.
Список литературы
Safronova T.V. // Inorg. Mater. 2021. V. 57. № 5. P. 443. [Сафронова Т.В. // Неорган. материалы. 2021. Т. 57. № 5. С. 467.]https://doi.org/10.1134/S002016852105006X
Dorozhkin S.V. // Ceram. Int. 2016. V. 42. № 6. P. 6529. https://doi.org/10.1016/j.ceramint.2016.01.062
Sakae T., Nakada H., LeGeros J.P. // J. Hard. Tis Biol. 2015. V. 24. № 2. P. 111. https://doi.org/10.2485/jhtb.24.111
Данильченко С.Н. // Вісник СумДУ. 2007. № 2. С. 33.
Destainville A., Champion E., Bernache-Assollant D. et al. // Mater. Chem. Phys. 2003. V. 80. № 1. P. 269. https://doi.org/10.1016/S0254-0584(02)00466-2
Uskoković V. // RSC Adv. 2015. V. 5. № 46. P. 36614. https://doi.org/10.1039/C4RA17180B
Borisov S.V., Magarill S.A., Pervukhina N.V. // J. Struct. Chem. 2019. V. 60. P. 1191. https://doi.org/10.1134/S0022476619080018
Ishikawa K., Ducheyne P., Radin S. // J. Mater. Sci. Mater. Med. 1993. V. 4. № 2. P. 165. https://doi.org/10.1007/BF00120386
Vignoles M., Bonel. G., Young R.A. // Calcified Tis. Int. 1983. V. 40. № 2. P. 64. https://doi.org/10.1007/BF02555707
Krut’ko V.K., Kulak A.I., Musskaya O.N. // Inorg. Mater. 2017. V. 53. № 4. P. 429. [Крутько В.К., Кулак А.И., Мусская О.Н. // Неорган. материалы. 2017. Т. 53. № 4. С. 427.]
Makarova S.V., Bulina N.V., Prosanov I.Y. et al. // Russ. J. Inorg. Chem. 2020. V. 65. P. 1831. https://doi.org/10.1134/S0036023620120116
Murzakhanov F.F., Mamin G.V., Goldberg M.A. et al. // Russ. J. Coord. Chem. 2020. V. 46. № 11. P. 729. https://doi.org/10.1134/S1070328420110044
Zhang H., Zhang M. // Mater. Chem. Phys. 2011. V. 126. № 3. P. 642. https://doi.org/10.1016/j.matchemphys.2010.12.067
Nikolenko M.V., Vasylenko K.V., Myrgorodska V.D. et al. // Processes. 2020. V. 8. № 9. P. 1009. https://doi.org/10.3390/pr8091009
Martin R.I., Brown P.W. // J. Biomed. Mater. Res. 1997. V. 35. № 3. P. 299. https://doi.org/10.1002/(SICI)1097-4636(19970605)35:3<299::AID-JBM4>3.0.CO;2-C
Trubitsyn M.A., Hung H.V., Furda L.V. et al. // Russ. J. Inorg. Chem. 2021. V. 66. P. 654. https://doi.org/10.1134/S0036023621050211
Combes C., Rey C. // Acta Biomater. 2010. V. 6. № 9. P. 3362. https://doi.org/10.1016/j.actbio.2010.02.017
Koutsopoulos S. // J. Biomed. Mater. Res. 2002. V. 62. № 4. P. 600. https://doi.org/10.1002/jbm.10280
Tsuber V.K., Lesnikovich L.A., Kulak A.I. et al. // Pharm. Chem. J. 2006. V. 40. № 8. P. 455. [Цубер В.К., Лесникович Л.А., Кулак А.И. и др. // ХФЖ. 2006. Т. 40. № 8. С. 48].https://doi.org/10.1007/s11094-006-0151-2
Krut’ko V.K., Kulak A.I., Lesnikovich L.A. et al. // Russ. J. Gen. Chem. V. 77. № 3. 2007. P. 336. [Крутько В.К., Кулак А.И., Лесникович Л.А. и др. // Журн. общей химии. 2007. Т. 77. № 3. С. 366.]https://doi.org/10.1134/S1070363207030036
Musskaya O.N., Kulak A.I., Krut’ko V.K. et al. // Inorg. Mater. 2018. V. 54. № 2. P. 117. [Мусская О.Н., Кулак А.И., Крутько В.К. и др. // Неорган. материалы. 2018. Т. 88. № 2. С. 146.]
Somrani S., Rey C., Jemal M. // J. Mater. Chem. 2003. V. 13. № 4. P. 888. https://doi.org/10.1039/B210900J
Carrodeguas R.G., De Aza S. // Acta Biomater. 2011. V. 7. № 10. P. 3536. https://doi.org/10.1016/j.actbio.2011.06.019
Markovic M., Fowler B.O., Tung M.S. // J. Res. NIST. 2004. V. 109. № 6. P. 553. https://doi.org/10.6028/jres.109.042
Jillavenkatesa A., Condrate Sr. R.A. // Spectroscopy Lett. 1998. V. 31. № 8. P. 1619. https://doi.org/10.1080/00387019808007439
Boonchom B., Nart P. // Mater. Lett. 2009. V. 63. № 20. P. 1709. https://doi.org/10.1016/j.matlet.2009.05.026
Gross K.A., Gross V., Berndt C.C. // J. Am. Ceram. Soc. 1998. V. 81. № 1. P. 106. https://doi.org/10.1111/j.1151-2916.1998.tb02301.x
Bethe H.A. // Proc. R. Soc. London, Ser. A: Math. Phys. Sci. 1935. V. 150. № 871. P. 552. https://doi.org/10.1098/rspa.1935.0122
Chaair H., Heughebaert J.C., Heughebaert M. // J. Mater. Chem. 1995. V. 5. № 6. P. 895. https://doi.org/10.1039/JM9950500895
Hurle K., Neubauer, Bohner M. et al. // Acta Biomater. 2015. V. 23. P. 338. https://doi.org/10.1016/j.actbio.2015.05.026
Huang Y., Huang W., Sun L. et al. // Int. J. App. Cer. Tech. 2010. V. 7. № 2. P. 184. https://doi.org/10.1111/j.1744-7402.2009.02384.x
Glazov I.E., Krut’ko V.K., Kulak A.I. et al. // Mater. Today Comm. 2021. V. 27. P. 102224. https://doi.org/10.1016/j.mtcomm.2021.102224
Глазов И.Е., Власов Р.А., Крутько В.К. и др. // Весці НАН Беларусі. Сер. хім. навук. 2019. Т. 55. № 2. С. 135. https://doi.org/10.29235/1561-8331-2019-55-2-135-141
Pleshko N., Boskey A., Mendelsohn R. // Biophys. J. 1991. V. 60. № 4. P. 786. https://doi.org/10.1016/S0006-3495(91)82113-0
Глазов И.Е., Крутько В.К., Мусская О.Н., Кулак А.И. // Весці НАН Беларусі. Сер. хім. навук. 2019. Т. 55. № 4. С. 391. https://doi.org/10.29235/1561-8331-2019-55-4-391-399
Schrödter K., Bettermann G., Staffel T. et al. // Ullmann’s Encyclopedia of Industrial Chemistry. 2012. V. 26. P. 679. https://doi.org/10.1002/14356007.a19_465.pub3
Hill R.M., Dissado L.A. // J. Phys. C: Sol. State Phys. 1982. V. 15. № 25. P. 5171. https://doi.org/10.1088/0022-3719/15/25/010
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии