

Журнал неорганической химии, 2022, T. 67, № 1, стр. 54-102
Комплексы металлов платиновой группы с ациклическими диаминокарбеновыми лигандами. Синтез и современные области применения (обзор)
М. А. Кинжалов a, *, К. В. Лузянин b, **
a Санкт-Петербургский государственный университет
199034 Санкт-Петербург, Университетская наб., 7–9, Россия
b Химический факультет Ливерпульского университета
L69 7ZD Ливерпуль, Краун-стрит, Великобритания
* E-mail: m.kinzhalov@spbu.ru
** E-mail: Konstantin.Luzyanin@liverpool.ac.uk
Поступила в редакцию 06.07.2021
После доработки 14.09.2021
Принята к публикации 15.09.2021
- EDN: ZIREJI
- DOI: 10.31857/S0044457X22010068
Аннотация
Комплексы металлов платиновой группы с ациклическими диаминокарбеновыми лигандами находят применение в таких разнообразных областях, как металлокомплексный катализ, фотолюминесцентные материалы, хемосенсоры и противоопухолевые препараты. В этом обзоре систематизированы исследования, опубликованные до настоящего времени, проведен анализ свойств и методов получения комплексов металлов платиновой группы с ациклическими диаминокарбеновыми лигандами и рассмотрены последние достижения в использовании этих соединений для разработки функциональных материалов.
ВВЕДЕНИЕ
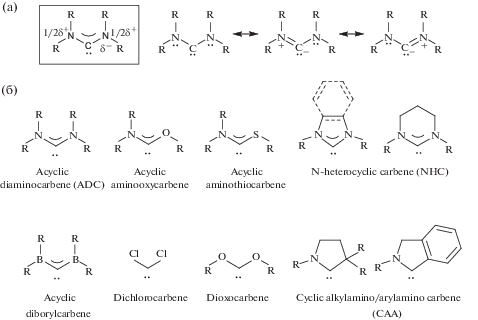
Открытые в конце прошлого века стабильные аминокарбены, а именно N-гетероциклические карбены (NHC) и их ациклические диаминокарбеновые аналоги (ADC), ассоциируются с высокой донорной способностью, что позволяет им, выступая в качестве лигандов, эффективно стабилизировать электронно- и координационно-ненасыщенные соединения переходных металлов. Эти свойства в сочетании с низкой токсичностью и умеренной химической устойчивостью NHC и ADC привели к вытеснению их комплексами традиционно используемых фосфиновых производных с доминирующих позиций в металлокомплексном катализе. Уникальные каталитические свойства, проявляемые комплексами металлов платиновой группы с аминокарбеновыми лигандами, привели к открытию важных каталитических реакций, что позволило минимизировать количество промежуточных стадий в ряде процессов органического синтеза. Помимо каталитических процессов, комплексы металлов платиновой группы с аминокарбенами активно используются в медицинской химии, в создании светоизлучающих систем и в сенсорных приложениях.
В то время как синтез, свойства и применение комплексов с NHC обсуждаются в ряде обзоров (релевантные обзоры по структуре и синтезу [1–7], каталитическое [8–15], люминесцентное [16–19], медицинское [20–24] применение и использование в супрамолекулярной химии [25]), соответствующие реферативные работы по комплексам с ациклическими диаминокарбенами чрезвычайны редки и охватывают только их применение в качестве катализаторов в органическом синтезе (до 2012 года) [26, 27] и реакционную способность ациклических диаминокарбеновых лигандов [28]. По сравнению с NHC ациклические диаминокарбены обладают уникальными, как правило, более сильными донорными свойствами, в то время как отсутствие циклической системы допускает вращение по связи азот–углерод, приводя к существенным изменениям в стерических и электронных характеристиках лиганда, что отражается, в частности, на фотоэмиссионных свойствах и каталитической активности комплексов.
Количество публикаций в настоящее время достигло уровня, когда новое исследование является своевременным (более 50 работ, посвященных ADC комплексам металлов платиновой группы только за последние 5 лет). Разработанные в последние года высокоэффективные каталитические системы для проведения реакций кросс-сочетания [29–31], Хека [32], борилирования [33] и гидросилилирования [34–36], в том числе в водной среде [37] и в условиях фотокатализа видимым светом [38], примеры использования в создании фотолюминесцирующих материалов [39–42], хемосенсоров [39, 43, 44] и потенциальных лекарственных препаратов [36, 45–47] требуют систематизации и критического анализа.
Основные цели этого обзора: анализ современных подходов к синтезу новых типов комплексов металлов платиновой группы с ADC лигандами; анализ различий между NHC и ADC; обобщение и анализ примеров применения комплексов металлов платиновой группы с ADC лигандами в катализе, материаловедении, фотофизике и других областях современной науки.
СТРОЕНИЕ АЦИКЛИЧЕСКИХ ДИАМИНОКАРБЕНОВ И ИХ ЛИГАНДНЫЕ СВОЙСТВА
В соответствии с номенклатурой IUPAC карбен (H2C:) или его производные (X2C:) – это электронейтральные частицы, в которых атом углерода ковалентно связан с двумя одновалентными группами любого типа или с двухвалентной группой и несет два несвязывающих электрона [48]. Геометрия карбенового атома углерода может быть линейной или уголковой [1]; линейная геометрия, основанная на sp-гибридизованном атоме углерода, несущем две несвязывающие вырожденные p-орбитали, практически не встречается. Большинство карбенов имеют уголковую конфигурацию, в которой атом углерода находится в промежуточной между sp и sp2 степенями гибридизации. При переходе от линейной конфигурации к изогнутой происходит снятие вырождения p-орбиталей: одна из p-орбиталей остается неизменной по энергии (ее условно обозначают pπ), в то время как другая p-орбиталь приобретает частичный s-характер и таким образом энергетически стабилизируется относительно исходной (условно обозначают σ). Образованные σ- и pπ-орбитали сравнительно близки по энергии, но отделены значительной энергетической щелью от занятых и вакантных скелетных МО. Поскольку карбеновый атом углерода несет два не участвующих в образовании связей электрона, карбеновая частица может находиться в триплетном (два несвязывающих электрона занимают σ- и pπ-орбитали с параллельной ориентацией спина) или в синглетном (два несвязывающих электрона занимают орбиталь с антипараллельной ориентацией спина) состоянии.
Триплетные карбены по химическим свойствам близки к свободным радикалам и обладают даже большей реакционной способностью по сравнению с последними из-за наличия двух полузаполненных МО вместо одной [49]. Синглетные карбены могут обладать двойственной реакционной способностью: дважды занятая и неглубокая по энергии МО придает им нуклеофильные анионоподобные свойства, а вакантная МО невысокой энергии обусловливает электрофильные катионоподобные свойства. Поскольку карбен в целом электронейтрален, энергия его ВЗМО ниже, чем у соответствующих анионов; аналогично, энергия НСМО – выше, чем у соответствующих катионов. Это предопределяет, с одной стороны, умеренную (по сравнению с катионами и анионами в газовой фазе) реакционную способность карбенов, а с другой – значительное влияние заместителей при карбеновом атоме углерода на энергию и электронную плотность обеих граничных МО, что, в свою очередь, обусловливает большое разнообразие свойств карбенов.
Мультиплетность основного состояния карбена определяется относительными энергиями σ и pπ-орбиталей. Синглетное основное состояние стабилизируется при наличии значительной разницы энергий между σ- и pπ-орбиталями. Квантово-химические расчеты показали, что для стабилизации синглетного состояния требуется разница энергий около 2 эВ [50]. Разница энергий менее 1.5 эВ между относительными энергиями σ и pπ-орбиталей благоприятствует триплетному состоянию [50].
В простейшем карбене – метилене (H2C:) – орбитали близки по энергии, поэтому заселение электронами происходит в соответствии с принципом Паули; для метилена триплет – основное состояние, а синглет – возбужденное [51]. Присоединение к карбеновому центру заместителей изменяет относительную устойчивость этих состояний. Электроноакцепторные заместители индуктивно стабилизируют связывающую σ-орбиталь, увеличивая ее s-характер, при этом энергия p-орбитали остается практически неизменной. Таким образом, разрыв σ–pπ увеличивается, и синглетное состояние становится предпочтительным (рис. 1). Как в случае дихлоркарбенов, для которых синглетное состояние основное [52, 53], так и в случае диаминокарбенов два электроотрицательных атома азота стабилизирует сиглетное состояние и повышают устойчивость карбена.
Рис. 1.
Корреляционные диаграммы орбитальных энергий для молекул синглетных и триплетных карбенов, показывающие влияние индуктивных и мезомерных эффектов заместителей.
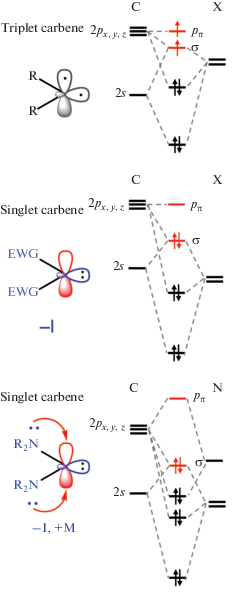
Мезомерные эффекты также играют значительную роль в стабилизации карбена. Они заключаются во взаимодействии углеродных pπ-орбиталей и соответствующих p- или π-орбиталей двух заместителей [50, 54]. В случае π-электронодонорных групп, таких как галогениды, NR2, PR2, OR, SR, SR3, энергия вакантной pπ-орбитали увеличивается благодаря взаимодействию с симметричной комбинацией неподеленных пар заместителей [55–57]. Поскольку σ-орбиталь остается практически неизменной, то разрыв по σ–pπ увеличивается и синглетное состояние становится более предпочтительным. Влияние π-донорной способности заместителей для стабилизации карбенов можно продемонстрировать сравнением ациклических диаминокарбенов и диалкоксикарбенов: аминогруппы по сравнению с алкоксигруппами обладают более высокой π-донорной способностью. Экспериментально показано, что бис(N-пиперидил)карбен (СH2)5N–C–N(СH2)5 существует в растворе при комнатной температуре в течение суток [58], тогда как время жизни диметоксикарбена MeO–C–OMe в растворе при комнатной температуре всего 2 мс [59].
Взаимодействие π-электронов заместителей с pπ-орбиталью карбенового атома углерода приводит к образованию четырехэлектронной трехцентровой π-системы, в которой связи C–X приобретает частичный π-характер; такие карбены лучше всего описываются суперпозицией двух цвиттер-ионных структур с отрицательным зарядом в центре карбена (рис. 2а). Данные рентгеноструктурных исследований ациклических диаминокарбенов указывают на планарную структуру атомов азота и на частичный π-характер C–N связей [58, 60, 61]; последнее подтверждается большими барьерами вращения заместителей вокруг связи C–N в растворе (13 ккал/моль), определенными для ациклических диаминокарбенов в температурных экспериментах методом спектроскопии ЯМР [60, 62].
Рис. 2.
Мезомерные эффекты в стабилизации диаминокарбеновой структуры (а) и примеры гетероатом-стабилизированных синглетных карбенов (б).
Примеры стабилизированных гетероатомом синглетных карбенов представлены на рис. 2б, среди которых диаминокарбены – наиболее важные представители данного класса соединений [1].
Важная особенность диаминокарбенов – их ярко выраженные лигандные свойства в комплексах переходных металлов [63]. Склонность ADC и их аналогов к комплексообразованию связана с возможностью образования σ связи М–Сcarbene путем передачи пары несвязывающих электронов с нуклеофильной σ-орбитали карбенового атома углерода на атом металла М. Одновременно возможно образование и π связи в результате взаимодействия подходящих по симметрии d-AO металла с вакантной электрон-дефицитной pπ- орбиталью карбенового атома углерода. Как правило, ADC лиганд – эффективный σ-донор и слабый π-акцептор (рис. 3) [64].
Рис. 3.
Ациклические диаминокарбены (ADC) и их металлокомплексы (а); взаимодействие металл–лиганд в комплексах с ациклическими диаминокарбенами (б).
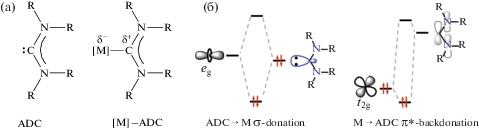
Cильные σ-донорные и слабые π-акцепторные свойства делают ADC схожими по координационным свойствам с фосфиновыми лигандами, однако диаминокарбены – более сильные электронодоноры, чем фосфины [65–67]. Это выражается в более прочной связи металл–лиганд, что подтверждается более высокими энергиями диссоциации связи и более короткими длинами связей металл–лиганд для диаминокарбеновых комплексов по сравнению с их фосфиновыми аналогами. Из-за более низкой электроотрицательности углерода по сравнению с азотом неподеленная электронная пара на атоме углерода карбена приобретает значительно большую энергию, чем в обычных N-донорах, например, в пиридиновых лигандах.
В настоящее время известны ADC комплексы многих переходных металлов, включая комплексы с кинетически лабильными в реакциях лигандного обмена ранними переходными элементамами, такими как хром [68], марганец [69–72], железо [73–79], кобальт [80], медь [81] (а также ртуть [82]), однако наибольшее практическое значение представляют координационные соединения металлов платиновой группы.
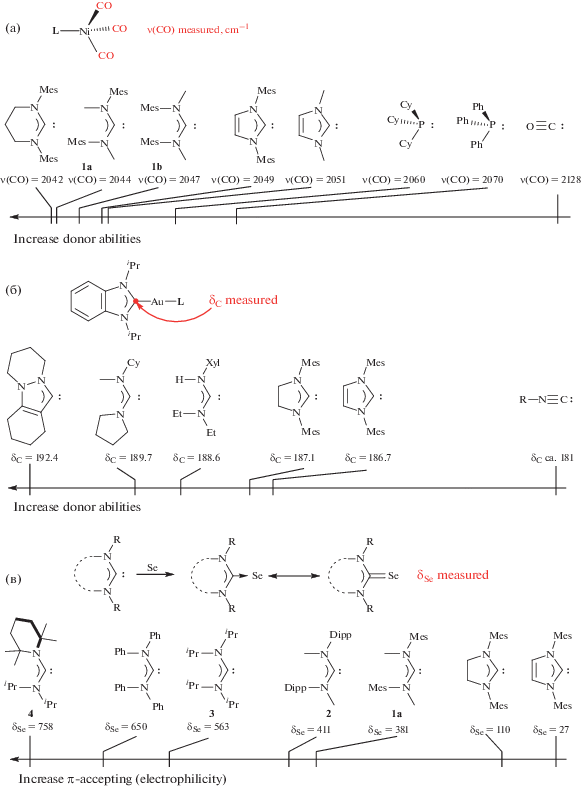
Высокие электронодонорные свойства ADC лигандов следуют из их электронных параметров. Электронный параметр Толмана [83], основанный на измерении изменения частоты валентных колебаний карбонильного лиганда ν(СО) в [Ni(CO)3(L)] в зависимости от электронодонорной способности лиганда L, а также аналогичные по принципу системы, основанные на карбонильных комплексах родия(I) (транс-[RhCl(CO)2(L)]) [84] и иридия(I) (транс-[IrCl(CO)2(L)]) [85], подтверждают, что диаминокарбены – более сильные доноры, чем фосфины и монооксид углерода (рис. 4a ). Из-за отсутствия делокализации электронной плотности по гетероароматическому фрагменту ADC, как правило, более сильные электронодонорные лиганды, чем пятичленные ароматические NHC. Кроме того, донорная способность ADC выше, чем у пятичленных насыщенных NHC, так как больший угол связи NCкарбенN уменьшает s-характер неподеленной электронной пары на карбеновом атоме углерода [60, 63, 86]. Согласно величине электронного параметра Толмана, донорная способность ADC сопоставима с донорной способностью шестичленных NHC [87, 88]. Из анализа электронного параметра Толмана для несимметрично замещенных ADC (соединения 1a и 1b на рис. 4a ) наглядно следует, что донорная способность ациклического диаминокарбена зависит от его конформации [88]. Альтернативный подход к измерению электронодонорных свойств, основанный на измерении δС карбенового атома углерода NHC лиганда, расположенного в транс-положении к измеряемому лиганду, в комплексах палладия(II) и золота(I) [2, 89, 90] также подтверждает высокую донорную способность ациклических диаминокарбеновых лигандов (рис. 4 б) [91].
Хотя диаминокарбены позиционируются как сильные σ-донорные лиганды с незначительным вкладом π-дативного взаимодействия, исследования показали, что диаминокарбены могут в определенной степени принимать электронную плотность по механизму обратного π-донирования [64, 88, 92]. Метод оценки π-акцепторных свойств диаминокарбеновых лигандов, основанный на измерении химического сдвига 77Se в аддуктах диаминокарбенов с селеном [93], показывает, что ADC, как правило, являются более сильными π-акцепторами, чем NHC (рис. 4 в) [93, 94]. π-Акцепторная способность ADC зависит в первую очередь от объема заместителей при атомах азота. ADC, имеющие хотя бы один необъемистый заместитель при каждом из атомов азота, например диаминокарбены, такие как (Mes(Me)N)2C: (соединение 1a) и (Dipp(Me)N)2C: (соединение 2 на рис. 4 в) [88, 95], проявляют умеренные электрофильные свойства. Тетраизопропилзамещенный диаминокарбен Альдера (iPr2N)2C: (соединение 3 на рис. 4 в) [60] имеет промежуточную электрофильность и по своим π-акцепторным свойствам близок к циклическим алкиламинокарбенам (CAAC) [96]. Карбен (iPr2N)2C: демонстрирует амбифильные свойства, что подтверждается его химической активностью, в частности, способностью активировать небольшие молекулы, например, монооксид углерода [97]. На сегодняшний день ADC с объемистным 2,2,6,6-тетраметилпиперидиновым заместителем (соединение 4 на рис. 4 в) – наиболее электрофильный диаминокарбен из исследованных. По результатам квантово-химических расчетов, минимуму энергии соответствует конформация, в которой 2,2,6,6-тетраметилпиперидиновый фрагмент перпендикулярен плоскости N2C. В этой конформации невозможно π-сопряжение неподелённой пары электронов атома азота с карбеновым атомом углерода, энергия НСМО понижается и, следовательно, повышается электрофильность [94]. В большинстве случаев синергетический характер взаимодействия металл–лиганд не позволяет оценить индивидуальный вклад σ-донорных и π-акцепторных свойств карбеновых лигандов.
Рис. 4.
Оценка электронодонорных свойств ADC лигандов и сравнение с другими лигандами: а – электронный параметр Толмана; б – δС карбенового атома углерода в смешаннолигандных NHC комплексах; в – δSe в аддуктах с селеном.
Ключевое отличие ациклических диаминокарбенов от циклических аналогов заключается в отсутствии связывающего оба атома азота ковалентного фрагмента. Отсутствие цикла в ADC допускает вращение заместителей вокруг связей Cкарбен–N, однако свободное вращение вокруг этих связей ограничивает их частичный π-характер, появляющийся вследствие сопряжения неподеленных электронных пар атомов азота и вакантной p-орбитали карбенового атома углерода. Как показывают экспериментальные [78] и теоретические [98, 99] исследования, в ациклических диаминокарбенах с небольшими заместителями и в их комплексах с переходными металлами барьер вращения вокруг связей Cкарбен–N составляет не более 13–20 ккал/моль. У монодентатных ациклических диаминокарбенов, с различными заместителями при атомах азота возможны 4 конформера (рис. 5).
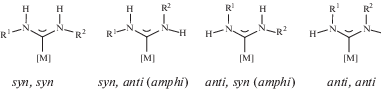
Геометрия наиболее предпочтительного конформера в отсутствие специфических нековалентных взаимодействий зависит от того, насколько в нем сведены к минимуму неблагоприятные стерические взаимодействия; изменения в относительных объемах заместителей при атоме азота могут сдвигать конформационное равновесие в пользу одного из конформеров. В качестве иллюстрации можно привести данные из работы [100], в которой установлено, что несимметрично замещенные ADC комплексы палладия(II) 5, имеющие в качестве одного из заместителей при карбеновом атоме углерода эфир пирролидинкарбоновой кислоты, существуют в растворе в виде равновесной смеси конформеров (схема 1 ).
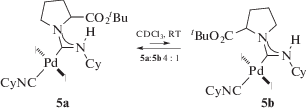
Схема 1 . Равновесная смесь конформеров, зафиксированная в растворе несимметрично замещенного ADC комплекса палладия(II).
Стабилизация определенной конформерной формы ADC лиганда может происходить за счет нековалентного связывания с участием донорных и акцепторных центров нековалентных взаимодействий в структуре комплекса. ADC комплексы, в которых атомы азота связаны с одним или несколькими атомами водорода, могут выступать донорами водородной связи и образовывать нековалентные взаимодействия с различными нуклеофильными частицами [28]. Образование водородных связей блокирует вращение вокруг связей C–N в диаминокарбеновом фрагменте, что приводит к стабилизации определенной конформации (рис. 6). В частности, катионные ADC комплексы с внешнесферными галогенидными анионами могут быть стабилизированы в твердой фазе и в растворе в син,син-конфигурации за счет образования трехцентровой (бифуркатной) водородной связи N–H⋅⋅⋅Cl⋅⋅⋅Н–N между NH-фрагментами диаминокарбенового лиганда и галогенидным анионом (структура 6) [101–105].
Рис. 6.
Примеры нековалентных взаимодействий, участвующих в стабилизации конформерной формы ADC лиганда.
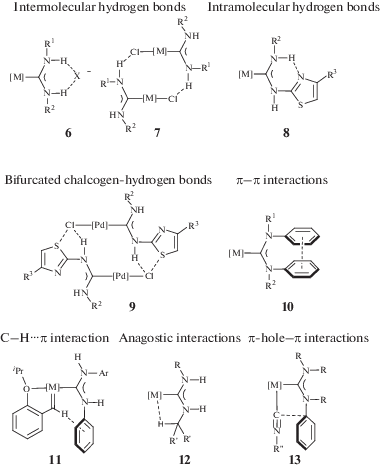
Анти-конфигурация при одном из атомов азота может достигаться за счет межмолекулярных парных водородных связей N–H⋅⋅⋅Cl между NH-фрагментами диаминокарбенового лиганда и галогенидным лигандом при атоме металла соседней молекулы (структура 7) [106]. В работе [106] установлено, что в соединении 8 реализуется парное кооперативное межмолекулярное взаимодействие с образованием халькогенных и водородных связей S⋅⋅⋅Cl/H⋅⋅⋅Cl, которое стабилизирует конформацию ADC лиганда не только в твердой фазе, но и в растворе.
При введении в структуру диаминокарбенового фрагмента заместителей с акцепторами водородных связей может происходить образование внутримолекулярной водородной связи, фиксирующей диаминокарбеновый фрагмент в амфи-конформации, как в случае ADC комплексов палладия(II) c 4-арилзамещенным тиазол-2-ильным заместителем (структура 8) [106]. Аналогичная стабилизация диаминокарбенового фрагмента в амфи-конформации также происходит в случае ADC комплексов других поздних переходных металлов с пиридин-2-ильным заместителем, а именно в комплексах золота [107–112] и железа [113]. Стабилизация определенной конформации также может достигаться за счет π–π взаимодействия между арильными заместителями (структура 10) [88, 114], С–H⋅⋅⋅π взаимодействий (структура 11) [99] и анагостических взаимодействий H⋅⋅⋅М (структура 12) [101]. В смешанолигандных диаминокарбеновых/изоцианидных комплексах 13 с арильными заместителями возможна стабилизация определенной конформации за счет образования внутримолекулярного взаимодействия π-дырка⋅⋅⋅π [115]. Эти взаимодействия также контролируют конформационную структуру ациклических диаминокарбеновых комплексов как в твердой фазе, так и в растворе. Образование водородных связей с NH-фрагментами диаминокарбенового лиганда может не только изменять конформационную стабильность, но и приводить к изомеризации координационного полиэдра [116].
Наличие в диаминокарбеновом лиганде дополнительных донорных центров может приводить к образованию С,Х-хелатных и Х,С,Х-пинцерных комплексов, в которых зафиксированы конформации у одного или обоих атомов азота соответственно. В качестве дополнительных донорных центров Х в С,Х-хелатных комплексах могут выступать атомы азота, углерода (примеры С,N- и С,С-хелатных комплексов представлены в разделе 3.4), серы [117] и фосфора (репрезентативные примеры C,P-хелатов (соединения 14 и 15) [118–120] и P,C,P-пинцеров (соединение 16) [102] представлены на риc. 7а).
Принципиально важно, что конформационные переходы вызывают изменения энергии граничных σ- и pπ-орбиталей и приводят к существенным изменениям в стерических и электронных характеристиках лиганда, в частности, конформационные изомеры ADC комплексов демонстрируют разную энергию 3MLCT-перехода, что отражается на их фотоэмиссионных свойствах (репрезентативный пример представлен на рис. 7б) [121], а также проявляют различную каталитическую активность [122].
МЕТОДЫ СИНТЕЗА КОМПЛЕКСОВ МЕТАЛЛОВ ПЛАТИНОВОЙ ГРУППЫ С АЦИКЛИЧЕСКИМИ ДИАМИНОКАРБЕНОВЫМИ ЛИГАНДАМИ
Значимые свойства комплексов с диаминокарбеновыми лигандами, обусловливающие каталитические, фотофизические и другие приложения, определяются характеристиками металлоцентра, а также балансом между донорными и стерическими свойствами диаминокарбенового лиганда. Для каждой области применения требуются комплексы с лигандами, обладающими различным набором параметров (свойств). Лиганды подбирают, как правило, эмпирическим способом, что накладывает жесткие требования к методу их синтеза. Синтетический подход должен быть модульным, позволяя осуществлять легкое введение нужных функциональных групп, универсальным, дающим возможность получать широкую гамму производных, и эффективным, гарантирующим высокий реакционный выход при низкой сложности процесса.
Предложен ряд экспериментальных подходов к синтезу комплексов с диаминокарбеновыми лигандами (рис. 8) [26–28], основные из которых – (1) депротонирование солей формамидиния с последующей координацией сгенерированного карбена к металлоцентру (Direct complexation), (2) окислительное присоединение солей C-хлориминия к металлоцентру (Oxidative addition), (3) литий-галогенный обмен с последующим трансметаллированием (Transmetalation), (4) металлопромотируемое нуклеофильное присоединение к изоцианидным лигандам (Metal-mediated addition) и (5) внедрение изоцианида по связи М–N (Insertion).
Рис. 8.
Экспериментальные подходы к синтезу комплексов с ациклическими диаминокарбеновыми лигандами.
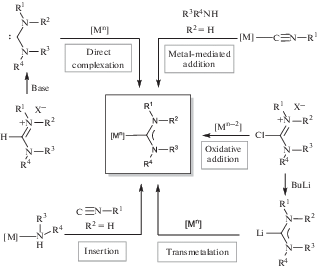
Метод прямой координации
Прямое комплексообразование между металлоцентром и свободным карбеном, полученным из прекурсоров in situ, – основной метод синтеза N-гетероциклических карбеновых комплексов [123, 124]. В качестве прекурсоров для синтеза их ациклических аналогов ([M]-ADC), как правило, используют N,N,N',N'-тетразамещенные формамидиниевые соли, депротонирование которых приводит к образованию свободного карбена [27, 125]. Для депротонирования N,N,N',N'-тетразамещенных формамидиниевых солей необходимы сильные основания, такие как LDA, KH/KOtBu или LiN(SiMe3)2 [60, 67, 126]. На схеме 2 представлены репрезентативные примеры комплексов родия(I) и иридия(I), синтезированные методом прямой координации диаминокарбена, полученного из соответствующих формамидиниевых прекурсоров [87].
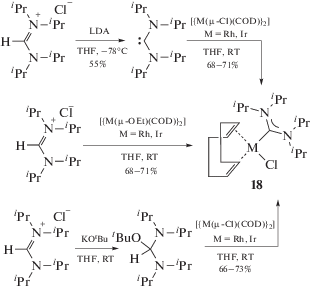
Схема 2 . Репрезентативные примеры комплексов родия(I) и иридия(I), синтезированных методом прямой координации диаминокарбена.
Основной недостаток этого метода – ограниченная синтетическая доступность солей N,N,N',N'-тетраалкилформамидиния, получение которых сопряжено со значительными проблемами [127, 128]. Кроме того, к недостатку метода относится способность образующегося свободного карбена, если он не стабилизирован объемистыми заместителями, вступать в побочные реакции окисления, димеризации, элиминирование алкена и другие [58, 61, 119, 120, 125]. В качестве прекурсоров также могут выступают замещенные тиомочевины, образующие диаминокарбен при восстановлении, однако этот подход ограничен всего несколькими примерами [129, 130].
Метод окислительного присоединения
Реакция окислительного присоединения формамидиниевых солей может протекать только на металлоцентрах в нулевой степени окисления, и использование данного метода ограничивается всего несколькими примерами синтеза ADC комплексов палладия(II) (схема 3 ) [131, 132].
Схема 3 . Реакция окислительного присоединения формамидиниевых солей, приводящая к получению ADC комплексов палладия(II).
Метод трансметаллирования
Альтернативный подход к получению ADC комплексов металлов платиновой группы основан на взаимодействии предварительно литиированных формамидиниевых солей с координационно-ненасыщенными металло-прекурсорами (схема 4 ) [132, 133]. Однако высокая реакционная способность литийорганических соединений накладывает существенные ограничения на природу заместителей в диаминокарбеновых фрагментах. Поэтому, насколько нам известно, использование данного метода представлено всего несколькими примерами получения комплексов палладия(II), родия(I) и иридия(I) [132, 133].
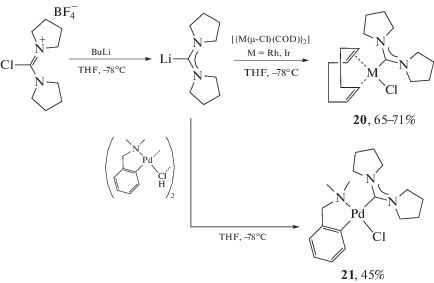
Схема 4 . Подход к получению ADC комплексов путем взаимодействия литиированных формамидиниевых солей с координационно-ненасыщенными металлопрекурсорами.
Метод металлопромотируемого присоединения к изоцианидным лигандам
Металлопромотируемое сочетание изоцианидов с N-центрированными нуклеофилами (NH-нуклеофилами) является наиболее перспективным методом синтеза комплексов с ациклическими диаминокарбеновыми лигандами [134, 135], так как при использовании этого метода можно удобно настраивать электронодонорные и стерические свойства диаминокарбенового лиганда путем варьирования заместителей, что достигается подбором подходящей пары изоцианид–нуклеофил. Металлопромотируемое взаимодействие изоцианидов с NH-нуклеофилами открыто чуть более ста лет назад профессором Санкт-Петербургского университета, в то время носившего название Императорский Петроградский университет, Л.А. Чугаевым при изучении промотируемого платиной(II) взаимодействия метилизоцианида с гидразином [136]. Благодаря своей универсальности за последние 20 лет “изоцианидный” подход расширен на другие металлоцентры и нуклеофилы [135, 137].
В свободном состоянии изоцианидный атом углерода обладает нуклеофильным характером, поэтому в отсутствие электрофильных частиц, включая соединения переходных металлов, изоцианиды не взаимодействуют с аминами и спиртами [63]. Координация изоцианидов к металлоцентрам, находящимся в высоких степенях окисления и обедненным электронами, увеличивает частичный положительный заряд на изоцианидном атоме углерода, приводя к увеличению его электрофильности [134, 135]. Для осуществления металлопромотируемого сочетания изоцианидов с NH-нуклеофилами необходим металлоцентр, гарантирующий достаточную активацию изоцианида. К таким металлоцентрами относятся поздние переходные металлы в относительно высоких степенях окисления: палладий(II), платина(II), золото (I/III) и другие. Другое необходимое условие – использование сильных и умеренно-сильных нуклеофилов.
Подавляющее большинство известных в литературных источниках примеров нуклеофильного присоединения к координированным изоцианидам сводится к взаимодействию с N-нуклеофилами (аминами) [134, 138] и О-нуклеофилами (спиртами) [76, 139–144]. Эти реакции приводят к образованию “классических” монодентатных диаминокарбеновых (при атаке координированного изоцианида N-донорным центром) и амино(оксо)карбеновых лигандов (при атаке координированного изоцианида O-донорным центром). В то же время при наличии в молекуле присоединяющегося N-нуклеофила дополнительного N'-нуклеофильного центра возможно образование соединений более сложной структуры, в том числе содержащих С,N- и С,С'-хелатные диаминокарбеновые лиганды [138]. Зачастую такие комплексы обладают повышенной стабильностью по сравнению с комплексами, содержащими “классические” монодентатные С-координированные диаминокарбеновые лиганды.
Важно отметить, что наше рассмотрение не будет сосредоточено на энциклопедическом перечислении имеющихся данных. Вместо этого мы будем стремиться проанализировать основные движущие силы образования соединений того или иного типа, которые, несомненно, будут способствовать и стимулировать дальнейшую исследовательскую деятельность в этой области. Последовательность раздела основана на типе нуклеофила, а именно на количестве N-центрированных нуклеофильных центров и их взаимном расположении.
Присоединение монофункциональных NH-нуклеофилов. Взаимодействие изоцианидных комплексов цис-[MCl2(CNR)2] (M = Pd, Pt) с различными монофункциональными NH-нуклеофилами, такими как алифатические [101, 145–151] и ароматические амины [140, 152–154], включая производные аминокислот [100], как правило, протекает только по одному изоцианидному лиганду и приводит к комплексам с одним ациклическим диаминокарбеновым и одним изоцианидным лигандами. Репрезентативные примеры таких реакций приведены на схеме 5 . Взаимодействие обычно протекает в мягких условиях с хорошими скоростями и высокими выходами. Использование хиральных изоцианидов или нуклеофилов позволяет синтезировать оптически активные диаминокарбеновые комплексы [100], перспективные для использования в качестве катализаторов в асимметрическом синтезе [26, 27].
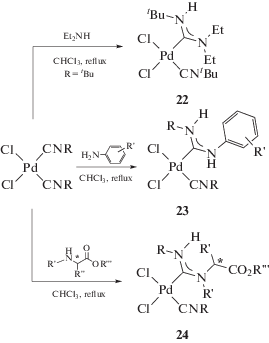
Схема 5 . Примеры взаимодействия изоцианидных комплексов цис-[MCl2(CNR)2] (M = Pd, Pt) с монофункциональными NH-нуклеофилами, приводящие к получению моно-ADC комплексов.
Эмпирическим показателем электрофильного характера изоцианидного лиганда в его металлокомплексах является силовая постоянная колебания связи CN, которая коррелирует с величиной относительного положительного заряда на атоме углерода (чем выше силовая постоянная, тем больше положительный заряд). В обзорной работе [134] на основании анализа экспериментальных данных сделан вывод, что атаке должен подвергаться комплекс, в котором Δν = ν(CN)коорд − ν(CN)свободн ≥ ≥ 40 см–1. По данным ИК-спектроскопии, ν(C≡N) в цис-[MCl2(CNR)2] больше, чем в свободных изоцианидах, на 90–115 см–1, что указывает на произошедшее при координации значительное повышение электрофильности изоцианидного атома углерода и, таким образом, косвенно свидетельствует о существенном возрастании реакционной способности по отношению к нуклеофилам. Высокая электрофильная активация изоцианидов объясняет способность комплексов цис-[MCl2(CNR)2] реагировать с разными по своей основности нуклеофилами.
Отсутствие реакции по второму изоцианидному лиганду в цис-[MCl2(CNR)2] (M = Pd, Pt) при использовании избытка амина связано с тем, что образующийся диаминокарбеновый лиганд – более сильный донор, чем изоцианидный [148] и поэтому дезактивирует изоцианидный лиганд в реакциях нуклеофильного присоединения. Нуклеофильная атака обоих изоцианидных лигандов в реакции с монофункциональными нуклеофилами реализуется только сильными нуклеофилами, например, алифатическими [155–157] или циклоалифатическими аминами [158], и/или при активировации изоцианиадов электронакцепторными заместителями (схема 6) [159]. В зависимости от природы заместителя в изоцианиде взаимодействие цис-[PdCl2(CNR)2] с аминами может приводить как к монокарбеновым (25), так и к бис-карбеновым комплексам (26). Взаимодействие комплексов с ароматическими изоцианидами реализуется одновременно по обоим изоцианидным лигандам, в то время как при использовании менее активного в реакциях с нуклеофилами трет-бутилизоцианидного комплекса реакция протекает только по одному из них [158].
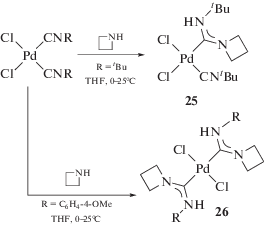
Схема 6 . Пример нуклеофильной атаки на оба изоцианидных лиганда, приводящей к генерации бис-карбеновых производных.
Взаимодействие транс-[PtI2(CNAr)2] с первичными и вторичными аминами протекает по обоим изоцианидным лигандам и приводит к бис-карбеновым комплексам 27–29. В зависимости от условий реакции и природы амина нуклеофильное присоединение может сопровождаться дальнейшей реакцией орто-металлирования арильного фрагмента с образованием С,C-хелатных диаминокарбеновых комплексов платины(II) и платины(IV) (схема 7) [155–157, 160]. В частности, при проведении реакции с первичными аминами процесс сопровождается стадией орто-металлирования арильного фрагмента одного из образованных ADC лигандов и приводит к соединению платины(II) 28. Дважды циклометаллированный комплекс платины(IV) 29 может быть синтезирован при проведении реакции транс-[PtI2(CNAr)2] с избытком первичного амина в хлороформе, последний выступает не только растворителем, но и окислителем, восстанавливаясь до дихлорметана.
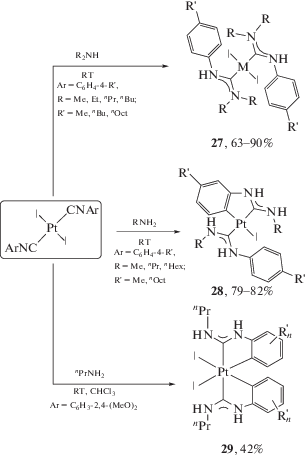
Схема 7 . Взаимодействие транс-[PtI2(CNAr)2] с аминами, проводящее к получению бис-карбеновыых производных 27–29; последующее орто-металлирование арильного фрагмента с образованием С,C-хелатных диаминокарбеновых комплексов.
Взаимодействие N-изоцианодиалкиламинов в координационной сфере палладия(II) и платины(II) с аминами протекает в мягких условиях, приводя к получению бис-диаминокарбеновых комплексов 30 с хорошим препаративным выходом (схема 8) [159]. Следует отметить, что использование N-изоцианодиалкиламинов имеет хорошие перспективы для получения новых диаминокарбеновых комплексов ввиду их высокой реакционной способности [159, 161–164].
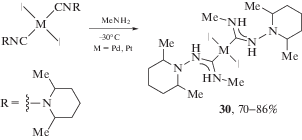
Схема 8 . Взаимодействие N-изоцианодиалкиламинов в координационной сфере палладия(II) и платины(II).
Гомолептические тетракис(ациклические диаминокарбеновые комплексы) палладия(II) и платины(II) 31 могут быть синтезированы взаимодействием тетрахлорпалладата/тетрахлорплатината калия с метилизоцианидом и метиламином (схема 9) [165–167]. Несмотря на кажущуюся простоту метода, известные в настоящее время примеры ограничены соединениями с метильными заместителями, поэтому синтетический потенциал метода оценить затруднительно.
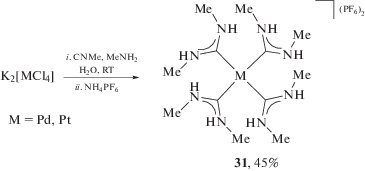
Схема 9 . Гомолептические тетракис-ADC комплексы палладия(II) и платины(II).
Взаимодействие моноизоцианидных комплексов палладия(II) и платины(II) с аминами, как и следовало ожидать, приводит к образованию монодентатных ациклических диаминокарбеновых комплексов (репрезентативные примеры приведены на схеме 10 ) [33, 43, 47, 168, 169]. Благодаря высокой электрофильной активации изоцианидов указанными металлоцентрами, реакции, как правило, протекают в мягких условиях и приводят к широкому спектру ациклических диаминокарбеновых комплексов с хорошим препаративным выходом.
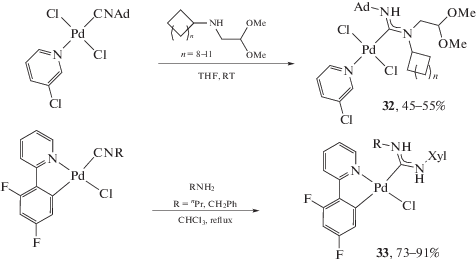
Схема 10 . Примеры взаимодействия моноизоцианидных комплексов палладия(II) и платины(II) с аминами.
Метилизоцианидные лиганды в циклопентадиенильных комплексах рутения(II) присоединяют метиламин и диметиламин с образованием ациклических диаминокарбеновых лигандов (схема 11) [170]. При проведении реакции при низкой температуре образующиеся монокарбеновые комплексы 34 могут быть выделены с хорошим препаративным выходом; при проведении реакции при более высокой температуре в большом избытке амина происходит образование бис-карбеновых комплексов, однако последние нестабильны в отсутствие избытка амина и при стоянии образуют монокарбеновые соединения 34 [28, 170].
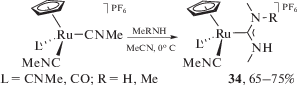
Схема 11 . Взаимодействие метилизоцианидных лигандов в циклопентадиенильных комплексах рутения(II) с аминами.
Порфириновые комплексы иридия(III) с аксиальными изоцианидными лигандами вступают в сочетание с аминами с образованием соответствующих ADC комплексов (схема 12а) [171]. При стехиометрическом соотношении реагентов реакция протекает только по одному изоцианидному лиганду и приводит к соединению 35 с хорошим препаративным выходом. Авторы отмечают, что превращение второго изоцианидного лиганда возможно в присутствии десятикратного избытка амина, однако образовавшийся бис-ADC комплекс был зафиксирован только в растворе и не был выделен в чистом виде. ADC производные порфириновых комплексов родия(III) 36 могут быть получены в результате кипячения соответствующих бис-изоцианидных комплексов в метаноле [172]. Аксиально расположенный изоцианидный лиганд в комплексе иридия(III) с тетрадентатным пиридин-карбоксамидным лигандом реагирует с аминами схожим образом (схема 12 б) [173].
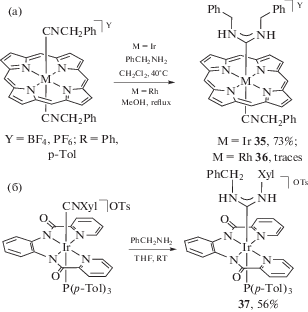
Схема 12 . Взаимодействие порфириновых (а) и пиридин-карбоксамидных (б) комплексов иридия(III), содержащими аксиальные изоцианидные лиганды, с аминами.
Вследствие влияния циклометаллирующих σ-донорных лигандов иридий(III) в составе бис-циклометаллированных комплексов является слабым электрофильным активатором изоцианидов (Δν = ν(СN)изоцианидного лиганда – – ν(СN)некоординированного изоцианида = 4–32 см–1 [174–177]), поэтому в реакции нуклеофильного присоединения вступают только изоцианидные лиганды, активированные акцепторными заместителями. Взаимодействие бис-изоцианидных комплексов 38 с аммиаком приводит к количественному образованию бис(диаминокарбеновых) комплексов 40 (схема 13) [178]. При этом присоединение аммиака к первому и второму изоцианидным лигандам в 38 происходит со схожими скоростями, что уже на начальном этапе приводит к образованию неразделимой смеси соединений 39 и 40 и делает невозможным получение чистых комплексов 39 по этой реакции. Интересно отметить, что дальнейшее выдерживание соединений 40 в атмосфере аммиака приводит к фрагментации одного из диаминокарбеновых лигандов с образованием цианидного комплекса (схема 13 , соединение 41).
Взаимодействие моноизоцианидных комплексов иридия(III) 42 с избытком газообразного аммиака приводит к образованию диаминокарбеновых комплексов 43 (схема 14) [39]. В то же время при использовании в качестве нуклеофилов первичных и вторичных алифатических аминов реакция нуклеофильного присоединения сопровождается металлированием ароматического заместителя с образованием C,N-хелатных диаминокарбеновых комплексов 44 [41].
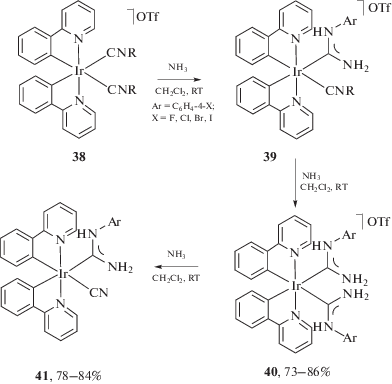
Схема 13 . Взаимодействие циклометаллированных комплексов иридия(III), содержащих изоцианидные лиганды, с аминами.
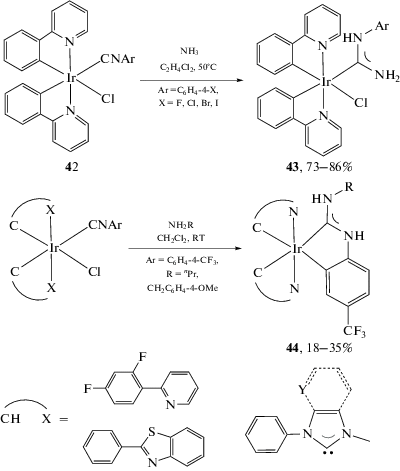
Схема 14 . Взаимодействие моноизоцианидных комплексов иридия(III) 42 с аммиаком и аминами.
Экспериментальные кинетические исследования [140, 143, 179–183] и квантово-химические расчеты [184–186] указывают на то, что реакция, приводящая к образованию ациклических диаминокарбеновых лигандов в результате атаки NH‑нуклеофилов на координированные изоцианиды имеет общий второй порядок [179–181], т.е. образование ассоциата происходит на стадии, определяющей скорость реакции. Небольшое увеличение энтальпии и уменьшение энтропии активации реакции (ΔH = 9.4 ± 0.1 ккал/моль, ΔS = –35.1 ± 1.7 ккал/моль для реакции цис-[PdCl2(CNPh)(PPh3)] c пара-толуидином) также указывают на протекание реакции через образование ассоциата [140]. На лимитирующей стадии скорость реакции должна зависеть от природы заместителей как в изоцианидном лиганде, так и в атакующем нуклеофиле: введение электроноакцепторных заместителей в молекулу изоцианида и/или электронодонорных заместителей в молекулу нуклеофила должно приводить к увеличению скорости реакции, что и наблюдается экспериментально [181]. Увеличение полярности растворителя замедляет скорость реакции, что, вероятно, связано с большей стабилизацией атакующего амина по сравнению с ведущим к ассоциату переходным состоянием в полярных растворителях, по всей видимости, за счет образования водородных связей [183].
По данным теоретических исследований методом теории функционала плотности, механизм сочетания CNXyl-лиганда в комплексе цис-[PtCl2(CNXyl)(CNMe)] с различными NH-нуклеофилами (HNMe2, HN=CPh2, H2N–N=CPh2) является ступенчатым процессом ассоциативного типа и включает присоединение нуклеофила к изоцианидному атому углерода, депротонирование полученного промежуточного соединения 45 (например, второй молекулой нуклеофила или другим основанием из реакционной смеси) и протонирование атома азота изоцианида с образованием продукта реакции 46 (схема 15) [184]. Нуклеофильное присоединение является лимитирующей стадией всего процесса. По данным квантово-химических расчетов, выполненных для разных типов нуклеофилов (HNMe2, HN=CPh2, H2N–N=CPh2), общая энергия активации почти не зависит от природы нуклеофила и составляет 19.8–22.4 ккал/моль [184]. Перенос протона при переходе от интермедиата 45 к соединению 46 может происходить ступенчато или согласованно. По данным квантово-химических расчетов изученных нуклеофилов (HNMe2, HN=CPh2, H2N–N=CPh2), перенос протона реализуется как ступенчатый процесс, в то время как согласованный перенос протона (как через четырехцентровое переходное состояние, так и через шестичленное переходное состояние с участием дополнительной молекулы нуклеофила) термодинамически менее выгоден.
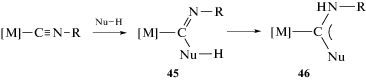
Схема 15 . Ступенчатый механизм сочетания CNXyl-лиганда в комплексе цис-[PtCl2(CNXyl)(CNMe)] с различными NH-нуклеофилами.
Следует отметить, что использование β-аминофункционализированных изоцианидов, претерпевающих самопроизвольную циклизацию при координации к металлоцентрам [164, 187], служит надежным методом синтеза комплексов с N-гетероциклическими карбеновыми лигандами (схема 16) [3].
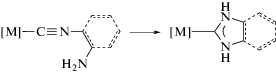
Схема 16 . Самопроизвольная циклизация β-аминофункционализированных изоцианидов.
Присоединение N,N'-полинуклеофилов. При наличии в молекуле присоединяющегося N-нуклеофила дополнительного N'-нуклеофильного центра возможно образование соединений более сложной структуры, в том числе с С,N- и С,С'-хелатными диаминокарбеновыми лигандами. Примеры сочетания N,N'-полинуклеофилов с изоцианидными лигандами в комплексах палладия(II) детально проанализированы в обзоре [138], поэтому ниже приведены только общие закономерности, характерные для всех металлов платиновой группы.
Взаимодействие N,N'-полинуклеофилов с изоцианидными комплексами логичнее всего систематизировать, опираясь на взаимное расположение нуклеофильных центров. Для N,N'-полинуклеофилов со смежными нуклеофильными центрами, обладающих одинаковой (гидразин, N,N'-диметилгидразин) или близкой нуклеофильностью атомов азота (N-моноалкилзамещенные гидразины), наиболее характерны реакции с цис-бис-изоцианидными или тетракис-изоцианидными комплексами, приводящие к образованию C,C'-хелатного бис-диаминокарбенового комплекса. Поскольку впервые такие соединения описаны профессором Л.А. Чугаевым [136], впоследствии они стали носить название комплексы “чугаевского типа” [188, 189]. Взаимодействие сгенерированного in situ тетракис(метилизоцианидного) комплекса платины(II) с гидразином приводит к образованию красного соединения 47 (красная соль Чугаева, схема 17 ), которое при действии раствора соляной кислоты переходит в желтый комплекс 48 (желтая соль Чугаева). Комплекс 48 может быть обратно превращен в 47 взаимодействием с метилизоцианидом [188].
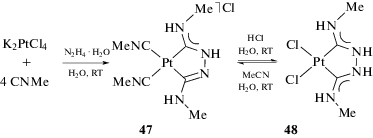
Схема 17 . Взаимодействие сгенерированного in situ тетракис(метилизоцианидного) комплекса платины(II) с гидразином.
Позднее в научных группах Балча (A. Balch) [73, 166, 188, 190–194], Слотера (L.M. Slaughter) [32, 195–198] и Титца (T.S. Teets) [40, 199] установлено, что реакция носит общий характер и синтетическая процедура может быть расширена на другие изоцианиды, замещенные гидразины и металлоцентры палладий(II) [32, 188, 193, 195, 198], иридий(III) [40, 199], золото(III) [200] и железо(II) [73, 79]. Легкость образования C,C'-хелатного продукта при использовании гидразина и монозамещенных гидразинов можно объяснить сочетанием нескольких сонаправленных факторов, а именно: 1) нуклеофильность гидразина выше, чем аминов и аммиака, что связано с α-эффектом [201, 202]; 2) сочетание второй изоцианидной группы происходит как внутримолекулярный процесс, что облегчает реакцию; 3) положительный заряд комплексного иона способствует нуклеофильной атаке на изоцианидный лиганд [203].
Производные гидразина с сильно пониженной нуклеофильностью одного из реакционных центров (гидразоны [34, 204–207], гидразиды карбоновых и сульфоновых кислот [29, 37, 208], N-Boc-гидразин [209], 4-нитрофенилгидразин [210, 211], N,N-дифенилгидразин [212], индазолы [213]) реагируют преимущественно с образованием продуктов моноприсоединения (схема 18). Несмотря на наличие двух нуклеофильных центров, активным является только один из них, тогда как второй центр дезактивирован структурным фрагментом или снижающим его нуклеофильность электроноакцепторным заместителем. Отдельно следует отметить, что присоединение индазола [213] к координированному изоцианиду является одним из малочисленных примеров [117, 213], когда NH-нуклеофильный центр включен в ароматическую систему. Соединения 53 имеют структуру, аналогичную ариламинокарбеновым комплексам [96], при этом стабилизация диаминокарбена происходит за счет частичного нарушения ароматической системы гетероарильного фрагмента [213].
Взаимодействие моноизоцианидных комплексов с производными гидразина, как и следовало ожидать, приводит к образованию монодентатных ациклических диаминокарбеновых комплексов, например, взаимодействие изоцианидных комплексов платины(II) c гидразином (cхема 19а) [214] и изоцианидного комплекса родия(III) c N-Boc-гидразином (cхема 19б) [209].
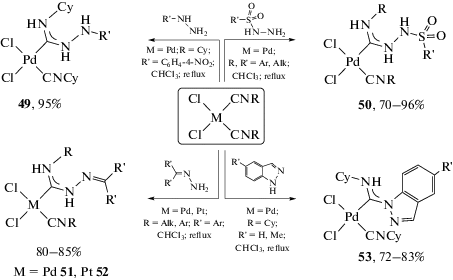
Схема 18 . Нуклеофильная атака изоцианидов, координированных к металлоцентрам палладий(II) и платина(II), производными гидразина.
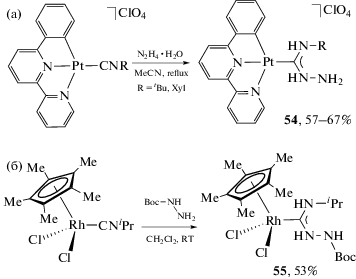
Схема 19 . Взаимодействие моноизоцианидных комплексов платины(II) (а) и родия(III) (б) с производными гидразина.
Симметричные и несимметричные N=C–N полинуклеофилы – 2-аминоазагетероциклы [38, 115, 215–221], 3-иминизоиндолин-1-оны [35, 222, 223], изоиндолин-1,3-диимины [224], 1H-пиррол-2,5-диимины [36, 225, 226], амидины [116, 227] и N,N'-дифенилгуанидин [228] – присоединяются к изоцианидным лигандам в цис-[MCl2(CNR)2] (M = Pd, Pt) только одним из своих нуклеофильных центров, тогда как вторым нуклеофильным атомом азота координируются к металлоцентру, образуя C,N-хелатный диаминокарбеновый лиганд (схема 20). Общей закономерностью реакций цис-[MCl2(CNR)2] (M = Pd, Pt) с указанными N=C–N полинуклеофилами является протекание взаимодействия в два этапа. Сначала происходит координация одного N-донорного центра к металлоцентру, как правило, путем замещения галогенидного лиганда, а затем протекает внутримолекулярная нуклеофильная атака другим нуклеофильным центром N-координированного полинуклеофила по тройной связи CN изоцианида.
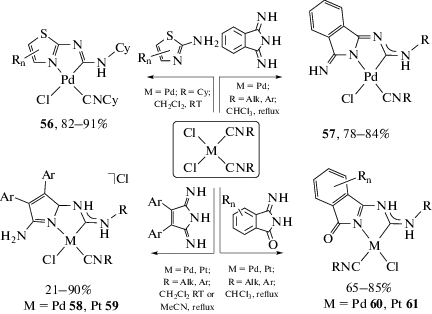
Схема 20 . Сочетание симметричных и несимметричных полинуклеофилов N=C–N с изоцианидами в комплексах цис-[MCl2(CNR)2] (M = Pd, Pt).
Сочетание несимметричных N=C–N полинуклеофилов, таких как N,N'-дифенилгуанидин [228] и N-фенилбензамидин [227], приводит к образованию региоизомерных диаминокарбеновых комплексов (схема 21). В растворе N,N'-дифенилгуанидин и N-фенилбензамидин существуют в таутомерном равновесии, которое представляет собой внутримолекулярный процесс переноса протона между N-центрами; вследствие такого таутомерного равновесия N‑донорные центры сходны по своей нуклеофильности.
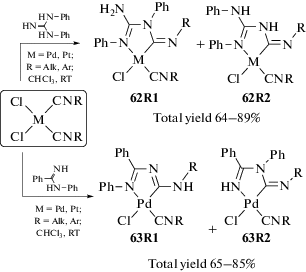
Схема 21 . Сочетание N,N'-дифенилгуанидина и N-фенилбензамидина с изоцианидами в комплексах палладия(II) и платины(II), приводящее к образованию региоизомерных диаминокарбеновых комплексов.
Координация N,N'-дифенилгуанидина к металлоцентру, протекающая на начальном этапе реакции с замещением хлоридного лиганда, во всех случаях происходит фрагментом PhN=C [228]. Последующая стадия нуклеофильного сочетания протекает внутримолекулярно при атаке атома углерода координированного изоцианида одним из двух оставшихся донорных центров – NHPh (региоизомер 62R1) или NH2 (региоизомер 62R2), что приводит к образованию региоизомерных С,N-хелатных диаминокарбеновых комплексов 62 (схема 21). Региоизомерный состав полностью определяется характером заместителя в изоцианидном лиганде; варьирование растворителя и/или условий не приводит к изменению изомерного состава реакционной смеси. Кроме того, для обоих металлоцентров сочетание приводит к одинаковым по составу региоизомерным смесям. Вовлечение в реакцию арилизоцианидов приводит преимущественно к образованию региоизомера 62R1, в то время как использование комплексов с алкилизоцианидами повышает долю региоизомера 62R2.
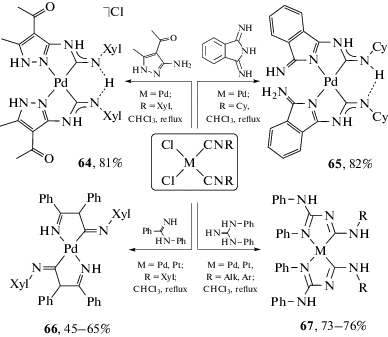
Схема 22 . Образование бис-диаминокарбеновых комплексов из бис-изоцианидных комплексов цис-[MCl2(CNR)2] (M = Pd, Pt).
В случае N-фенилбензамидина региоизомерный состав также определяется характером заместителя в изоцианидном лиганде [227]. При использовании ароматического изоцианида (R = = Xyl) N-фенилбензамидин координируется к металлоцентру центром HN=C, а нуклеофильная атака осуществляется за счет центра NHPh амидина (63R1). Если заместителем является трет-бутильная группа (R = t-Bu), наблюдается обратная ситуация: координация амидина происходит центром NHPh, а присоединение к изоцианиду – нуклеофильным центром HN=C (63R2). При использовании циклогексилизоцианида (R = Cy) наблюдается образование обоих региоизомеров (63R1 и 63R2).
Внутримолекулярный характер присоединения N=C–N полинуклеофилов не только ускоряет реакцию, но и делает возможным образование бис-диаминокарбеновых комплексов из бис-изоцианидных комплексов цис-[MCl2(CNR)2] (M = = Pd, Pt) при проведении реакции в избытке нуклеофила и/или при более жестких условиях (схема 22) [216, 224, 227, 228]. Бис-диаминокарбены 66 и 67 также могут быть получены при взаимодействии комплексов 62 и 63 с дополнительным количеством соответствующего нуклеофила [227, 228].
Диаминокарбеновые комплексы палладия(II) и платины(II) 68, образующиеся в результате реакции нуклеофильного сочетания арилизоцианидов в цис-[MCl2(CNAr)2] (M = Pd, Pt) с α-аминоазагетероциклами, могут реагировать со второй молекулой изоцианидного комплекса цис-[MCl2(CNAr)2] с образованием биядерных диаминокарбеновых комплексов 69 (схема 23, детально реакционная способность ациклических диаминокарбенов в комплексах переходных металлов описана в обзоре [28]) [38, 115, 215–217, 219–221, 229].
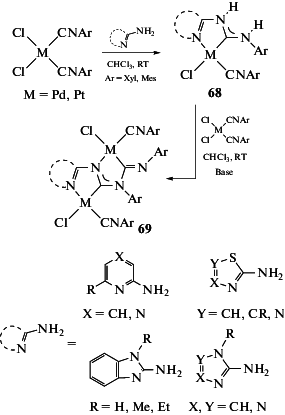
Схема 23 . Взаимодействие диаминокарбеновых комплексов палладия(II) и платины(II) с исходным изоцианидным комплексом цис-[MCl2(CNAr)2], приводящее к биядерным диаминокарбеновым производным.
Синтез комплекса палладия(II) 72 с диаминокарбеновым лигандом C,N,C-пинцерного типа также можно считать одним из примеров присоединения NCN-нуклеофилов (схема 24) [230]. Соединение 70, содержащее монодентатный лиганд с фрагментом мочевины, в основных условиях дважды аннелируется приводя к 72. Механизм превращения заключается в депротонировании основанием одного из кислых (NH) атомов водорода фрагмента мочевины и замещении иодидного лиганда с образованием интермедиата 71. В условиях реакции интермедиат 71 самопроизвольно циклизуется путем внутримолекулярного нуклеофильного присоединения второй N-донорной группы мочевины к координированному изоцианиду.
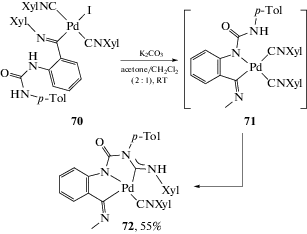
Схема 24 . Синтез комплекса палладия(II) с диаминокарбеновым лигандом C,N,C-пинцерного типа.
Взаимодействие изоцианидных комплексов иридия(III) c α-аминоазагетероциклами также приводит к образованию соединений 73 с C,N-хелатным диаминокарбеновым лигандом, однако в случае указанного металлоцентра необходимо дополнительное использование солей серебра для предварительного удаления галогенидных лигандов вследствие высокой кинетической инертности металлоцентра иридий(III) в реакциях лигандного обмена (схема 25) [121].
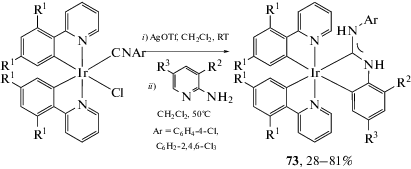
Схема 25 . Взаимодействие изоцианидных комплексов иридия(III) c α-аминоазагетероциклами.
В молекулах 1,2- и 1,3-диаминов нуклеофильные центры расположены у различных атомов углерода. При разделении нуклеофильных центров атомами углерода, с одной стороны, уменьшает их взаимное влияние друг на друга, а с другой – структура становится более гибкой, что делает возможным образование комплексов с C,N- или C,C'-хелатными диаминокарбеновыми лигандами при взаимодействии этих нуклеофилов с изоцианидными комплексами металлов платиновой группы. Взаимодействие изоцианидных комплексов палладия(II) цис-[PdCl2(CNR)2] и о-фенилендиаминов в зависимости от стехиометрии исходных веществ и условий реакции может приводить к соединениям трех типов (схема 26). Во всех случаях во взаимодействие вступают обе аминогруппы и продукт включает хелатный диаминокарбеновый лиганд [103]. На основе кинетических исследований в работе [154] установлено, что при взаимодействии комплексов цис-[PdCl2(CNR)2] с о-фенилендиаминами на первой стадии происходит обратимое замещение хлоридного лиганда, затем внутримолекулярно нуклеофильное присоединение.
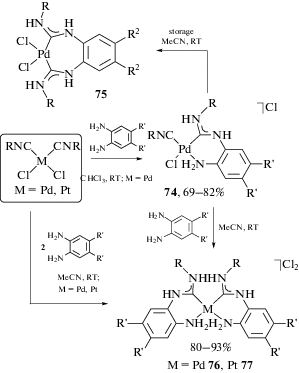
Схема 26 . Взаимодействие изоцианидных комплексов цис-[PdCl2(CNR)2] с орто-фенилендиаминами.
Благодаря внутримолекулярному характеру сочетания 1,2-диаминов с координированными изоцианидами процесс сочетания происходит даже в случае смешанных диаминокарбен/изоцианидных комплексов, которые, как правило, не реагируют с монофункциональными нуклеофилами. Бис-диаминокарбены 76 могут быть получены взаимодействием 74 с дополнительным количеством соответствующего о-фенилендиамина (схема 26 ) [103]. Взаимодействие диаминокарбенового комплекса 78 с 1,2-диамином приводит к бис-диаминокарбеновому комплексу 79 с двумя типами лигандов ADC, а именно, с монодентатным и C,N-хелатным (схема 27) [154].
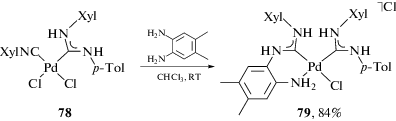
Схема 27 . Образование бис-диаминокарбенового комплекса палладия(II), содержащего два типа ADC лигандов.
Взаимодействие комплексов цис-[PdCl2(CNR)2] с о-аминофенолом и о-аминобензиловым спиртом с группой ОН в качестве второго атакующего центра (гораздо менее основную, чем NH2 в о-фенилендиаминах) приводит исключительно к монодентатным диаминокарбеновым комплексам 80 (схема 28). м-Фенилендиамин и п-фенилендиамин также взаимодействуют с цис-[PdCl2(CNR)2], однако реакция протекает только по одному изоцианидному лиганду (схема 28 ). м-Фенилендиамин реагирует только одной аминогруппой, образуя комплексы 81; п-фенилендиамин участвует в реакции обеими аминогруппами, каждая из которых вступает в сочетание с отдельной молекулой цис-[PdCl2(CNR)2], и образуются биядерные соединения 82.
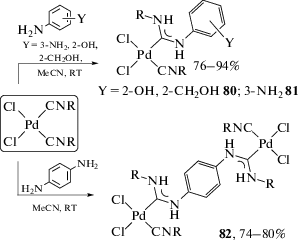
Схема 28 . Взаимодействие комплексов цис-[PdCl2(CNR)2] с орто-аминофенолом и орто-аминобензиловым спиртом.
Гомолептические комплексы палладия(II) и платины(II) 83, содержащие два С,С-хелатных бис-диаминокарбеновых лиганда, могут быть синтезированы посредством “one pot” взаимодействия соответствующей соли металла, изоцианида и 1,2-диаминоэтана или 1,3-диаминопропана (схема 29а) [231].
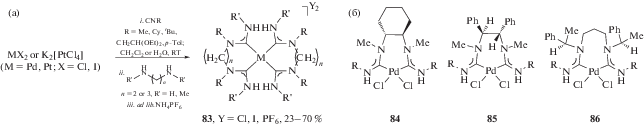
Схема 29 . Получение гомолептических комплексов палладия(II) и платины(II), содержащих два С,С-хелатных бис-диаминокарбеновых лиганда (а); примеры хиральных С,С-хелатных бис-диаминокарбеновых комплексов палладия(II) (б).
Взаимодействием оптически активных диаминов, а именно замещенных 1,2-диаминоэтанов [232, 233] и 1,3-диаминопропана [234] с цис-[PdCl2(CNC6H4-4-CF3)2] могут быть получены хиральные С,С-хелатные бис-диаминокарбеновые комплексы палладия(II) 84–86 (схема 29 б). В отличие от сочетания гидразинов, сочетание 1,2- и 1,3-диаминов с изоцианидными лигандами требует более жестких условий реакции (повышенная температура, большее время).
Алифатические диамины, у которых нуклеофильные центры отделены друг от друга более чем тремя атомами, могут вступать в реакцию с двумя эквивалентами изоцианидных комплексов с образованием биядерных соединений, в этом случае образовавшийся бис-диаминокарбеновый лиганд выступает линкером, связывающим два металлоцентра (схема 30) [214].
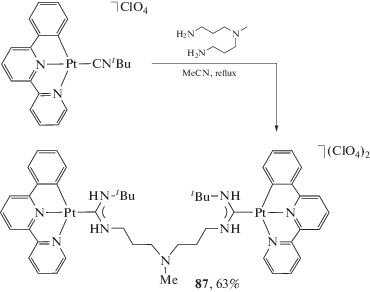
Схема 30 . Бис-диаминокарбеновый лиганд как линкер, связывающий два металлоцентра.
Таким образом, метод синтеза диаминокарбеновых комплексов, основанный на реакциях металлоактивированных изоцианидов с N-центрированными нуклеофилами, отличается простотой и универсальностью и позволяет получать ациклические диаминокарбеновые комплексы металлов платиновой группы с заданной структурой, которая, в свою очередь, напрямую связана со строением нуклеофила и условиями реакции. Данный метод соответствует принципам “зеленой химии”, так как является “атомэкономичным” и обеспечивает высокую скорость получения целевого продукта со значительным выходом. Отметим, что образующиеся в ходе металлопромотируемого сочетания комплексы содержат как минимум один водородный заместитель при атоме азота диаминокарбенового фрагмента, поэтому такие соединения могут в дальнейшем быть модернизированы при помощи реакций алкилирования или аннелирования (схема 31, детально реакционная способность ациклических диаминокарбенов в комплексах переходных металлов описана в обзоре [28]).
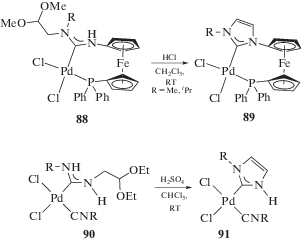
Схема 31 . Примеры аннелирования ациклических диаминокарбеновых комплексов.
Метод внедрения изоцианидов по связи металл–азот
Внедрение изоцианида по связи M–N приводит к образованию диаминокарбенового фрагмента, при этом из аминокомплексов с формально нейтральным N-донорным лигандом образуются классические диаминокарбены, а в случае амидокомплексов с координированным анионным N-донорным центром реакция внедрения изоцианида приводит к депротонированным диаминокарбенам. Следует отметить, что в среде металлокатализируемых и металлопромотируемых превращений изоцианидов их внедрение по связи металл–элемент является наиболее распространенной реакцией [135, 235]. Реакции внедрения изоцианидов важны не только в химии комплексов переходных элементов для получения библиотек различных металлоорганических соединений, но и в органическом синтезе, так как многие металлокатализируемые органические процессы образования азагетероциклических систем или полимеризации изоцианидов включают реакцию внедрения как промежуточную стадию [135, 235–240]. Введение изоцианидов по связи M–N в основном происходит через начальную координацию неподеленной пары атома углерода изоцианида к металлоцентру с последующим быстрым внедрением в связь металл–азот [135].
Комплексы палладия(II) 92 и 94, которые имеют в составе одного из лигандов фрагмент ароматического амина, взаимодействуют с изоцианидами в мягких условиях; реакция приводит к диаминокарбеновым комплексам 93 и 95 с хорошим выходом (схема 32) [241]. Биядерный комплекс 96, полученный в результате депротонирования 95, может быть окислен до соединения 97, представляющего собой диаминокарбеновый комплекс палладия(III).
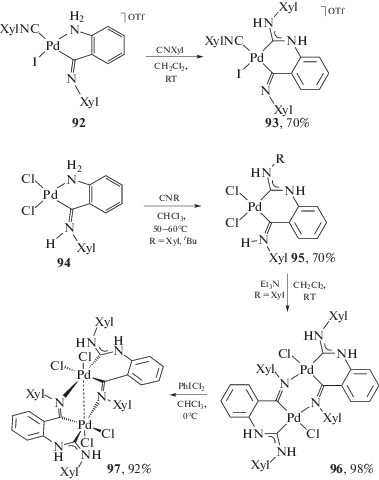
Схема 32 . Внедрение изоцианидов в комплексы палладия(II), содержащие ароматические амины.
Депротонированные диаминокарбеновые комплексы палладия(II) 99 [242] и платины(II) 101 [243] синтезированы путем внедрения изоцианида по связи M–N в соответствующие амидные комплексы 98 и 100 (схема 33). Необходимо отметить, что несмотря на то, что исходные соединения содержат несколько N-донорных лигандов, реакция внедрения протекает селективно только по одной связи M–N.
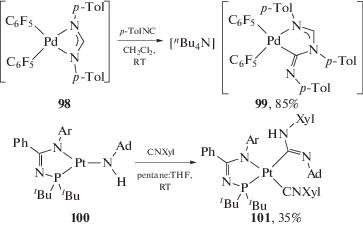
Схема 33 . Получение депротонированных диаминокарбеновых комплексов палладия(II) и платины(II) путем внедрения изоцианида по связи M–N в соответствующие амидные комплексы.
C,N-хелатный ADC комплекс платины(II) 102 был синтезирован из аминонитронного комплекса (схема 34) [244]. Реакция протекает формально путем внедрение изоцианида по связи Pt–N с последующей стадией деоксигенирования при помощи второй молекулы изоцианида. Недостатком данного метода является использование двухкратного избытка изоцианида и низкий препаративный выход продукта.
Пример синтеза ADC комплекса рутения(II) 106 представлен в работе [77] (схема 35). Синтетическая схема основана на N-металлировании ADC комплекса железа(II) 103 димером дихлорида рутения с последующим внедрением изоцианида по связи Ru–N. Гетеробиметаллический комплекс содержит два диаминокарбеновых фрагмента, связанный с атомом рутения ациклический диаминокарбеновый фрагмент является частью пятичленного металл-N-гетероциклического карбена, связанного с железом.
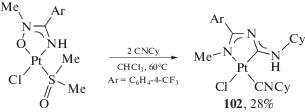
Схема 34 . Синтез C,N-хелатного ADC производного платины(II) из исходного аминонитронного комплекса.
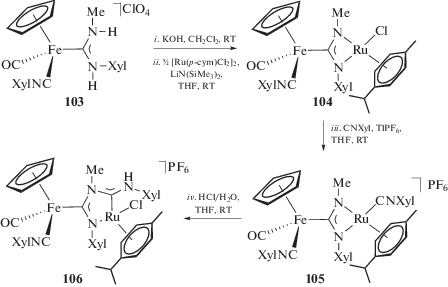
Схема 35 . N-металлирование ADC комплекса железа(II) 103 димером дихлорида рутения с последующим внедрением изоцианида по связи Ru–N.
ПРИМЕНЕНИЕ КОМПЛЕКСОВ МЕТАЛЛОВ ПЛАТИНОВОЙ ГРУППЫ С АЦИКЛИЧЕСКИМИ ДИАМИНОКАРБЕНОВЫМИ ЛИГАНДАМИ
Применение в катализе органических процессов
Первые работы, в которых сообщалось о применении ациклических диаминокарбеновых комплексов в качестве катализаторов, были опубликованы в 2005 г. и посвящены разработке каталитических систем палладий-катализируемых реакций арилгалогенидов, а именно кросс-сочетания Сузуки и реакции Хека [131, 195]. В этих исследованиях наглядно продемонстрировано сходство каталитических свойств комплексов с ациклическими и гетероциклическими диаминокарбеновыми лигандами, что привело к лавинообразному росту исследований в области каталитического применения ациклических диаминокарбеновых комплексов. В то время как некоторые исследователи открыто отказывались признавать перспективность комплексов с ациклическими диаминокарбенами (“There is little hope that acyclic carbenes could find applications as ligands in transition metal catalysts” [245]), другие совершенствовали каталитические системы. В 2012 г. одновременно вышли две обзорные работы, подготовленные в группе акад. РАН В.Ю. Кукушкина [26] и в группе проф. Л.М. Слотера [27], в которых суммированы результаты исследования каталитической активности ациклических диаминокарбеновых комплексов в период с 2005 по 2011 г. Основные отличия циклических и ациклических диаминокарбеновых комплексов в свете каталитического применения, сделанные авторами упомянутых обзоров, заключаются в конформационной гибкости ациклических диаминокарбенов, позволяющей лиганду принимать несколько конформаций за счет вращения вокруг связей C–N, и в модульном методе синтеза, делающем синтетически доступной библиотеку M‑ADC комплексов с широким набором донорных и стерических характеристик, а также функционализированных соединений. Для предотвращения дублирования информации в данном обзоре акцент сделан на рассмотрении опубликованных с 2012 г. по настоящее время каталитических систем.
Открытые в 70-х годах прошлого века палладий-катализируемые реакции кросс-сочетания с участием металлоорганических соединений и арил/алкенилгалогенидов, позволяющие создавать связи Csp2–Csp2 и Csp2–Csp, а также связи Csp2–X углерод–гетероатом, в настоящее время – один из наиболее используемых инструментов органического синтеза [246–248]. Несмотря на то, что реакции кросс-сочетания могут катализироваться широким спектром катализаторов, в том числе соединениями менее дорогих металлов, таких как никель [249–253], кобальт [254], железо [255–257], катализ палладиевыми катализаторами обладает рядом преимуществ: более мягкие условия реакции, высокая селективность, возможность использования дезактивированных субстратов и проведения кооперативного катализа [258, 259].
Реакция кросс-сочетания Сузуки–Мияура (Suzuki–Miyaura), представляющая собой сочетание арилборных кислот с арил/алкенилгалогенидами, зарекомендовала себя как один из наиболее удобных и простых методов создания биарильных/арилалкенильных фрагментов [260, 261]. Борорганические соединения отличаются от литий-, магний- и цинкорганических соединений толерантностью к функциональным группам и большей устойчивостью, при этом их сочетание с арил/винилгалогенидами протекает с высоким выходом. В работах [222, 224] исследована каталитическая активность в реакции кросс-сочетания Сузуки смешанолигандных диаминокарбен/изоцианидных комплексов 57 и 60 и диаминокарбен/фосфиновых комплексов 107 (схема 36). Катализаторы 60 и 107 продемонстрировали схожую высокую каталитическую активность. В оптимизированных условиях (этанол, K2CO3 или Cs2CO3, 2–2.5 ч, 80°С) выход биарилов составил 58–99%, при этом оборотные числа катализаторов достигали 5.8 × 104 для арилбромидов и 9.8 × × 104 для замещенных арилиодидов. Как и в случае других каталитических систем на основе Pd-ADC [206], для систем на основе 60 и 107 отсутствуют различия между экспериментами, проведенными в сухом растворителе и в атмосфере инертного газа, с аналогичными экспериментами, проведенными на воздухе и в неосушенном EtOH. Таким образом, каталитические системы на основе 60 и 107 могут успешно использоваться на воздухе и в неосушенном растворителе, не требующем дополнительной дегазации и азеотропной осушки. Стоит отметить, что диаминокарбен/фосфиновые производные 107 являются более эффективными катализаторами реакций Сузуки по сравнению со структурно схожими диаминокарбен/изоцианидными производными 57, которые содержат вместо фосфинового лиганда изоцианидный; кроме того, в условиях эксперимента катализаторы 107 на порядок активнее, чем цис-[PdCl2(PPh3)2]. Комплексы палладия 49 [210], 50 [37], 51 [205] и 63 [31] также являются эффективными катализаторами реакции Сузуки.
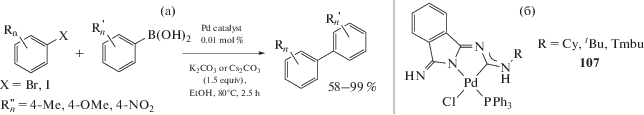
Схема 36 . Кросс-сочетание Сузуки арилбромидов и арилиодидов с замещенными фенилборными кислотами (а); каталитически высокоактивные производные палладия(II), содержащие диаминокарбен/фосфиновые лиганды (б).
Важная модификация реакции Сузуки – ее проведение в водной среде, что особенно важно для модификации биомолекул, а также перспективно с экономической и экологической точек зрения [262, 263]. В работе [37] описано использование катализаторов на основе Pd-ADC для проведения реакции кросс-сочетания Сузуки в водной среде. Соединения 50 и 108 умеренно растворимы в воде (0.01–0.02 ммоль/л) и могут быть извлечены из водных растворов в неизменном виде, что свидетельствует об их гидролитической стабильности. Вероятно, причиной умеренной водорастворимости соединений 50 и 108 является их способность к внутримолекулярной циклизации в водных растворах с образованием производных, содержащих шестичленный С,О-хелатный цикл (схема 37) [264]. Кросс-сочетание арилбромидов и арилиодидов с замещенными фенилборными кислотами протекает при использовании 0.01 мол. % палладиевых катализаторов 50 или 108 и приводит к биарильным производным с выходом до 98% (схема 38). Спектр применимости данной каталитической системы продемонстрирован на различных производных бромбензола и арилборных кислотах, в том числе на пространственно-затрудненных. Активированные арилхлориды также могут быть превращены в диарилпроизводные в подобных условиях (100°C, 3 ч), в то же время неактивированные арилхлориды в реакцию не вступают даже при увеличении загрузки катализатора до 1 мол. %. Следует отметить, что на момент выхода указанной работы Pd-ADC катализаторы 50 и 108 – самые эффективные для проведения реакции Сузуки в водной среде [265–268]. Эти катализаторы могут применяться в спиртовой среде, демонстрируя сравнимую и более высокую каталитическую активность.
Схема 37 . Водорастворимость диаминокарбеновых производных вследствие предполагаемой внутримолекулярной циклизации.
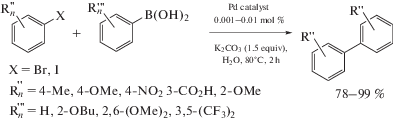
Схема 38 . Кросс-сочетание Сузуки арилбромидов и арилиодидов с замещенными фенилборными кислотами, катализируемое водорастворимыми диаминокарбеновыми комплексами палладия(II).
Катализируемая соединениями палладия реакция кросс-сочетания терминальных ацетиленов с арил/алкенилгалогенидами – реакция Соногаширы (Sonogashira) – удобный и эффективный метод синтеза интернальных алкинов и енинов [269–271]. Реакция Соногаширы в классическом варианте катализируется одновременно соединениями двух металлов, а именно палладия и меди, однако использование такой биметаллической системы негативно сказывается на селективности реакции из-за образования побочного продукта гомосочетания – бутадиина [26, 27, 272]. Pd-ADC комплексы 60 и 108 проявили высокую каталитическую активность в реакции Соногаширы в “безмедном” исполнении (схема 39) [29]. Реакция протекает в неосушенном EtOH в присутствии K2CO3 в качестве основания и при загрузке катализаторов 60 и 108 0.01 мол. %. Арилиодидные субстраты, включающие заместители с различными электронными и стерическими свойствами, вступают в реакцию с разнообразными терминальными алкинами, образуя соответствующие интернальные алкины. Таким образом, демонстрируется универсальность каталитической системы. Соединения палладия 49 [212], 51 [204] и 63 [227] также являются эффективными катализаторами реакции Соногаширы в схожих условиях.
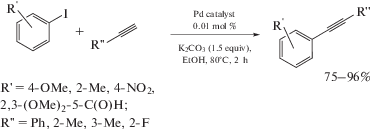
Схема 39 . Высокая каталитическая активность Pd-ADC комплексов в реакции Соногаширы в “безмедном” исполнении.
Используемый в каталитических реакциях комплекс представляет собой вещество определенной структуры, однако в большинстве случаев реальная структура каталитически активных частиц неизвестна [273–275]. Исследования в работе [29] механизма каталитического процесса свидетельствуют о том, что в системе на основе соединений 60 и 108 каталитический цикл основан на молекулярном механизме (схема 40). Образование каталитически активных частиц происходит с отрывом галогеновых лигандов, в то время как изоцианид и диаминокарбен выступают в качестве лигандов-стабилизаторов. Деактивация катализаторов связана с образованием каталитически неактивных палладиевых кластеров, стабилизированных диаминокарбеновыми и изоцианидными лигандами.
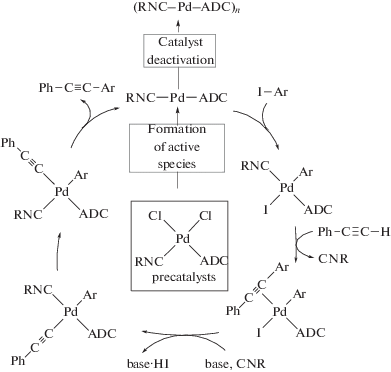
Схема 40 . Предполагаемый механизм каталитической активности Pd-ADC комплексов в реакции Соногаширы в “безмедном” исполнении.
В работах [276, 277] исследована возможность получения Pd-ADC катализаторов in situ путем нуклеофильного присоединения NH-нуклеофилов к изоцианидным лигандам в комплексах палладия(II). На примере модельной реакции Сузуки с использованием 4-иоданизола или 4-броманизола и фенилборной кислоты продемонстрировано, что Pd-ADC комплексы, образованные in situ путем взаимодействия 4-нитрофенилгидразина (комплекс 49), бензгидразида (комплекс 108а, R = Cy, R' = Ph) и морфолина (комплекс 109) с цис-[PdCl2(CNCy)2], позволяют проводить реакцию кросс-сочетания в мягких условиях (EtOH, кипячение с обратным холодильником в течение 2 ч в присутствии K2CO3) [277]. Синтезированные in situ Pd-ADC комплексы проявляют схожую каталитическую активность с предварительно полученным аналитически чистым соединением 109; морфолин оказался лучшим среди изученных нуклеофилов для активации изоцианидного комплекса палладия(II) в реакции кросс-сочетания Сузуки. Тот же подход успешно применен для реакции кросс-сочетания Соногаширы с использованием 4-иоданизола и фенилацетилена; в этом случае наибольший выход продукта кросс-сочетания достигнут при использовании бензгидразида в качестве “активатора” изоцианидного комплекса [276].
Реакция кросс-сочетания Хияма основана на использовании кремнийорганических соединений [278, 279] и может служить важным дополнением реакций Сузуки и Соногаширы. В работе [148] продемонстрировано, что Pd-ADC комплексы 110–112 могут быть использованы в качестве катализаторов реакции кросс-сочетания Хияма. В оптимизированных условиях, а именно при использовании в качестве основания NaOH, а в качестве растворителя смеси 1,4-диоксан/вода (2 : 1 об.) каталитическая система позволяет получать дизамещенные ацетилены из иодбензола и различных триэтоксисилилалкинов (схема 41). Катализаторы 110–112 также могут применяться в тандемной реакции кросс-сочетания Хияма с последующей циклизацией между 2-иодфенолом и триэтоксисилилалкинами, обеспечивая удобный синтетический путь к биологически релевантным производным бензофурана.
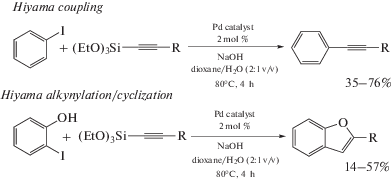
Схема 41 . Применение Pd-ADC комплексов в качестве катализаторов реакции кросс-сочетания Хияма.
Оптически активные Pd-ADC комплексы 113–115 исследованы в качестве катализаторов реакции межмолекулярного асимметричного аллильного алкилирования (схема 42) [145]. Использование катализатора 114 позволяет проводить каталитическую реакцию за короткое время с высоким выходом и умеренной энантиоселективностью (катализатор 113: выход 87% (энантиомерный избыток (э. и.) 6%, S) через 18 ч; катализатор 114: выход 100% (э. и. 45%, S) через 2 ч). Каталитическая активность бис-диаминокарбенового комплекса 115 в обоих реакциях оказалась существенно ниже, чем у 113 и 114. Соединения 113 и 114 также оказались каталитически активными в реакции Сузуки с использованием замещенных бромбензолов (1 мол. % катализатора, комнатная температура, 20 ч, выход биарила 75–84%).
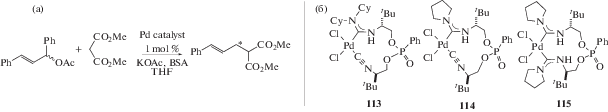
Схема 42 . Применение диаминокарбеновых комплексов палладия(II) в качестве катализаторов реакции межмолекулярного асимметричного аллильного алкилирования (а); структура соединений 113–115 (б).
Палладий-катализируемое взаимодействие арил- и алкенилгалогенидов с алкенами – реакция Хека (Heck) – является одним из лучших и продуктивных современных способов получения терминальных и интернальных алкенов [280, 281]. Диаминокарбеновые комплексы палладия(II) “чугаевского” типа предложены в качестве катализаторов реакции Хека (схема 43) [32]. Проведенный скрининг серии из 10 структурно похожих катализаторов в модельной реакции 4-бромацетофенона и стирола (1 мол. %, AcONa, ДМФ, 100°С) выявил большие вариации каталитической активности в зависимости от структуры катализатора (выход стильбена 16–98%). Комплексы дибромида палладия (Y = Br) проявляют значительно большую каталитическую активность (36–98%) по сравнению с хлоридными аналогами (Y = Cl, 16–59%), что, по мнению авторов работы, связано с большей лабильностью бромидных лигандов в каталитических условиях. Pd-ADC комплекс 116а (R, R' = Me) оказался наилучшим катализатором (выход стильбена 98%).
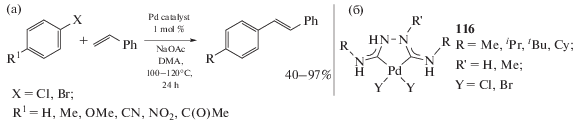
Схема 43 . Применение диаминокарбеновых комплексов палладия(II) “чугаевского” типа в качестве катализаторов реакции Хека (а); Pd-ADC, обладающий наиболее высокой каталитической активностью (б).
Данную каталитическую систему можно использовать для арилбромидов, содержащих как электронодонорные, так и электроноакцепторные заместители, которые могут быть преобразованы в соответствующие стильбены с хорошим выходом. Активированные сильными электроноакцепторными заместителями арилхлориды (4-нитрохлоранилин) также могут быть превращены в соответствующие стильбены. В то же время арилхлориды, содержащие менее электроноакцепторные заместители, приводят к продукту с низким выходом или не вступают в реакцию вовсе.
При проведении реакции в неосушенном и недеаэрированном растворителе выход целевых стильбенов снижается на 34–40% по сравнению с реакциями, проводимыми в атмосфере азота и в предварительно осушенном растворителе. Отметим, что в отличие от каталитической системы для реакции Хека ранее опубликованная каталитическая система для реакции Сузуки, основанная на соединениях 116, не чувствительна к влаге и кислороду воздуха [197].
Эфиры арилборных кислот часто используются в органическом синтезе в реакциях кросс-сочетания для образования связей С–С, С–О, С–N и C–S [282]. Современным наиболее общим методом синтеза эфиров арилборных кислот является относительно недавно открытый метод, основанный на катализируемом комплексами палладия взаимодействии арилгалогенидов и производных диборана (борилирование по Мияура (Miyaura)) [283]. В работе [33] показана возможность использования Pd-ADC комплексов 117 в качестве катализаторов реакции борилирования арилбромидов методом Мияура (схема 44). Реакция протекает с хорошим выходом целевого арилборного эфира с практически полным отсутствием продуктов гомосочетания биарилов. Использованные в качестве катализатора комплексы 117 превзошли эталонный катализатор [PdCl2(dppf)] как по активности, так и по селективности.
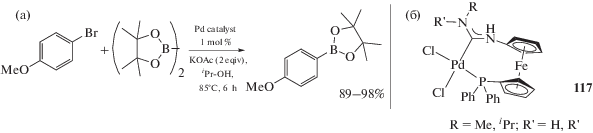
Схема 44 . Катализируемое комплексами палладия взаимодействие арилгалогенидов и производных диборана (реакция Мияура) (а); высокоэффективный и селективный катализатор реакции Мияура, содержащий диаминокарбеновый лиганд (б).
Создание регенерируемых катализаторов путем иммобилизации высокоактивных гомогенных комплексов на подходящем носителе является актуальным направлением исследований в области палладий-катализируемых реакций кросс-сочетания [284, 285]. При использовании иммобилизованного катализатора возможно просто его удаление из реакционной смеси и повторное использование, при этом продукт не будет загрязнен соединениями переходного металла, что особенно важно при разработке лекарственных препаратов [286]. Ациклические диаминокарбеновые комплексы палладия(II) 118, иммобилизованные на полистирольном носителе, синтезированы в работе [101] по реакции присоединения NH2-группы коммерчески доступной бензгидриламидной полистирольной смолы к изоцианидному лиганду в комплексах [PdCl2(CNR)2] (13% модификации аминогрупп носителя). Иммобилизованные соединения 118 апробированы качестве катализаторов в реакции кросс-сочетании Сузуки с использованием ряда арилиодидов и арилбромидов (схема 45). Оптимальными условиями для проведения кросс-сочетания с использованием 118 являются система ДМФА–вода (5–10 об. % воды) в качестве растворителя и K2CO3 в качестве основания. Авторы показали возможность отделения иммобилизованных катализаторов фильтрованием и их многократного использования (до трех раз) без потери каталитической активности. Исследование катализаторов методом РФЭС до и после каталитических экспериментов показало сохранение степени окисления палладия, что является косвенным свидетельством сохранения структуры иммобилизованного комплекса в неизменном виде в процессе реакции.
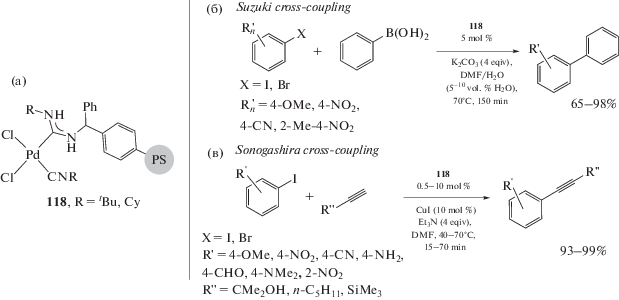
Схема 45. Ациклические диаминокарбеновые комплексы палладия(II), иммобилизованные на полистирольном носителе (а) в качестве катализаторов реакций кросс-сочетании Сузуки (б) и Соногаширы (в).
Катализаторы 118 продемонстрировали также высокую каталитическую активность в реакции кросс-сочетания Соногаширы для широкого набора субстратов (65–98%) с возможностью рецикла до 8 раз без значимой потери каталитической активности (схема 45). При исследовании образцов 118 методом РФЭС обнаружено, что после проведения реакции Соногаширы степень окисления иммобилизованного на носителе палладия изменяется от +2 до 0, таким образом, соединения 118 в этой реакции правильнее называть прекатализаторами.
В работе [119] сообщается о возможном применении Pd-ADC комплексов в реакции межмолекулярного аминирования алкинов, однако реакция протекает с низким выходом (20% гидроаминированного продукта при общей конверсии алкина 77%).
Гидросилилирование ненасыщенных соединений – один из фундаментальных подходов к лабораторному и промышленному синтезу кремнийорганических соединений [287, 288]. PtII-ADC комплексы 52 и 61 исследованы в качестве катализаторов реакции гидросилилирования терминальных и интернальных алкинов с гидросиланами [34, 35]. Каталитическую реакцию проводили при 80–100°С в течение 3–6 ч в толуоле (схема 46); наибольшая каталитическая активность (выход 96%) была достигнута при использовании соединения 61а (R = C6H3–2-Cl–6-Me; R'n = 3-Me).
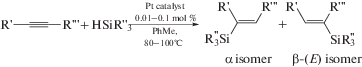
Схема 46 . Реакция гидросилилирования терминальных и интернальных алкинов гидросиланами, катализируемая комплексами платины(II) с ADC лигандами.
Гидросилилирование терминальных алкинов (PhC≡CH, tBuC≡CH, 4-(tBu)C6H4C≡CH) гидросиланами (Et3SiH, Pr3SiH, iPr3SiH, PhMe2SiH) во всех случаях протекает с образованием смеси α/β-изомеров силилированных алкенов (соотношение α/β от 81 : 19 до 5 : 95) с суммарным выходом 48–96%. Соотношение изомеров α/β зависит от заместителей в обоих субстратах, например, реакция iPr3SiH с 4-(tBu)C6H4C≡CH дает α-изомер силилированного алкена в качестве основного продукта (81 : 19), тогда как реакция PhMe2SiH с tBuC≡CH дает β-изомер в качестве основного продукта (5 : 95). Гидросилилирование интернальных алкинов (PhC≡CPh, Me(CH2)2C≡C(CH2)2Me и PhC≡CMe) Et3SiH и PhMe2SiH приводит к соответствующим тризамещенным силилированным алкенам с выходом 86–94%. Комплексы платины 59 [36] также являются эффективными катализаторами реакции гидросилилирования алкинов в схожих условиях.
Реакции, протекающие под действием видимого света, стали мощным инструментом современного органического синтеза. В этом контексте в последние годы набирает популярность фотокатализ соединениями переходных металлов, в котором металлокомплексный катализатор одновременно служит и светопоглощающим веществом, и каталитическим центром [289, 290]. Процесс образования/разрыва связи для этих индуцированных видимым светом превращений, катализируемых соединениями переходных металлов, происходит при непосредственном участии металлического центра. Превращения осуществляются в едином каталитическом цикле, где комплекс переходного металла выполняет две функции одновременно: поглощает свет и выступает каталитически активной частицей. Хотя преимущества каталитической системы последнего типа очевидны, точная настройка таких систем в данный момент затруднена по сравнению с кооперативными системами из-за ограниченного круга известных фотокаталитически активных лигандов и комплексов, а также из-за отсутствия знаний о механизме их каталитического действия.
В работе [38] впервые продемонстрировано, что комплексы платины(II) c ациклическими диаминокарбеновыми лигандами могут использоваться в качестве фотокатализаторов (рис. 10). Катализируемая фотоактивными соединениями 119 и 120 реакция гидросилилирования протекает при облучении синим светом (400–500 нм, 36 Вт) и приводит к силилированным алкенам с хорошим выходом (59–96%).
Проведенные исследования механизма показали, что облучение видимым светом необходимо на двух стадиях каталитического цикла. Во-первых, облучение светом способствует диссоциации изоцианидных лигандов от 119 и 120, делая металлоцентр способным к восстановлению силановым субстратом (схема 47). Окислительное присоединение силана приводит к продукту типа 121. Обратимая фотодиссоциация пиридиновой части C,N-бидентатного аминокарбенового лиганда приводит к изменению координационного режима на C-монодентатный, потенциально облегчая связывание субстрата в ходе катализа. За этим следует π-координация алкина, его 1,2-мигрирующее внедрение по связи Pt–H либо восстановительное отщепление соответствующих силилированных продуктов. Как в 121, так и в 122 платиновый центр имеет степень окисления +2, что согласуется с начальной потерей изоцианидных и хлоридных лигандов из исходного предкатализатора 119 и восстановлением металлического центра до платины(0). Расчетный квантовый выход для фотокаталитического процесса равен 0.2, что исключает возможность радикального цепного пути.
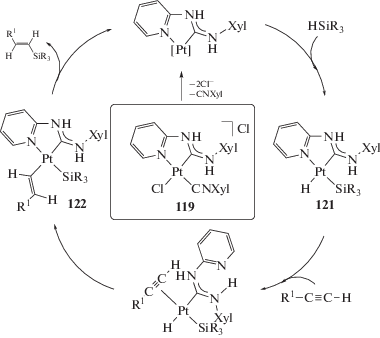
Схема 47 . Предполагаемый механизм фотокаталитического действия комплексов платины(II) c ациклическими диаминокарбеновыми лигандами в реакции гидросилилирования алкинов.
Эластичные кремнийорганические полимеры, так называемые силиконовые эластомеры, в последние десятилетия находят широчайшее применение как в промышленности, так и в высокоточных приложениях благодаря тому, что затвердевшие силиконовые покрытия обладают значительной устойчивостью к термическому, коррозионному и фрикционному воздействию. Обычное приготовление силиконового покрытия включает катализируемое металлами гидросилилирующее сшивание виниленда и полидиметилсилоксанов с водородными функциональными группами. В идеале состав покрытия остается жидким при комнатной температуре, но быстро затвердевает при температуре выше 100°C, что позволяет использовать покрытое изделие в дальнейшей обработке без охлаждения. В работе [36] комплексы платины(II) 59 предложены в качестве эффективных катализаторов вулканизации силоксановых эластомеров, сшивка протекает при температуре 85°С в присутствии 5 × 10–5–1 × × 10–3 М катализатора за 8–15 ч (схема 48). Производное платины(II) 59а (R; Ar = C6H4–4-OMe) обладает наибольшей каталитической активностью, что, вероятно, связано с присутствием электронодонорного метоксильного заместителя. Кроме того, силиконовые материалы, полученные с использованием катализаторов 59, обладают люминесцентными свойствами.
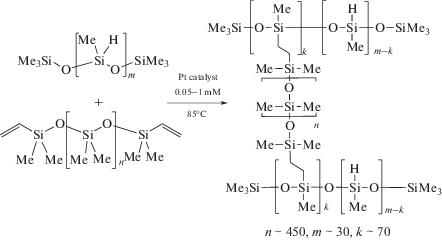
Схема 48 . Вулканизация силоксановых эластомеров с использованием комплексов платины(II).
Люминесцентные системы
Координационные соединения металлов платиновой группы с органическими лигандами проявляют яркую и эффективную флуоресценцию, фосфоресценцию, а также термически активированную замедленную флуоресценцию. Наличие этих свойств делает указанные соединения привлекательными объектами для применения в различных областях оптоэлектроники, в том числе при создании эффективных OLED (релевантные обзорные работы по этой теме см. [291–299]). Несмотря на то, что люминесцентные свойства ациклических диаминокарбеновых комплексов детально проанализированы в недавно вышедшей работе [300], мы считаем, что настоящий обзор будет неполным без текущей главы, поэтому приведем тезисно ключевые моменты.
В то время как органические люминофоры преимущественно флуоресцентные (образующиеся триплетные экситоны дезактивируются тепловыми процессами), сильное спин-орбитальное взаимодействие в комплексах переходных металлов допускает переход из синглетного в триплетное состояние с последующей фосфоресцентной релаксацией. Триплетное возбужденное состояние может эффективно генерироваться за счет индуцированного тяжелыми атомами спин-орбитального взаимодействия, поэтому при использовании металлоорганических соединений удается создать фосфоресцирующие OLED (PHOLED), которые генерируют свет как от триплетного, так и от синглетного экситонов [301–303]. Среди комплексов переходных металлов фосфоресцирующие соединения иридия [304–306] и платины [307] наиболее эффективны из-за высокой заселенности триплетных возбужденных состояний, подвергающихся радиационному распаду.
Цвет эмиссии, а также ее эффективность во многом определяются природой органического лиганда, связанного с атомом металла. Однако для некоторых типов триплетных люминофоров могут происходить вредные процессы фоторазложения, протекающие в результате диссоциации лиганда после термического заселения металл-центрированных возбужденных состояний, расположенных по энергии выше эмиссионного уровня. Проблема наиболее существенна для синих люминофоров, у которых обозначенные состояния имеют близкую энергию. Внедрение в молекулу люминофора на основе комплекса переходного металла сильных σ-донорных лигандов, в частности диаминокарбенов, приводит к увеличению расщепления энергии d-орбиталей полем лигандов и позволяет получать фосфоресцирующие люминофоры с высоким квантовым выходом.
Фотофизические свойства диаминокарбеновых комплексов определяются не только выбором подходящего металлоцентра, но и балансом между донорными свойствами циклометаллированных и вспомогательных карбеновых лигандов. Для каждой области применения светоизлучающего устройства требуются люминофоры с различным набором параметров (свойств), которые подбирают, как правило, эмпирическим способом. Описанная в работе стратегия получения ациклических диаминокарбеновых комплексов оказалась успешной в разработке металлоорганических люминофоров на основе соединений платины(II) и иридия(III) для применения в создании светоизлучающих систем, где необходима тонкая настройка фотофизических параметров; репрезентативные примеры люминесцирующих комплексов платины(II) [43, 47, 214, 308, 309] и иридия(III) [39–42, 121, 178, 199] и их основные характеристики приведены на рис. 11.
Сенсоры
Одними из наиболее чувствительных методов количественного анализа в химии являются методы, основанные на измерении люминесценции [310–314]. Высокая чувствительность люминесцентных методов анализа обусловливает их особую роль в количественном определении микропримесей в высокочистых веществах, в токсикологических исследованиях и в анализе фармацевтических препаратов. В контексте разработки хемосенсорных материалов металлоорганические фосфоресцентные эмиттеры обладают уникальными преимуществами, такими как 1) высокий квантовый выход люминесценции благодаря сильному спин-орбитальному взаимодействию вследствие “эффекта тяжелого атома”, 2) большое время жизни возбужденного состояния фосфоресценции, позволяющее отличить эмиссию хемосенсоров от фоновой флуоресценции образца и 3) большой стоксов сдвиг, необходимый для эффективной дискриминации длин волн возбуждения и излучения.
В работе [39] предложен новый класс металлоорганических хемосенсоров для количественного определения ртути на основе ациклических диаминокарбеновых комплексов иридия. Комплексы 41 обладают эффективной фосфоресценцией в растворе (Ф = 0.45), интенсивность которой линейно уменьшается при добавлении катионов ртути(II) вплоть до соотношения 41 : Hg2+ = 1 : 2, после чего дальнейшее добавление катионов ртути(II) практически не влияет на эмиссию. Предел обнаружения токсина составляет около 0.04 мг/л (2.63 × 10−7 M), присутствие катионов других тяжелых металлов (меди, цинка, свинца, кадмия, серебра и других) не сказывается на обнаружении катионов ртути. Исследование механизма показало, что тушение фосфоресценции при добавлении катиона ртути(II) происходит из-за образования биядерных комплексов иридия(III) 129 c мостиковым дицианортутным фрагментом (схема 49, рис. 12).
Схема 49 . Тушение фосфоресценции при добавлении катиона ртути(II), происходящее из-за образования биядерных комплексов иридия(III) 41 c мостиковым дицианортутным фрагментом.
Рис. 12.
Изменение интенсивности люминесценции при добавлении соединений ртути(II) к раствору диаминокарбенового комплекса иридия(III) 41. Воспроизведено с разрешения из работы [39]. Авторское право (2020) Американское химическое общество.
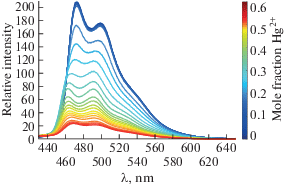
Примерами фосфоресцентных pH-сенсоров являются диаминокарбеновые комплексы “чугаевского типа”. Комплекс платины(II) 130, содержащий монодепротонированный C,C-хелатный бис-диаминокарбеновый лиганд, проявляет слабоинтенсивную фосфоресценцию в растворе (MeCN, λmax = 490 нм, Φ < 5 × 10−4) [43]. При увеличении кислотности среды происходит протонирование диаминокарбенового фрагмента (схема 50), приводящее к увеличению донорных свойств последнего, что сопровождается изменением его фотофизических свойств, а именно увеличением интенсивности эмиссии в 12 раз с одновременным коротковолновым сдвигом полосы фосфоресценции (Δλmax = 48 нм). Изменение фотофизических свойств является обратимым, и добавление основания к 131 приводит к регенерации соединения 130. Зависимая от pH фосфоресценция характерна также для других комплексов “чугаевского типа”, а именно комплексов платины(II) 125 [43] и иридия(III) 127 [199].
Схема 50 . Пример фосфоресцентных pH-сенсоров на основе диаминокарбеновых комплексов “чугаевского типа”.
Противоопухолевые препараты
Разработка препаратов на основе комплексов переходных металлов для клинической терапии рака началась с открытия американским химиком Розенбергом (Barnett Rosenberg) в 1969 г. цитотоксического действия соединений платины [315], а имеено соли Пейроне – цис-диамминдихлороплатины(II). Препараты на основе соединений платиновых металлов считаются одними из наиболее эффективных из используемых для терапии онкологических заболеваний, однако лечение известными препаратами платины всегда сопровождается (помимо возникновения внутренней или приобретенной резистентности) тяжелыми побочными эффектами вследствие неселективности действия и высокой общей токсичности [316, 317]. Поэтому перспективна разработка новых стратегий для улучшения направленности действия на опухоль (и, следовательно, уменьшения побочных эффектов) препаратов платины, а также их аналогов на основе других переходных металлов.
Устойчивость в физиологических условиях – критически важный параметр для создаваемого препарата. Особенно важна стабильность в физиологических условиях для металлоорганических препаратов, так как они могут быть склонны к реакциям лигандного обмена с биомолекулами. В этом отношении комплексы с диаминокарбеновыми лигандами перспективны для создания металлорганических противораковых препаратов ввиду прочной связи металл–карбен [318, 319]. Хотя NHC комплексы активно исследовались в течение приблизительно двух десятилетий как потенциальные антибактериальные и противоопухолевые препараты (релевантные обзоры [20–22, 24, 320–322]), ADC комплексы изучены в существенно меньшей степени. В литературе известно всего несколько примеров потенциальных терапевтических агентов на основе ADC комплексов платины(II) [36, 45–47], палладия(II) [46] и золота(I/III) [323–325].
В работе [45] изучена противоопухолевая активность ADC комплекса платины(II) “чугаевского” типа. Соединение 133 (рис. 13а) обладает схожей цитотоксической активностью с цисплатином по отношению к аденокарциномным клеткам протоков молочной железы человека (MCF7, IC50 = 4.5 мкМ; IC50 = 4.9 мкМ для цисплатина), однако его активность в 5 раз ниже, чем цисплатина, в случае линии клеток с фенотипом множественной лекарственной устойчивости (MCF7R, IC50 = 14.6 мкМ для 133; IC50 = 3.1 мкМ для цисплатина). На основе экспериментов по взаимодействию комплекса 133 с гуанидином и ДНК авторы предполагают, что первоначальное связывание с биомолекулами происходит за счет образования водородных связей между биомолекулой и диаминокарбеновыми (NH) атомами водорода, после чего реализуется замена лабильных хлоридных лигандов на тиольные фрагменты белков.
Рис. 13.
ADC комплексы платины(II), проявляющие цитотоксическую активность; нековалентное взаимодействие ADC комплексов с ДНК (б).
Противоопухолевый потенциал комплексов палладия(II) 76 и платины(II) 77 изучен на трех линиях раковых клеток человека (HT-29, MDA-MB-231, MCF-7) в работе [46]. Все протестированные соединения оказались цитотоксичными по отношению к опухолевым клеткам, однако ксилилзамещенные комплексы 76a (R = Xyl, R' = = H) и 77a (R = Xyl, R' = H) проявили наиболее высокую цитотоксическую активность (IC50 = = 10 мкМ для клеточной линии MCF-7) по отношению ко всем изученным клеточным линиям.
С помощью спектроскопических и гидродинамических методов установлено, что комплексы 76а и 77а взаимодействуют с ДНК преимущественно с образованием монофункциональных аддуктов в основной бороздке макромолекулы. Взаимодействие происходит посредством комбинации нековалентного и ковалентного связывания. Нековалентное взаимодействие ADC комплексов с ДНК реализуется через нековалентное связывание с фосфатными группами, известное как “фосфатный зажим” (рис. 13б) [326] и ранее описанное для плоскоквадратных комплексов тетраамминплатины(II) [327]. Один фрагмент O=P фосфатной группы ДНК действует как акцептор двух водородных связей, образованных с участием атомов водорода (по одному от каждого из двух цис-ориентированных диаминокарбеновых лигандов) [328]. Соединения 76а и 77а также способны образовывать межспиральные перекрестные сшивки. Ковалентное связывание комплексов 76а и 77а с ДНК происходит за счет нуклеофильного замещения азотистыми основаниями ДНК лабильных групп NH2, ослабленных сильным трансвлиянием ADC лигандов. Как подтверждают результаты исследования протонирования ДНК, координация металлоцентра происходит по атому N7 гуанина. Соединение 76а взаимодействует с ДНК намного быстрее, чем 77а, что можно объяснить более быстрым обменом лигандами с соединением 76а.
Антипролиферативное действие C,N-циклометаллированных комплексов платины(II) 33 и 134 с ациклическим диаминокарбеновым лигандом исследовано в работе [47]. Комплексы проявляют сопоставимую с цисплатином цитотоксическую активность на клеточных линиях карциномы легкого (A549) и шейки матки (HeLa). Соединения с бензильным заместителем 33a (рис. 11) и 134a (рис. 13) демонстрируют лучшую цитотоксическую активность по сравнению с соединениями 33b (cхема 10, R = nPr) и 134b, содержащими пропильный заместитель. Интересно отметить, что варьирование циклометаллированного лиганда не оказывает влияния на значение цитотоксической активности. Лучшие показатели наблюдаются для соединения 134b (IC50 = 4.47 (A549), 4.44 мкМ (HeLa)), которое оказалось более цитотоксично, чем цисплатин (IC50 = 6.45 (A549), 13.60 мкМ (HeLa)). Электрофоретические исследования указывают на то, что комплексы 33a, 33b и 134a могут изменять третичную структуру ДНК, поэтому ДНК может быть возможной целью этих комплексов. Напротив, добавление 134b к ДНК не вызывает конформационные изменения ДНК, это позволяет предположить, что цитотоксический эффект данного комплекса не основан на связывании с ДНК.
Цитотоксические свойства диаминокарбеновых комплексов платины(II) 59 изучены in vitro в работе [36]. Соединения обладают цитотоксичностью по отношению к клеточным линиям тератокарцинома яичника (CH1/PA-1), карциномы толстой кишки (SW480) и аденокарциномы альвеолярных базальных эпителиальных клеток человека (A549), однако их эффективность в несколько раз ниже, чем цисплатина.
ЗАКЛЮЧЕНИЕ
В представленном обзоре мы постарались максимально полно систематизировать опубликованные литературные данные, касающиеся синтеза и изучения свойств комплексов металлов платиновой группы с ациклическими диаминокарбеновыми лигандами. Уникальные σ-донорные свойства аминокарбенов (ADC и NHC) приводят к прочной связи металл–лиганд, обусловливая высокую устойчивость их комплексов. В то же время отсутствие в ADC ковалентного фрагмента, связывающего оба атома азота, как в случае NHC, допускает вращение заместителей вокруг связей Cкарбен–N, обусловливая уникальные стерические свойства этих лигандов.
Свободные ациклические диаминокарбены (ADC) могут быть синтезированы в лаборатории и использованы для прямой координации к металлоцентрам в той же степени, что и их гетероциклические аналоги (NHC). В то же время наибольшее распространение получил подход, основанный на металлопромотируемом нуклеофильном присоединении к координированным изоцианидам и приводящий к получению диаминокарбеновых комплексов металлов без выделения свободного карбена. Атом-экономичное присоединение N-нуклеофилов к координированным изоцианидам позволяет получать широкую гамму диаминокарбеновых комплексов, включая монодентатные, хелатные и пинцерные производные, в зависимости от количества донорных центров и их взаимного расположения в структуре нуклеофила.
Наибольшее применение данный подход нашел в синтезе комплексов поздних переходных металлов, в частности металлов платиновой группы, во многих случаях это обусловлено последующим применением указанных соединений. Комплексы палладия с диаминокарбеновыми лигандами зарекомендовали себя как высокоактивные катализаторы в реакциях кросс-сочетания и реакции Хека; при этом функционализация диаминокарбенового лиганда позволила сделать такие соединения водорастворимыми с сохранением их высокой каталитической активности. Значительная кинетическая устойчивость комплексов платины с диаминокарбеновыми лигандами позволила их использовать в качестве удобной модели для изучения каталитических процессов, в том числе металлокатализируемых органических процессов с участием изоцианидов и азот-центрированных нуклеофилов, многие из которых протекают через образование ациклического диаминокарбенового фрагмента. Уникальная способность платины к взаимодействию с π-системами непредельных соединений, в частности алкенов и алкинов, делает Pt-ADC комплексы перспективными катализаторами гидросилилирования кратных связей, в том числе в условиях фотокатализа видимым светом.
Несмотря на многообразие комплексов металлов платиновой группы с ациклическими диаминокарбеновыми лигандами, их потенциальная биологическая активность изучена в меньшей степени. Тем не менее на известных примерах установлено, что комплексы палладия(II) и платины(II) активны как противоопухолевые препараты; ациклический диаминокарбеновый фрагмент выступает центром связывания металлоорганических соединений с биомолекулами. В этом направлении целесообразно провести более систематические исследования с целью установления корреляции структура–свойство и создания новых фармацевтических препаратов.
Химия ациклических диаминокарбеновых комплексов металлов является динамически развивающимся направлением, поэтому нет сомнений, что в ближайшие годы появятся еще более впечатляющие примеры использования этих соединений как в области фундаментальных исследований, так и в решении прикладных задач. В связи с этим авторы надеются, что представленный обзор окажется полезным исследователям, работающим в областях металлоорганической химии, металлокомплексного катализа и бионеорганической химии.
Список литературы
Bourissou D., Guerret O., Gabbaï F., Bertrand G. // Chem. Rev. 2000. V. 100. P. 39. https://doi.org/10.1021/cr940472u
Huynh H.V. // Chem. Rev. 2018. V. 118. P. 9457. https://doi.org/10.1021/acs.chemrev.8b00067
Kuwata S., Hahn F.E. // Chem. Rev. 2018. V. 118. P. 9642. https://doi.org/10.1021/acs.chemrev.8b00176
Nesterov V., Reiter D., Bag P. et al. // Chem. Rev. 2018. V. 118. P. 9678. https://doi.org/10.1021/acs.chemrev.8b00079
Sipos G., Dorta R. // Coord. Chem. Rev. 2018. V. 375. P. 13. https://doi.org/10.1016/j.ccr.2017.10.019
Trose M., Nahra F., Cazin C.S.J. // Coord. Chem. Rev. 2018. V. 355. P. 380. https://doi.org/https://doi.org/10.1016/j.ccr.2017.10.013
Danopoulos A.A., Simler T., Braunstein P. // Chem. Rev. 2019. V. 119. P. 3730. https://doi.org/10.1021/acs.chemrev.8b00505
Zhang L., Hou Z. // Chem. Sci. 2013. V. 4. P. 3395. https://doi.org/10.1039/C3SC51070K
Lazreg F., Nahra F., Cazin C.S.J. // Coord. Chem. Rev. 2015. V. 293–294. P. 48. https://doi.org/10.1016/j.ccr.2014.12.019
Gafurov Z.N., Kantyukov A.O., Kagilev A.A. et al. // Russ. Chem. Bull. 2017. V. 66. P. 1529. https://doi.org/10.1007/s11172-017-1920-7
Gardiner M.G., Ho C.C. // Coord. Chem. Rev. 2018. V. 375. P. 373. https://doi.org/10.1016/j.ccr.2018.02.003
Iglesias M., Oro L.A. // Chem. Soc. Rev. 2018. V. 47. P. 2772. https://doi.org/10.1039/C7CS00743D
Peris E. // Chem. Rev. 2018. V. 118. P. 9988. https://doi.org/10.1021/acs.chemrev.6b00695
Roland S., Suarez J.M., Sollogoub M. // Chem. Eur. J. 2018. V. 24. P. 12464. https://doi.org/10.1002/chem.201801278
Wang W., Cui L., Sun P. et al. // Chem. Rev. 2018. V. 118. P. 9843. https://doi.org/10.1021/acs.chemrev.8b00057
Mercs L., Albrecht M. // Chem. Soc. Rev. 2010. V. 39. P. 1903. https://doi.org/10.1039/B902238B
Visbal R., Gimeno M.C. // Chem. Soc. Rev. 2014. V. 43. P. 3551. https://doi.org/10.1039/C3CS60466G
Elie M., Renaud J.L., Gaillard S. // Polyhedron. 2018. V. 140. P. 158. https://doi.org/10.1016/j.poly.2017.11.045
Bonfiglio A., Mauro M. // Eur. J. Inorg. Chem. 2020. V. 2020. P. 3427. https://doi.org/10.1002/ejic.202000509
Gautier A., Cisnetti F. // Metallomics. 2012. V. 4. P. 23. https://doi.org/10.1039/C1MT00123J
Ott I. Metal N-heterocyclic carbene complexes in medicinal chemistry // Medicinal Chemistry / Eds. Sadler P.J., VanEldik R. 2020. P. 121.
Patil A., Hoagland A.P, Patil S.A, Bugarin A. // Future Med. Chem. 2020. V. 12. P. 2239. https://doi.org/10.4155/fmc-2020-0175
Smith C.A., Narouz M.R., Lummis P.A. et al. // Chem. Rev. 2019. V. 119. P. 4986. https://doi.org/10.1021/acs.chemrev.8b00514
Liu W., Gust R. // Coord. Chem. Rev. 2016. V. 329. P. 191. https://doi.org/10.1016/j.ccr.2016.09.004
Ibáñez S., Poyatos M., Peris E. // Acc. Chem. Res. 2020. V. 53. P. 1401. https://doi.org/10.1021/acs.accounts.0c00312
Boyarskiy V.P., Luzyanin K.V., Kukushkin V.Y. // Coord. Chem. Rev. 2012. V. 256. P. 2029. https://doi.org/10.1016/j.ccr.2012.04.022
Slaughter L.M. // ACS Catal. 2012. V. 2. P. 1802. https://doi.org/10.1021/cs300300y
Kinzhalov M.A., Luzyanin K.V. // Coord. Chem. Rev. 2019. V. 399. P. 213014. https://doi.org/10.1016/j.ccr.2019.213014
Timofeeva S.A., Kinzhalov M.A., Valishina E.A. et al. // J. Catal. 2015. V. 329. P. 449. https://doi.org/10.1016/j.jcat.2015.06.001
Mikhaylov V., Sorokoumov V., Liakhov D. et al. // Catalysts. 2018. V. 8. P. 141. https://doi.org/10.3390/catal8040141
Fang Y., Wang S.-Y., Ji S.-J. // Tetrahedron. 2015. V. 71. P. 9679. https://doi.org/https://doi.org/10.1016/j.tet.2015.10.033
Owusu M.O., Handa S., Slaughter L.M. // Appl. Organomet. Chem. 2012. V. 26. P. 712. https://doi.org/10.1002/aoc.2915
Škoch K., Schulz J., Císařová I., Štěpnička P. // Organometallics. 2019. V. 38. P. 3060. https://doi.org/10.1021/acs.organomet.9b00398
Rocha B.G.M., Valishina E.A., Chay R.S. et al. // J. Catal. 2014. V. 309. P. 79. https://doi.org/10.1016/j.jcat.2013.09.003
Chay R.S., Rocha B.G.M., Pombeiro A.J.L. et al. // ACS Omega. 2018. V. 3. P. 863. https://doi.org/10.1021/acsomega.7b01688
Afanasenko A.M., Chulkova T.G., Boyarskaya I.A. et al. // J. Organomet. Chem. 2020. V. 923. P. 121435. https://doi.org/https://doi.org/10.1016/j.jorganchem.2020.121435
Kinzhalov M.A., Luzyanin K.V., Boyarskiy V.P. et al. // Organometallics. 2013. V. 32. P. 5212. https://doi.org/10.1021/om4007592
Gee J.C., Fuller B.A., Lockett H.-M. et al. // Chem. Commun. 2018. V. 54. P. 9450. https://doi.org/10.1039/C8CC04287J
Eremina A.A., Kinzhalov M.A., Katlenok E.A. et al. // Inorg. Chem. 2020. V. 59. P. 2209. https://doi.org/10.1021/acs.inorgchem.9b02833
Na H., Lai P.N., Cañada L.M., Teets T.S. // Organometallics. 2018. V. 37. P. 3269. https://doi.org/10.1021/acs.organomet.8b00446
Na H., Teets T.S. // J. Am. Chem. Soc. 2018. V. 140. P. 6353. https://doi.org/10.1021/jacs.8b02416
Na H., Cañada Louise M., Wen Z. et al. // Chem. Sci. 2019. V. 10. P. 6254. https://doi.org/10.1039/C9SC01386E
Lai S.-W., Chan M.C.W., Wang Y. et al. // J. Organomet. Chem. 2001. V. 617–618. P. 133. https://doi.org/10.1016/S0022-328X(00)00723-3
Ng C.-O., Cheng S.-C., Chu W.-K. et al. // Inorg. Chem. 2016. V. 55. P. 7969. https://doi.org/10.1021/acs.inorgchem.6b01017
Alves G., Morel L., El-Ghozzi M. et al. // Chem. Commun. 2011. V. 47. P. 7830. https://doi.org/10.1039/C1CC12228B
Serebryanskaya T.V., Kinzhalov M.A., Bakulev V. et al. // New J. Chem. 2020. V. 44. P. 5762. https://doi.org/10.1039/D0NJ00060D
Moreno M.T., Lalinde Peña E., Martínez-Junquera M. et al. // Dalton Trans. 2021. V. 50. P. 4539. https://doi.org/10.1039/D1DT00480H
McNaught A.D., Wilkinson A., IUPAC. Compendium of Chemical Terminology. Oxford: Blackwell Scientific Publications, 1997.
Нефедов О.М., Иоффе А.И., Мечников Л.Г. Химия карбенов. М.: Химия, 1990.
Hoffmann R., Zeiss G.D., Vandine G.W. // J. Am. Chem. Soc. 1968. V. 90. P. 1485. https://doi.org/10.1021/ja01008a017
Gleiter R., Hoffmann R. // J. Am. Chem. Soc. 1968. V. 90. P. 5457. https://doi.org/10.1021/ja01022a023
Moss R.A., Mallon C.B. // J. Am. Chem. Soc. 1975. V. 97. P. 344. https://doi.org/10.1021/ja00835a020
Koda S. // Chem. Phys. Lett. 1978. V. 55. P. 353. https://doi.org/10.1016/0009-2614(78)87037-7
Baird N.C., Taylor K.F. // J. Am. Chem. Soc. 1978. V. 100. P. 1333. https://doi.org/10.1021/ja00473a001
Schoeller W.W. // Chem. Commun. 1980. P. 124. https://doi.org/10.1039/c39800000124
Pauling L. // Chem. Commun. 1980. P. 688. https://doi.org/10.1039/c39800000688
Irikura K.K., Goddard W.A., Beauchamp J.L. // J. Am. Chem. Soc. 1992. V. 114. P. 48. https://doi.org/10.1021/ja00027a006
Alder R.W., Blake M.E. // Chem. Commun. 1997. V. P. 1513. https://doi.org/10.1039/a703610h
Moss R.A., Wlostowski M., Shen S. et al. // J. Am. Chem. Soc. 1988. V. 110. P. 4443. https://doi.org/10.1021/ja00221a071
Alder R.W., Allen P.R., Murray M., Orpen A.G. // Angew. Chem., Int. Ed. 1996. V. 35. P. 1121. https://doi.org/10.1002/anie.199611211
Wallbaum L., Weismann D., Löber D. et al. // Chem. Eur. J. 2019. V. 25. P. 1488. https://doi.org/10.1002/chem.201805307
Alder R.W., Butts C.P., Orpen A.G. // J. Am. Chem. Soc. 1998. V. 120. P. 11526. https://doi.org/10.1021/ja9819312
Frémont P., Marion N., Nolan S.P. // Coord. Chem. Rev. 2009. V. 253. P. 862. https://doi.org/10.1016/j.ccr.2008.05.018
Jacobsen H., Correa A., Poater A. et al. // Coord. Chem. Rev. 2009. V. 253. P. 687. https://doi.org/10.1016/j.ccr.2008.06.006
Crabtree R.H. // J. Organomet. Chem. 2005. V. 690. P. 5451. https://doi.org/10.1016/j.jorganchem.2005.07.099
Antonova N.S., Carbo J.J., Poblet J.M. // Organometallics. 2009. V. 28. P. 4283. https://doi.org/10.1021/om900180m
Lee M.T., Hu C.H. // Organometallics. 2004. V. 23. P. 976. https://doi.org/10.1021/om0341451
Herrmann W.A., Ofele K., von Preysing D., Herdtweck E. // J. Organomet. Chem. 2003. V. 684. P. 235. https://doi.org/10.1016/s0022-328x(03)00754-x
Fischer H., Schleu J., Troll C. // J. Organomet. Chem. 1994. V. 464. P. 83. https://doi.org/10.1016/0022-328X(94)87013-6
Ruiz J., Perandones B.F. // Organometallics. 2009. V. 28. P. 830. https://doi.org/10.1021/om800888r
Ruiz J., Sol D., Mateo M.A., Vivanco M. // Dalton Trans. 2018. V. 47. P. 6279. https://doi.org/10.1039/C8DT01200H
Ruiz J., García L., Mejuto C. et al. // Organometallics. 2012. V. 31. P. 6420. https://doi.org/10.1021/om3006454
Balch A.L., Miller J. // J. Am. Chem. Soc. 1972. V. 94. P. 417. https://doi.org/10.1021/ja00757a019
Costanzo L.L., Giuffrida S., De Guidi G. et al. // J. Organomet. Chem. 1985. V. 289. P. 81. https://doi.org/10.1016/0022-328X(85)88029-3
Angelici R.J., Christian P.A., Dombek B.D., Pfeffer G.A. // J. Organomet. Chem. 1974. V. 67. P. 287. https://doi.org/10.1016/S0022-328X(00)82356-6
Yu I., Wallis C.J., Patrick B.O. et al. // Organometallics. 2010. V. 29. P. 6065. https://doi.org/10.1021/om100841j
Ruiz J., Garcia L., Vivanco M. et al. // Dalton Trans. 2017. V. 46. P. 10387. https://doi.org/10.1039/C7DT02049J
Johnson B.V., Shade J.E. // J. Organomet. Chem. 1979. V. 179. P. 357. https://doi.org/10.1016/S0022-328X(00)91751-0
Paulus B.C., Nielsen K.C., Tichnell C.R. et al. // J. Am. Chem. Soc. 2021. V. 143. P. 8086. https://doi.org/10.1021/jacs.1c02451
Werner H., Heiser B., Otto H. // Chem. Ber. 1985. V. 118. P. 3932. https://doi.org/doi:10.1002/cber.19851181006
Hirsch-Weil D., Snead D.R., Inagaki S. et al. // Chem. Commun. 2009. P. 2475. https://doi.org/10.1039/b821169h
Schöllkopf U., Gerhart F. // Angew. Chem. 1967. V. 6. P. 990. https://doi.org/10.1002/ange.19670792232
Tolman C.A. // Chem. Rev. 1977. V. 77. P. 313. https://doi.org/10.1021/cr60307a002
Wolf S., Plenio H. // J. Organomet. Chem. 2009. V. 694. P. 1487. https://doi.org/10.1016/j.jorganchem.2008.12.047
Kelly R.A., III, Clavier H., Giudice S. et al. // Organometallics 2008. V. 27. P. 202. https://doi.org/10.1021/om701001g
Martin D., Canac Y., Lavallo V., Bertrand G. // J. Am. Chem. Soc. 2014. V. 136. P. 5023. https://doi.org/10.1021/ja412981x
Denk K., Sirsch P., Herrmann W.A. // J. Organomet. Chem. 2002. V. 649. P. 219. https://doi.org/10.1016/S0022-328X(02)01133-6
Collins M.S., Rosen E.L., Lynch V.M., Bielawski C.W. // Organometallics. 2010. V. 29. P. 3047. https://doi.org/10.1021/om1004226
Meier M., Tan T.T.Y., Hahn F.E., Huynh H.V. // Organometallics. 2017. V. 36. P. 275. https://doi.org/10.1021/acs.organomet.6b00736
Huynh H.V., Han Y., Jothibasu R., Yang J.A. // Organometallics. 2009. V. 28. P. 5395. https://doi.org/10.1021/om900667d
Singh C., Kumar A., Huynh H.V. // Inorg. Chem. 2020. V. 59. P. 8451. https://doi.org/10.1021/acs.inorgchem.0c00886
Jacobsen H., Correa A., Costabile C., Cavallo L. // J. Organomet. Chem. 2006. V. 691. P. 4350. https://doi.org/10.1016/j.jorganchem.2006.01.026
Liske A., Verlinden K., Buhl H. et al. // Organometallics. 2013. V. 32. P. 5269. https://doi.org/10.1021/om400858y
Schulz T., Weismann D., Wallbaum L. et al. // Chem. Eur. J. 2015. V. 21. P. 14107. https://doi.org/10.1002/chem.201502315
Rosen E.L., Sanderson M.D., Saravanakumar S., Bielawski C.W. // Organometallics. 2007. V. 26. P. 5774. https://doi.org/10.1021/om7007925
Jazzar R., Soleilhavoup M., Bertrand G. // Chem. Rev. 2020. V. 120. P. 4141. https://doi.org/10.1021/acs.chemrev.0c00043
Siemeling U., Färber C., Bruhn C. et al. // Chem. Sci. 2010. V. 1. P. 697. https://doi.org/10.1039/C0SC00451K
Alder R.W., Blake M.E., Oliva J.M. // J. Phys. Chem. A. 1999. V. 103. P. 11200. https://doi.org/10.1021/jp9934228
Pazio A., Woźniak K., Grela K., Trzaskowski B. // Organometallics. 2015. V. 34. P. 563. https://doi.org/10.1021/om5006462
Anisimova T.B., Guedes da Silva M.F.C., Kukushkin V.Y. et al. // Dalton Trans. 2014. V. 43. P. 15861. https://doi.org/10.1039/c4dt01917b
Mikhaylov V.N., Sorokoumov V.N., Korvinson K.A. et al. // Organometallics. 2016. V. 35. P. 1684. https://doi.org/10.1021/acs.organomet.6b00144
Zhang L., Yu W., Liu C. et al. // Organometallics. 2015. V. 34. P. 5697. https://doi.org/10.1021/acs.organomet.5b00746
Kinzhalov M.A., Timofeeva S.A., Luzyanin K.V. et al. // Organometallics. 2016. V. 35. P. 218. https://doi.org/10.1021/acs.organomet.5b00936
Kinzhalov M.A., Novikov A.S., Chernyshev A.N., Suslonov V.V. // Z. Kristallogr. – Cryst. Mater. 2017. V. 232. P. 299. https://doi.org/10.1515/zkri-2016-2018
Ivanov D.M., Kinzhalov M.A., Novikov A.S. et al. // Cryst. Growth Des. 2017. V. 17. P. 1353. https://doi.org/10.1021/acs.cgd.6b01754
Mikherdov A.S., Novikov A.S., Kinzhalov M.A. et al. // Inorg. Chem. 2018. V. 57. P. 3420. https://doi.org/10.1021/acs.inorgchem.8b00190
Ramiro Z., Bartolomé C., Espinet P. // Eur. J. Inorg. Chem. 2014. V. 2014. P. 5499. https://doi.org/10.1002/ejic.201402744
Wang Y.-M., Kuzniewski C.N., Rauniyar V. et al. // J. Am. Chem. Soc. 2011. V. 133. P. 12972. https://doi.org/10.1021/ja205068j
Bartolome C., Carrasco-Rando M., Coco S. et al. // Inorg. Chem. 2008. V. 47. P. 1616. https://doi.org/10.1021/ic702201e
Bartolomé C., Carrasco-Rando M., Coco S. et al. // Organometallics. 2006. V. 25. P. 2700. https://doi.org/10.1021/om0601753
Bartolomé C., Ramiro Z., Pérez-Galán P. et al. // Inorg. Chem. 2008. V. 47. P. 11391. https://doi.org/10.1021/ic801446v
Bartolomé C., García-Cuadrado D., Ramiro Z., Espinet P. // Inorg. Chem. 2010. V. 49. P. 9758. https://doi.org/10.1021/ic101059c
Shi Y.-C., Wang S., Xie S. // J. Coord. Chem. 2015. V. 68. P. 3852. https://doi.org/10.1080/00958972.2015.1079312
Rosen E.L., Sung D.H., Chen Z. et al. // Organometallics. 2010. V. 29. P. 250. https://doi.org/10.1021/om9008718
Mikherdov A.S., Kinzhalov M.A., Novikov A.S. et al. // Inorg. Chem. 2018. V. 57. P. 6722. https://doi.org/10.1021/acs.inorgchem.8b01027
Mikhaylov V.N., Sorokoumov V.N., Novikov A.S. et al. // J. Organomet. Chem. 2020. V. 912. P. 121174. https://doi.org/10.1016/j.jorganchem.2020.121174
Popov R.A., Mikherdov A.S., Novikov A.S. et al. // New J. Chem. 2021. V. 4. P. 1785. https://doi.org/10.1039/D0NJ05386D
Eberhard M.R., van Vliet B., Durán Páchon L. et al. // Eur. J. Inorg. Chem. 2009. V. 2009. P. 1313. https://doi.org/10.1002/ejic.200801067
Marchenko A., Koidan G., Hurieva A. et al. // Eur. J. Inorg. Chem. 2018. V. 2018. P. 652. https://doi.org/10.1002/ejic.201701342
Marchenko A., Koidan G., Hurieva A. et al. // Dalton Trans. 2016. V. 45. P. 1967. https://doi.org/10.1039/C5DT02250A
Han J., Tang K.-M., Cheng S.-C. et al. // Inorg. Chem. Front. 2020. V. 7. P. 786. https://doi.org/10.1039/C9QI01278H
Handa S., Slaughter L.M. // Angew. Chem. Int. Ed. 2012. V. 51. P. 2912. https://doi.org/10.1002/anie.201107789
Scattolin T., Nolan S.P. // Trends in Chemistry. 2020. V. 2. P. 721. https://doi.org/10.1016/j.trechm.2020.06.001
Benhamou L., Chardon E., Lavigne G. et al. // Chem. Rev. 2011. V. 111. P. 2705. https://doi.org/10.1021/cr100328e
Vignolle J., Catton X., Bourissou D. // Chem. Rev. 2009. V. 109. P. 3333. https://doi.org/10.1021/cr800549j
Herrmann W.A., Schutz J., Frey G.D., Herdtweck E. // Organometallics. 2006. V. 25. P. 2437. https://doi.org/10.1021/om0600801
Alder R.W., Blake M.E., Bufali S. et al. // J. Chem. Soc., Perkin Trans. 1. 2001. P. 1586. https://doi.org/10.1039/b104110j
Alder R.W., Chaker L., Paolini F.P.V. // Chem. Commun. 2004. P. 2172. https://doi.org/10.1039/B409112D
Schoultz X., Gerber T.I.A., Hosten E.C. // Inorg. Chem. Commun. 2016. V. 68. P. 13. https://doi.org/10.1016/j.inoche.2016.03.020
Bulak E., Dogan I., Varnali T. et al. // Eur. J. Inorg. Chem. 2021. V. 2021. P. 2425. https://doi.org/10.1002/ejic.202100277
Kremzow D., Seidel G., Lehmann C.W., Furstner A. // Chem. Eur. J. 2005. V. 11. P. 1833.
Snead D.R., Inagaki S., Abboud K.A., Hong S. // Organometallics. 2010. V. 29. P. 1729. https://doi.org/10.1021/om901112n
Snead D.R., Chiviriga I., Abboud K.A., Hong S. // Org. Lett. 2009. V. 11. P. 3274. https://doi.org/10.1021/ol9013156
Michelin R.A., Pombeiro A.J.L., Guedes da Silva M.F.C. // Coord. Chem. Rev. 2001. V. 218. P. 75. https://doi.org/10.1016/S0010-8545(01)00358-7
Boyarskiy V.P., Bokach N.A., Luzyanin K.V., Kukushkin V.Y. // Chem. Rev. 2015. V. 115. P. 2698. https://doi.org/10.1021/cr500380d
Chugaev L., Skanavy-Grigorjeva M.J. // Russ. Chem. Soc. 1915. V. 47. P. 776.
Luzyanin K.V., Pombeiro A.J.L. Carbene complexes derived from metal-bound isocyanides // Isocyanide chem. / Ed. Nenajdenko V. Wiley-VCH, 2012, P. 531.
Kinzhalov M.A., Boyarskii V.P. // Russ. J. Gen. Chem. 2015. V. 85. P. 2313. https://doi.org/10.1134/S1070363215100175
Fehlhammer W.P., Bartel K., Metzner R., Beck W. // Z. Anorg. Allg. Chem. 2008. V. 634. P. 1002. https://doi.org/10.1002/zaac.200700540
Crociani B., Boschi T., Belluco U. // Inorg. Chem. 1970. V. 9. P. 2021. https://doi.org/10.1021/ic50091a013
Arias J., Bardají M., Espinet P. // Inorg. Chem. 2008. V. 47. P. 3559. https://doi.org/10.1021/ic701831v
Michelin R.A., Zanotto L., Braga D. et al. // Inorg. Chem. 1988. V. 27. P. 85. https://doi.org/10.1021/ic00274a019
Canovese L., Visentin F., Uguagliati P., Crociani B. // J. Organomet. Chem. 1997. V. 543. P. 145. https://doi.org/10.1016/s0022-328x(97)00095-8
Badley E.M., Chatt J., Richards R.L., Sim G.A. // Chem. Commun. 1969. P. 1322. https://doi.org/10.1039/c29690001322
Knorn M., Lutsker E., Reiser O. // Organometallics. 2015. V. 34. P. 4515. https://doi.org/10.1021/acs.organomet.5b00516
Rassadin V.A., Yakimanskiy A.A., Eliseenkov E.V., Boyarskiy V.P. // Inorg. Chem. Commun. 2015. V. 61. P. 21. https://doi.org/10.1016/j.inoche.2015.08.008
Hashmi A.S.K., Lothschutz C., Boohling C., Rominger F. // Organometallics. 2011. V. 30. P. 2411. https://doi.org/10.1021/om200141s
Singh C., Prakasham A.P., Gangwar M.K. et al. // ACS Omega. 2018. V. 3. P. 1740. https://doi.org/10.1021/acsomega.7b01974
Hashmi A.S.K., Lothschütz C., Böhling C. et al. // Adv. Synth. Catal. 2010. V. 352. P. 3001. https://doi.org/10.1002/adsc.201000472
Riedel D., Wurm T., Graf K. et al. // Adv. Synth. Catal. 2015. V. 357. P. 1515. https://doi.org/10.1002/adsc.201401131
Lothschütz C., Wurm T., Zeiler A. et al. // Chem. Asian J. 2016. V. 11. P. 342. https://doi.org/10.1002/asia.201500353
Kinzhalov M.A., Kashina M.V., Mikherdov A.S. et al. // Angew. Chem. Int. Ed. 2018. V. 57. P. 12785. https://doi.org/10.1002/anie.201807642
Baikov S.V., Trukhanova Y.A., Tarasenko M.V., Kinzhalov M.A. // Russ. J. Gen. Chem. 2020. V. 90. P. 1892. https://doi.org/10.1134/S1070363220100126
Kinzhalov M.A., Starova G.L., Boyarskiy V.P. // Inorg. Chim. Acta. 2017. V. 455. P. 607. https://doi.org/10.1016/j.ica.2016.05.014
Zhang S.-W., Motoori F., Takahashi S. // J. Organomet. Chem. 1999. V. 574. P. 163. https://doi.org/https://doi.org/10.1016/S0022-328X(98)00937-1
Zhang S.-W., Ishii R., Motoori F. et al. // Inorg. Chim. Acta. 1997. V. 265. P. 75. https://doi.org/10.1016/S0020-1693(97)05673-9
Zhang S.-W., Kaharu T., Pirio N. et al. // J. Organomet. Chem. 1995. V. 489. P. C62. https://doi.org/10.1016/0022-328X(94)05287-L
Bertani R., Mozzon M., Benetollo F. et al. // Dalton Trans. 1990. V. 1990. P. 1197. https://doi.org/10.1039/dt9900001197
Fehlhammer W.P., Metzner R., Luger P., Dauter Z. // Chem. Ber. 1995. V. 128. P. 1061. https://doi.org/10.1002/cber.19951281102
Zhang S.-W., Takahashi S. // Organometallics. 1998. V. 17. P. 4757. https://doi.org/10.1021/OM9805077
Golubev P., Krasavin M. // Tetrahedron Lett. 2018. V. 59. P. 3532. https://doi.org/10.1016/j.tetlet.2018.08.025
Fehlhammer W.P., Metzner R., Sperber W. // Chem. Ber. 1994. V. 127. P. 829. https://doi.org/10.1002/cber.19941270507
Moderhack D. // Tetrahedron. 2012. V. 68. P. 5949. https://doi.org/10.1016/j.tet.2012.04.099
Giustiniano M., Basso A., Mercalli V. et al. // Chem. Soc. Rev. 2017. V. 46. P. 1295. https://doi.org/10.1039/C6CS00444J
Goldberg S.Z., Eisenberg R., Miller J.S. // Inorg. Chem. 1977. V. 16. P. 1502. https://doi.org/10.1021/ic50172a053
Miller J.S., Balch A.L. // Inorg. Chem. 1972. V. 11. P. 2069. https://doi.org/10.1021/ic50115a017
Stork J.R., Rios D., Pham D. et al. // Inorg. Chem. 2005. V. 44. P. 3466. https://doi.org/10.1021/ic048333a
Zeiler A., Rudolph M., Rominger F., Hashmi A.S.K. // Chem. Eur. J. 2015. V. 21. P. 11065. https://doi.org/10.1002/chem.201500025
Tanase T., Urabe M., Mori N. et al. // J. Organomet. Chem. 2019. V. 879. P. 47. https://doi.org/10.1016/j.jorganchem.2018.10.005
Steinmetz A.L., Johnson B.V. // Organometallics. 1983. V. 2. P. 705. https://doi.org/10.1021/om00078a002
Anding B.J., Ellern A., Woo L.K. // Organometallics. 2014. V. 33. P. 2219. https://doi.org/10.1021/om500081w
Boschi T., Licoccia S., Paolesse R. et al. // Organometallics. 1989. V. 8. P. 330. https://doi.org/10.1021/om00104a010
Wong K.-H., Cheung W.-M., Sung H.H.Y. et al. // Eur. J. Inorg. Chem. 2020. V. 2020. P. 2085. https://doi.org/10.1002/ejic.202000208
Na H., Maity A., Teets T.S. // Dalton Trans. 2017. V. 46. P. 5008. https://doi.org/10.1039/C7DT00694B
Maity A., Le L.Q., Zhu Z. et al. // Inorg. Chem. 2016. V. 55. P. 2299. https://doi.org/10.1021/acs.inorgchem.5b02691
Katlenok E.A., Kinzhalov M.A., Eremina A.A. et al. // Opt. Spectrosc. 2017. V. 122. P. 723. https://doi.org/10.1134/S0030400X17050113
Islamova R.M., Dobrynin M.V., Vlasov A.V. et al. // Catal. Sci. Technol. 2017. V. 7. P. 5843. https://doi.org/10.1039/C7CY02013A
Kinzhalov M.A., Eremina A.A., Smirnov A.S. et al. // Dalton Trans. 2019. V. 48. P. 7571. https://doi.org/10.1039/C9DT01138B
Canovese L., Visentin F., Uguagliati P. et al. // J. Organomet. Chem. 1997. V. 535. P. 69. https://doi.org/10.1016/s0022-328x(96)06943-4
Crociani B., Boschi T., Nicolini M., Belluco U. // Inorg. Chem. 1972. V. 11. P. 1292. https://doi.org/10.1021/ic50112a028
Calligaro L., Uguagliati P., Crociani B., Belluco U. // J. Organomet. Chem. 1975. V. 92. P. 399. https://doi.org/10.1016/S0022-328X(00)85689-2
Belluco U., Michelin R.A., Uguagliati P., Crociani B. // J. Organomet. Chem. 1983. V. 250. P. 565. https://doi.org/10.1016/0022-328X(83)85078-5
Crociani B., Uguagliati P., Belluco U. // J. Organomet. Chem. 1976. V. 117. P. 189. https://doi.org/10.1016/S0022-328X(00)87271-X
Kuznetsov M.L., Kukushkin V.Y. // Molecules. 2017. V. 22. P. 1141. https://doi.org/10.3390/molecules22071141
Zhang X., Wu X., Lei Y. // J. Mol. Model. 2019. V. 25. P. 261. https://doi.org/10.1007/s00894-019-4145-x
Casella G., Casarin M., Kukushkin V.Y., Kuznetsov M.L. // Molecules. 2018. V. 23. P. 2942. https://doi.org/10.3390/molecules23112942
Tamm M., Ekkehardt Hahn F. // Coord. Chem. Rev. 1999. V. 182. P. 175. https://doi.org/10.1016/S0010-8545(98)00233-1
Burke A., Balch A.L., Enemark J.H. // J. Am. Chem. Soc. 1970. V. 92. P. 2555. https://doi.org/10.1021/ja00711a063
Rouschias G., Shaw B.L. // J. Chem. Soc. A. Inorg. Phys. Theor. 1971. P. 2097. https://doi.org/10.1039/J19710002097
Butler W.M., Enemark J.H., Parks J., Balch A.L. // Inorg. Chem. 1973. V. 12. P. 451. https://doi.org/10.1021/ic50120a042
Stork J.R., Olmstead M.M., Balch A.L. // J. Am. Chem. Soc. 2005. V. 127. P. 6512. https://doi.org/10.1021/ja050014a
Stork J.R., Olmstead M.M., Fettinger J.C., Balch A.L. // Inorg. Chem. 2006. V. 45. P. 849. https://doi.org/10.1021/ic051252+
Balch A.L., Parks J.E. // J. Am. Chem. Soc. 1974. V. 96. P. 4114. https://doi.org/10.1021/ja00820a009
Butler W.M., Enemark J.H. // Inorg. Chem. 1971. V. 10. P. 2416. https://doi.org/10.1021/ic50105a010
Moncada A.I., Khan M.A., Slaughter L.M. // Tetrahedron Lett. 2005. V. 46. P. 1399. https://doi.org/10.1016/j.tetlet.2005.01.033
Moncada A.I., Tanski J.M., Slaughter L.M. // J. Organomet. Chem. 2005. V. 690. P. 6247. https://doi.org/10.1016/j.jorganchem.2005.07.019
Moncada A.I., Manne S., Tanski J.M., Slaughter L.M. // Organometallics. 2006. V. 25. P. 491. https://doi.org/10.1021/om050786f
Wanniarachchi Y.A., Slaughter L.M. // Organometallics. 2008. V. 27. P. 1055. https://doi.org/10.1021/om700866p
Na H., Maity A., Morshed R., Teets T.S. // Organometallics. 2017. V. 36. P. 2965. https://doi.org/10.1021/acs.organomet.7b00428
Usón R., Laguna A., Villacampa M.D. et al. // Dalton Trans. 1984. P. 2035. https://doi.org/10.1039/DT9840002035
Edwards J.O., Pearson R.G. // J. Am. Chem. Soc. 1962. V. 84. P. 16. https://doi.org/10.1021/ja00860a005
Carey F.A., Sundberg R.J. Advanced Organic Chemistry. Part A: Structure and Mechanisms. Berlin: Springer, 2007.
Кукушкин Ю.Н. Реакционная способность координационных соединений. Ленинград: Химия, 1987.
Valishina E.A., Guedes da Silva M.F.C., Kinzhalov M.A. et al. // J. Mol. Catal. A: Chem. 2014. V. 395. P. 162. https://doi.org/10.1016/j.molcata.2014.08.018
Valishina E.A., Buslaeva T.M., Luzyanin K.V. // Russ. Chem. Bull. 2013. V. 62. P. 1361. https://doi.org/10.1007/s11172-013-0193-z
Luzyanin K.V., Tskhovrebov A.G., Carias M.C. et al. // Organometallics. 2009. V. 28. P. 6559. https://doi.org/10.1021/om900682v
Yakimanskiy A., Boyarskaya I., Boyarskiy V. // J. Coord. Chem. 2013. V. 66. P. 3592. https://doi.org/10.1080/00958972.2013.847185
Boyarskaya D.V., Kinzhalov M.A., Suslonov V.V., Boyarskiy V.P. // Inorg. Chim. Acta. 2017. V. 458. P. 190. https://doi.org/10.1016/j.ica.2017.01.008
Tšupova S., Rudolph M., Rominger F., Hashmi A.S.K. // Adv. Synth. Catal. 2016. V. 358. P. 3999. https://doi.org/10.1002/adsc.201600615
Miltsov S.A., Karavan V.S., Boyarsky V.P. et al. // Tetrahedron Lett. 2013. V. 54. P. 1202. https://doi.org/10.1016/j.tetlet.2012.12.060
Ryabukhin D.S., Sorokoumov V.N., Savicheva E.A. et al. // Tetrahedron Lett. 2013. V. 54. P. 2369. https://doi.org/10.1016/j.tetlet.2013.02.086
Savicheva E.A., Kurandina D.V., Nikiforov V.A., Boyarskiy V.P. // Tetrahedron Lett. 2014. V. 55. P. 2101. https://doi.org/10.1016/j.tetlet.2014.02.044
Kinzhalov M.A., Boyarskiy V.P., Luzyanin K.V. et al. // Dalton Trans. 2013. V. 42. P. 10394. https://doi.org/10.1039/c3dt51335a
Lai S.-W., Chan M.C.-W., Cheung K.-K., Che C.-M. // Organometallics. 1999. V. 18. P. 3327. https://doi.org/10.1021/om990256h
Tskhovrebov A.G., Luzyanin K.V., Dolgushin F.M. et al. // Organometallics. 2011. V. 30. P. 3362. https://doi.org/10.1021/om2002574
Kinzhalov M.A., Luzyanin K.V., Boyarskiy V.P. et al. // Russ. Chem. Bull. 2013. V. 62. P. 758. https://doi.org/10.1007/s11172-013-0103-4
Mikherdov A.S., Kinzhalov M.A., Novikov A.S. et al. // J. Am. Chem. Soc. 2016. V. 138. P. 14129. https://doi.org/10.1021/jacs.6b09133
Mikherdov A., Novikov A., Kinzhalov M. et al. // Crystals. 2018. V. 8. P. 112. https://doi.org/10.3390/cryst8030112
Mikherdov A.S., Orekhova Y.A., Boyarskii V.P. // Russ. J. Gen. Chem. 2018. V. 88. P. 2119. https://doi.org/10.1134/s1070363218100158
Mikherdov A.S., Tiuftiakov N.Y., Polukeev V.A., Boyarskii V.P. // Russ. J. Gen. Chem. 2018. V. 88. P. 713. https://doi.org/10.1134/s1070363218040151
Singh C., Prakasham A.P., Gangwar M.K., Ghosh P. // Chem. Select. 2018. V. 3. P. 9361. https://doi.org/10.1002/slct.201801667
Chay R.S., Luzyanin K.V. // Inorg. Chim. Acta. 2012. V. 380. P. 322. https://doi.org/10.1016/j.ica.2011.09.047
Luzyanin K.V., Pombeiro A.J.L., Haukka M., Kukushkin V.Y. // Organometallics. 2008. V. 27. P. 5379. https://doi.org/10.1021/om800517c
Chay R.S., Luzyanin K.V., Kukushkin V.Y. et al. // Organometallics. 2012. V. 31. P. 2379. https://doi.org/10.1021/om300020j
Boyarskaya D.V., Chulkova T.G. // Russ. J. Org. Chem. 2020. V. 56. P. 1937. https://doi.org/10.1134/S107042802011007X
Boyarskaya D.V., Bulatov E., Boyarskaya I.A. et al. // Organometallics. 2019. V. 38. P. 300. https://doi.org/10.1021/acs.organomet.8b00725
Tskhovrebov A.G., Luzyanin K.V., Kuznetsov M.L. et al. // Organometallics. 2011. V. 30. P. 863. https://doi.org/10.1021/om101041g
Katkova S.A., Kinzhalov M.A., Tolstoy P.M. et al. // Organometallics. 2017. V. 36. P. 4145. https://doi.org/10.1021/acs.organomet.7b00569
Mikherdov A.S., Popov R.A., Kinzhalov M.A. et al. // Inorg. Chim. Acta. 2021. V. 514. P. 120012. https://doi.org/10.1016/j.ica.2020.120012
Vicente J., Abad J.-A., López-Serrano J. et al. // Organometallics. 2005. V. 24. P. 5044. https://doi.org/10.1021/om050451y
Fehlhammer W.P. // Z. Naturforsch., B. 1994. V. 49. P. 494. https://doi.org/doi:10.1515/znb-1994-0411
Wanniarachchi Y.A., Kogiso Y., Slaughter L.M. // Organometallics. 2008. V. 27. P. 21. https://doi.org/10.1021/om701029j
Wanniarachchi Y.A., Slaughter L.M. // Chem. Commun. 2007. V. 31. P. 3294. https://doi.org/10.1039/B703769D
Wanniarachchi Y.A., Subramanium S.S., Slaughter L.M. // J. Organomet. Chem. 2009. V. 694. P. 3297. https://doi.org/10.1016/j.jorganchem.2009.06.007
Qiu G., Ding Q., Wu J. // Chem. Soc. Rev. 2013. V. 42. P. 5257. https://doi.org/10.1039/C3CS35507A
Altundas B., Marrazzo J.-P.R., Fleming F.F. // Org. Biomol. Chem. 2020. V. 18. P. 6467. https://doi.org/10.1039/D0OB01340D
Lang S. // Chem. Soc. Rev. 2013. V. 42. P. 4867. https://doi.org/10.1039/C3CS60022J
Suginome M., Ito Y. // Polymer Synthesis. 2004. V. 171. P. 77. https://doi.org/10.1007/b95531
Collet J.W., Roose T.R., Ruijter E. et al. // Angew. Chem. Int. Ed. 2020. V. 59. P. 540. https://doi.org/10.1002/anie.201905838
Vlaar T., Ruijter E., Maes B.U.W., Orru R.V.A. // Angew. Chem., Int. Ed. 2013. V. 52. P. 7084. https://doi.org/10.1002/anie.201300942
Martínez-Martínez A.J., Chicote M.T., Bautista D., Vicente J. // Organometallics. 2012. V. 31. P. 3711. https://doi.org/10.1021/om300201s
Usón R., Fornies J., Espinet P., Lalinde E. // J. Organomet. Chem. 1987. V. 334. P. 399. https://doi.org/10.1016/0022-328X(87)80102-X
Kelly C.M., Kwon D.-H., Ferguson M.J. et al. // Angew. Chem. Int. Ed. 2015. V. 54. P. 14498. https://doi.org/10.1002/anie.201506871
Il'in M.V., Bolotin D.S., Novikov A.S. et al. // Inorg. Chim. Acta. 2019. V. 490. P. 267. https://doi.org/10.1016/j.ica.2019.03.026
Melaimi M., Soleilhavoup M., Bertrand G. // Angew. Chem. Int. Ed. 2010. V. 49. P. 8810. https://doi.org/10.1002/anie.201000165
Johansson Seechurn C.C.C., Kitching M.O., Colacot T.J., Snieckus V. // Angew. Chem. Int. Ed. 2012. V. 51. P. 5062. https://doi.org/10.1002/anie.201107017
Rayadurgam J., Sana S., Sasikumar M., Gu Q. // Org. Chem. Front. 2021. V. 8. P. 384. https://doi.org/10.1039/d0qo01146k
Buskes M.J., Blanco M.J. // Molecules. 2020. V. 25. 3493. https://doi.org/10.3390/molecules25153493
Diccianni J.B., Diao T.N. // Trends in Chemistry. 2019. V. 1. P. 830. https://doi.org/10.1016/j.trechm.2019.08.004
Tasker S.Z., Standley E.A., Jamison T.F. // Nature. 2014. V. 509. P. 299. https://doi.org/10.1038/nature13274
Phapale V.B., Cardenas D.J. // Chem. Soc. Rev. 2009. V. 38. P. 1598. https://doi.org/10.1039/b805648j
Han F.S. // Chem. Soc. Rev. 2013. V. 42. P. 5270. https://doi.org/10.1039/c3cs35521g
Soliev S.B., Astakhov A.V., Pasyukov D.V., Chernyshev V.M. // Russ. Chem. Bull. 2020. V. 69. P. 683. https://doi.org/10.1007/s11172-020-2818-3
Cahiez G., Moyeux A. // Chem. Rev. 2010. V. 110. P. 1435. https://doi.org/10.1021/cr9000786
Gudmundsson A., Bäckvall J.E. // Molecules. 2020. V. 25. P. 1349. https://doi.org/10.3390/molecules25061349
Furstner A. // Bull. Chem. Soc. Jpn. 2021. V. 94. P. 666. https://doi.org/10.1246/bcsj.20200319
Furstner A., Leitner A., Mendez M., Krause H. // J. Am. Chem. Soc. 2002. V. 124. P. 13856. https://doi.org/10.1021/ja027190t
Kantchev E.A.B., O’Brien C.J., Organ M.G. // Angew. Chem. Int. Ed. 2007. V. 46. P. 2768. https://doi.org/10.1002/anie.200601663
Fortman G.C., Nolan S.P. // Chem. Soc. Rev. 2011. V. 40. P. 5151. https://doi.org/10.1039/c1cs15088j
Martin R., Buchwald S.L. // Acc. Chem. Res. 2008. V. 41. P. 1461. https://doi.org/10.1021/ar800036s
Kotha S., Lahiri K., Kashinath D. // Tetrahedron. 2002. V. 58. P. 9633. https://doi.org/10.1016/s0040-4020(02)01188-2
Polshettiwar V., Decottignies A., Len C., Fihri A. // ChemSusChem. 2010. V. 3. P. 502. https://doi.org/10.1002/cssc.200900221
Hooshmand S.E., Heidari B., Sedghi R., Varma R.S. // Green Chem. 2019. V. 21. P. 381. https://doi.org/10.1039/c8gc02860e
Kinzhalov M.A., Borozdinova A.M., Boyarskaya I.A. et al. // Russ. J. Gen. Chem. 2014. V. 84. P. 2138. https://doi.org/10.1134/S1070363214110164
Casalnuovo A.L., Calabrese J.C. // J. Am. Chem. Soc. 1990. V. 112. P. 4324.
Wei J.-F., Jiao J., Feng J.-J. et al. // J. Org. Chem. 2009. V. 74. P. 6283.
Moseley J.D., Murray P.M., Turp E.R. et al. // Tetrahedron. 2012. V. 68. P. 6010.
Godoy F., Segarra C., Poyatos M., Peris E. // Organometallics. 2011. V. 30. P. 684.
Mohajer F., Heravi M.M., Zadsirjan V., Poormohammad N. // RSC Adv. 2021. V. 11. P. 6885. https://doi.org/10.1039/d0ra10575a
Kanwal I., Mujahid A., Rasool N. et al. // Catalysts. 2020. V. 10. P. 443. https://doi.org/10.3390/catal10040443
Chinchilla R., Najera C. // Chem. Soc. Rev. 2011. V. 40. P. 5084. https://doi.org/10.1039/c1cs15071e
Murashkina A.V., Mitrofanov A.Y., Beletskaya I.P. // Russ. J. Org. Chem. 2019. V. 55. P. 1445. https://doi.org/10.1134/s1070428019100014
Eremin D.B., Ananikov V.P. // Chem. Soc. Rev. 2017. V. 346. P. 2. https://doi.org/10.1016/j.ccr.2016.12.021
Chernyshev V.M., Khazipov O.V., Eremin D.B. et al. // Chem. Soc. Rev. 2021. V. 437. P. 213860. https://doi.org/10.1016/j.ccr.2021.213860
Chernyshev V.M., Denisova E.A., Eremin D.B., Ananikov V.P. // Chem. Sci. 2020. V. 11. P. 6957. https://doi.org/10.1039/d0sc02629h
Boyarskii V.P. // Russ. J. Gen. Chem. 2017. V. 87. P. 1663. https://doi.org/10.1134/s1070363217080035
Boyarskaya D.V., Boyarskii V.P. // Russ. J. Gen. Chem. 2016. V. 86. P. 2033. https://doi.org/10.1134/s1070363216090085
Handy C.J., Manoso A.S., McElroy W.T. et al. // Tetrahedron. 2005. V. 61. P. 12201. https://doi.org/https://doi.org/10.1016/j.tet.2005.08.057
Rendler S., Oestreich M. // Synthesis. 2005. V. 2005. P. 1727.
Beletskaya I.P., Cheprakov A.V. // Chem. Rev. 2000. V. 100. P. 3009. https://doi.org/10.1021/cr9903048
Paul D., Das S., Saha S. et al. // Eur. J. Org. Chem. 2021. V. 14. P. 2057. https://doi.org/10.1002/ejoc.202100071
Kadu B.S. // Catal. Sci. Technol. 2021. V. 11. P. 1186. https://doi.org/10.1039/D0CY02059A
Ishiyama T., Murata M., Miyaura N. // J. Organomet. Chem. 1995. V. 60. P. 7508. https://doi.org/10.1021/jo00128a024
Molnár Á. // Chem. Rev. 2011. V. 111. P. 2251. https://doi.org/10.1021/cr100355b
Mikhaylov V.N., Sorokoumov V.N., Balova I.A. // Russ. Chem. Rev. 2017. V. 86. P. 459. https://doi.org/10.1070/rcr4715
Magano J., Dunetz J.R. // Chem. Rev. 2011. V. 111. P. 2177. https://doi.org/10.1021/cr100346g
Beletskaya I.P., Najera C., Yus M. // Russ. Chem. Rev. 2020. V. 89. P. 250. https://doi.org/10.1070/rcr4916
Nakajima Y., Shimada S. // RSC Advances 2015. V. 5. P. 20603. https://doi.org/10.1039/C4RA17281G
Parasram M., Gevorgyan V. // Chem. Soc. Rev. 2017. V. 46. P. 6227. https://doi.org/10.1039/C7CS00226B
Chuentragool P., Kurandina D., Gevorgyan V. // Angew. Chem. Int. Ed. 2019. V. 58. P. 11586. https://doi.org/10.1002/anie.201813523
Lee S., Han W.-S. // Inorg. Chem. Front. 2020. V. 7. P. 2396. https://doi.org/10.1039/D0QI00001A
Chi Y., Chou P.-T. // Chem. Soc. Rev. 2010. V. 39. P. 638. https://doi.org/10.1039/B916237B
To W.P., Wan Q.Y., Tong G.S.M., Che C.M. // Trends Chem. 2020. V. 2. P. 796. https://doi.org/10.1016/j.trechm.2020.06.004
Bai R.B., Meng X.W., Wang X.X., He L. // Adv. Funct. Mater. 2020. V. 30. P. 1907169. https://doi.org/10.1002/adfm.201907169
Zhuang Y.L., Guo S., Deng Y.J. et al. // Chem. Asian. J. 2019. V. 14. P. 3791. https://doi.org/10.1002/asia.201901209
Housecroft C.E., Constable E.C. // Coord. Chem. Rev. 2017. V. 350. P. 155. https://doi.org/10.1016/j.ccr.2017.06.016
Zhang Q.-C., Xiao H., Zhang X. et al. // Chem. Soc. Rev. 2019. V. 378. P. 121. https://doi.org/10.1016/j.ccr.2018.01.017
Henwood A.F., Zysman-Colman E. // Top. Curr. Chem. 2016. V. 374. P. https://doi.org/10.1007/s41061-016-0036-0
Colombo A., Dragonetti C., Guerchais V. et al. // Coord. Chem. Rev. 2020. V. 414. P. https://doi.org/10.1016/j.ccr.2020.213293
Kinzhalov M.A., Grachova E.V., Luzyanin K.V. // 2021. V. -. P. in press.
Baldo M.A., Lamansky S., Burrows P.E. et al. // Appl. Phys. Lett. 1999. V. 75. P. 4. https://doi.org/10.1063/1.124258
O'Brien D.F., Baldo M.A., Thompson M.E., Forrest S.R. // Appl. Phys. Lett. 1999. V. 74. P. 442. https://doi.org/10.1063/1.123055
Vlcek A., Zalis S. // Coord. Chem. Rev. 2007. V. 251. P. 258. https://doi.org/10.1016/j.ccr.2006.05.021
Yang X.H., Neher D., Hertel D., Daubler T.K. // Adv. Mater. 2004. V. 16. P. 161. https://doi.org/10.1002/adma.200305621
Yang X., Xu X., Zhou G. // J. Mater. Chem. C. 2015. V. 3. P. 913. https://doi.org/10.1039/C4TC02474E
Yang X., Zhou G., Wong W.-Y. // Chem. Soc. Rev. 2015. V. 44. P. 8484. https://doi.org/10.1039/C5CS00424A
Baldo M.A., O’Brien D.F., You Y. et al. // Nature. 1998. V. 395. P. 151. https://doi.org/10.1038/25954
Lai S.W., Cheung K.K., Chan M.C.W., Che C.M. // Angew. Chem. Int. Ed. 1998. V. 37. P. 182. https://doi.org/10.1002/(SICI)1521-3773(19980202)37:1/2<182::AID-ANIE182>3.0.CO;2-X
Wu Y., Wen Z., Wu J.I.-C., Teets T.S. // Chem. Eur. J. 2020. V. 26. P. 16028. https://doi.org/10.1002/chem.202002775
Panchenko P.A., Fedorova O.A., Fedorov Y.V. // Russ. Chem. Rev. 2014. V. 83. P. 155. https://doi.org/10.1070/RC2014v083n02ABEH004380
Bren V.A. // Russ. Chem. Rev. 2001. V. 70. P. 1017. https://doi.org/10.1070/RC2001v070n12ABEH000667
Shi H., Wang Y., Lin S. et al. // Dalton Trans. 2021. V. 50. P. 6410. https://doi.org/10.1039/D1DT00592H
Ma D.-L., Lin S., Wang W. et al. // Chem. Sci. 2017. V. 8. P. 878. https://doi.org/10.1039/C6SC04175B
Ramón-Márquez T., Marín-Suárez M., Fernández-Gutiérrez A. et al. Iridium Complexes in the Development of Optical Sensors, in Iridium(III) // Optoelectronic and Photonics Applications / Ed. E. Zysman-Colman. John Wiley & Sons, 2017. 479 p.
Rosenberg B., Van Camp L., Krigas T. // Nature. 1965. V. 205. P. 698. https://doi.org/10.1038/205698a0
Rottenberg S., Disler C., Perego P. // Nat. Rev. Cancer. 2021. V. 21. P. 37. https://doi.org/10.1038/s41568-020-00308-y
Kelland L. // Nat. Rev. Cancer. 2007. V. 7. P. 573. https://doi.org/10.1038/nrc2167
Noffke A.L., Habtemariam A., Pizarro A.M., Sadler P.J. // Chem. Commun. 2012. V. 48. P. 5219. https://doi.org/10.1039/C2CC30678F
Hillard E.A., Jaouen G. // Organometallics. 2011. V. 30. P. 20. https://doi.org/10.1021/om100964h
Hussaini S.Y., Haque R.A., Razali M.R. // J. Organomet. Chem. 2019. V. 882. P. 96. https://doi.org/10.1016/j.jorganchem.2019.01.003
Budagumpi S., Haque R.A., Endud S. et al. // Eur. J. Inorg. Chem. 2013. V. 2013. P. 4367. https://doi.org/10.1002/ejic.201300483
Hindi K.M., Panzner M.J., Tessier C.A. et al. // Chem. Rev. 2009. V. 109. P. 3859. https://doi.org/10.1021/cr800500u
Montanel-Perez S., Elizalde R., Laguna A. et al. // Eur. J. Inorg. Chem. 2019. V. 39-40. P. 44273. https://doi.org/10.1002/ejic.201900606
Montanel-Pérez S., Herrera R.P., Laguna A. et al. // Dalton Trans. 2015. V. 44. P. 9052. https://doi.org/10.1039/C5DT00703H
Bertrand B., Romanov A.S., Brooks M. et al. // Dalton Trans. 2017. V. 46. P. 15875. https://doi.org/10.1039/C7DT03189K
Farrell N.P. // Chem. Soc. Rev. 2015. V. 44. P. 8773. https://doi.org/10.1039/C5CS00201J
Komeda S., Moulaei T., Chikuma M. et al. // Nucleic Acids Res. 2010. V. 39. P. 325. https://doi.org/10.1093/nar/gkq723
Komeda S., Moulaei T., Woods K.K. et al. // J. Am. Chem. Soc. 2006. V. 128. P. 16092. https://doi.org/10.1021/ja062851y
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии