

Журнал неорганической химии, 2022, T. 67, № 1, стр. 118-126
Синтез, фотокаталитические и электрофизические свойства керамических материалов в системе PbO–Bi2O3–Fe2O3
Д. С. Ершов a, *, Н. В. Беспрозванных a, О. Ю. Синельщикова a
a Институт химии силикатов им. И.В. Гребенщикова РАН
199034 Санкт-Петербург, наб. Макарова, 2, Россия
* E-mail: ershov.d.s@yandex.ru
Поступила в редакцию 30.03.2021
После доработки 15.07.2021
Принята к публикации 16.07.2021
- EDN: BTNQAD
- DOI: 10.31857/S0044457X22010032
Аннотация
Представлены результаты исследования фотокаталитической активности и общей электропроводности керамических материалов, формирующихся в двойной системе PbO–Bi2O3 и в частном разрезе (PbO)0.6(Bi2O3)0.4–BiFeO3. Образцы синтезированы твердофазным методом и пиролизом цитратно-нитратных композиций. Исследованные составы показали степень деградации метиленового оранжевого от 30 до 90% (С0 = 20 мг/л, концентрация катализатора 0.01 г/мл) при облучении в течение 3 ч люминесцентной ртутной лампой. Значения ширины запрещенной зоны, оцененные по функции Кубелки–Мунка, для синтезированных образцов находятся в диапазоне от 2.22 до 2.88 эВ. Общая электропроводность композитов в разрезе (PbO)0.6(Bi2O3)0.4–BiFeO3 с увеличением содержания Fe2O3 возрастает с 2.0 × 10–4 до 2.6 × 10–4 См/см при 500°C.
ВВЕДЕНИЕ
В системе PbO–Bi2O3–Fe2O3 формируются соединения, интересные как для практического использования, так и для понимания взаимосвязи электрофизических свойств с их структурными особенностями. Следует отметить, что двойная система PbO–Bi2O3 изучена довольно подробно [1, 2] и, согласно последним уточненным данным, в ней образуются следующие стабильные соединения: Bi6Pb2O11 (3 : 2), Bi12PbO19 (6 : 1), Bi8Pb5O17 (4 : 5), Bi2Pb3O6 (1 : 3), а также ряд твердых растворов со структурами α-, δ- и β-Bi2O3 [3, 4]. Несколько указанных составов имеют высокую проводимость по кислороду [5–9]. Проведено множество исследований по стабилизации высокопроводящей δ-фазы Bi2O3 типа флюорита за счет образования твердых растворов с другими оксидами, в том числе с использованием PbO [10]. В работе [5] исследована электропроводность соединения состава Bi8Pb5O17, подтвержден ее высокий уровень и показан кислородно-ионный характер. О положительном влиянии легирования Bi2O3 на механические свойства керамических таблеток PbO, используемых в качестве охлаждающего материала в твердофазных системах управления кислородом, говорилось в работе [11]. Было обнаружено, что легирование умеренным количеством Bi2O3 (3 мас. %) может увеличить твердость в четыре раза, а прочность – в два раза по сравнению с чистым PbO. Авторами [12] разработан и запатентован полупроводниковый элемент, в состав которого входят Bi2Pb3O6, Bi6Pb2O11, Bi0.57Pb1.43O2.29, Bi8Pb5O17, Bi1.23Pb1.43O2.29 и др. Согласно [12], этот полупроводниковый элемент, в котором на проводящей подложке сформирована пористая тонкая пленка из композитного оксидного полупроводника, чувствителен к видимому свету, обладает высокими рабочими характеристиками и уникальной способностью одновременно пропускать не только анодный, но и катодный фототок.
В двойной системе Bi2O3–Fe2O3 особый интерес для решения экологических проблем представляют соединения BiFeO3 и Bi25FeO40. Композитные фотокатализаторы на их основе способны инициировать разложение красителей под действием видимого света [13–15], в то время как наиболее применяемые в настоящее время фотокатализаторы на основе оксида титана могут быть активированы только ультрафиолетовым излучением, на которое приходится лишь около 4% спектра солнечного света [16]. Недавние исследования композитного фотокатализатора на основе BiFeO3, допированного свинцом с оксидом графена (Pb-BiFeO3/R-GO), показали, что он проявляет высокую антибактериальную эффективность в отношении грамотрицательных бактерий Escherichia coli и грамположительных золотистых стафилококков Staphylococcus aureus [17].
Наравне с поиском кристаллических твердых электролитов в тройной системе PbO–Bi2O3–Fe2O3 авторами [18, 19] была изучена электропроводность оксидных стекол данного состава и установлено, что она возрастает с увеличением содержания Fe2O3. Проводимость увеличивается с температурой, показывая общую полупроводниковую природу исследованных стекол.
Для улучшения свойств полученных материалов в качестве альтернативы традиционному твердофазному методу синтеза с целью увеличения удельной поверхности зачастую применяют методы “мягкой химии” [20, 21]. Одним из таких методов является пиролиз цитратно-нитратных композиций – простой и удобный метод получения разнообразных усовершенствованных керамических материалов, катализаторов и наночастиц [22].
Цель настоящей работы – поиск новых твердых электролитов и фотокатализаторов на основе висмутатов, определение условий их образования и исследование физико-химических свойств.
В рамках данной работы поставлены следующие задачи: определение оптимального метода синтеза и условий образования материалов, кристаллизующихся в тройной системе PbO–Bi2O3–Fe2O3; исследование их общей электропроводности и фотокаталитических свойств.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез образцов в системе PbO–Bi2O3–Fe2O3 осуществляли двумя методами: твердофазным и пиролизом цитратно-нитратных композиций.
В случае твердофазного метода в качестве исходных реактивов использовали Bi2O3 марки “ос. ч.”, Fe2O3 “ч. д. а.” и PbO “ос. ч.”.
После взвешивания каждого компонента в соответствии со стехиометрией получаемых сложных композиций смесь реагентов c добавлением этанола гомогенизировали в планетарной мельнице Fritsch Pulverisette 6 в течение 15 мин при скорости 350 об/мин.
После гомогенизации шихты под давлением 500 МПа из нее прессовали таблетки, которые затем обжигали в корундовых тиглях при температуре, рассчитанной в соответствии с фазовой диаграммой: образец І – при 640°C в течение 8 ч; образцы ІІ–V – при 610°C в течение 24 ч. Исходная смесь образца ІІІ также была подвергнута длительной термической обработке при 560°C в течение 72 ч для получения фазы φ-Bi8Pb5O17. Для лучшего взаимодействия реагирующих компонентов между обжигами проводили промежуточные перетирания и перепрессовывания синтезируемых образцов.
При синтезе методом пиролиза использовали предварительно приготовленные водные растворы нитратов висмута Bi(NO3)3 · 5H2O марки “ос. ч.”, железа Fe(NО3)3 · 9Н2О и свинца Pb(NO3)2 марки “ч. д. а.”. Для лучшего растворения нитрата висмута добавляли разбавленную азотную кислоту.
В ходе синтеза смесь растворов нитратов металлов добавляли к раствору лимонной кислоты марки “х. ч.”. Условное соотношение окислителя и восстановителя φ = 1 соответствует ситуации, когда композиция PbyBixFezO1.5(x+z) +y может быть получена непосредственно в результате реакции между топливом и окислителем без введения дополнительного кислорода:
В нашем случае соотношение нитратов (окислителей) было выбрано исходя из желаемого фазового состава, в то время как соотношение между лимонной кислотой (горючим) и окислителем (нитратами металлов) было подобрано экспериментально и соответствовало φ = 0.9. Таким образом, это позволило вести реакцию в режиме объемного горения [23].
К полученной смеси при постоянном перемешивании медленно добавляли сильно разбавленный раствор NH4OH до pH 5.5–6, при этом раствор становился прозрачным, что означало образование цитратных комплексов металлов.
Данный прозрачный раствор упаривали до получения ксерогеля путем термостатирования при 80°С. Полученный ксерогель прокаливали при 500°С в течение 1 ч, затем в зависимости от состава синтезируемых сложных оксидных соединений подвергали дополнительному обжигу при более высоких температурах: образец І – при 640°C в течение 8 ч; образцы ІІ–V – при 610°C в течение 12 ч.
Для получения фазы φ-Bi8Pb5O17 полученный ксерогель образца ІІІ без предварительного прокаливания при 500°С сжигали при 560°C с последующей выдержкой в течение 24 ч.
Фазовый состав образцов на каждой стадии синтеза определяли методом рентгеновской дифракции (дифрактометр ДРОН-3М, излучение CuKα). Идентификацию фаз исследуемых образцов осуществляли с помощью базы данных PDF-2.
Исследования микроструктуры шлифов керамических образцов проводили на сканирующем электронном микроскопе Tescan MIRA 3. Сканирующая электронная микроскопия сопровождалась микрорентгеноспектральным анализом.
Удельную поверхность образцов исследовали с помощью прибора СОРБИ-М по 4-точечному методу БЭТ. Измерение удельной поверхности включает в себя несколько циклов адсорбции-десорбции и проходит в автоматическом режиме. Для работы с прибором используется специализированное программное обеспечение SORBI-M.
Фотокаталитическую активность полученных материалов оценивали по реакции разложения органического красителя метилового оранжевого (МО). Выбор данного красителя обусловлен изученным ранее механизмом фотокаталитической деградации на висмутатах кальция под воздействием УФ-излучения [13]. В настоящей работе водный раствор МО с концентрацией 20 мг/л смешивали с тестируемым фотокатализатором исходя из расчета 0.01 г/мл, после чего облучали люминесцентными ртутными лампами низкого давления (UVB – 1%, UVA – 3.8 Вт) мощностью 20 Вт в течение 3 ч. Содержание красителя в растворе определяли с помощью спектрофотометра ПФ-5400 после отбора и центрифугирования аликвоты.
Эффективный диаметр и распределение частиц по размерам оценивали в водной суспензии с помощью метода динамического светорассеяния (NanoBrook 90 Plus Zeta), перед измерением порошки диспергировали в воде ультразвуком в течение 40 мин.
С помощью УФ-спектрометра с интегрирующей сферой Shimadzu UV2600 в интервале длин волн 220–850 нм были получены спектры диффузного отражения всех синтезированных материалов. Для пересчета спектров диффузного отражения в спектры поглощения применяли функцию Кубелки–Мунка, численно равную коэффициенту оптического поглощения материала:
где R – коэффициент диффузного отражения материала, измеренный относительно абсолютно белого тела.Для оценки ширины запрещенной зоны использовали построение Тауца [24]:
где hv – энергия фотонов, эВ; n – константа, зависящая от типа электронного перехода в материале; А – коэффициент пропорциональности; Eg – ширина запрещенной зоны.Аппроксимация линейных участков края оптического поглощения для модели прямых разрешенных переходов (n = 1/2) до пересечения с осью энергии фотона hv (эВ) позволила определить ширину запрещенной зоны Eg полученных материалов.
Определение общей электропроводности полученной керамики проводили двухконтактным методом в универсальной ячейке на постоянном токе с помощью RLC-метра PM6306 (рабочее напряжение 0.5 В) в диапазоне температур 20–600°C. Предварительно на торцевые поверхности цилиндрических таблеток наносили металлические контакты путем вжигания серебросодержащей проводниковой пасты при температуре 550°C (ПСН-1, производства ООО “Элма-Пасты”).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Результаты рентгенофазового анализа образцов, полученных двумя методами синтеза (твердофазным и пиролизом цитратно-нитратных композиций), представлены на рис. 1. В двойной системе PbO–Bi2O3 (рис. 1а) и в частном разрезе (PbO)0.6(Bi2O3)0.4–BiFeO3 (рис. 1б) тройной системы PbO–Bi2O3–Fe2O3 были синтезированы соединения следующей стехиометрии: І – (PbO)0.143(Bi2O3)0.857, ІІ – (PbO)0.55(Bi2O3)0.45, ІІІ – (PbO)0.6(Bi2O3)0.4, IV – (PbO)0.5(Bi2O3)0.42(Fe2O3)0.08, V – (PbO)0.4(Bi2O3)0.44(Fe2O3)0.16. Выбор составов в двойной системе PbO–Bi2O3 обусловлен формированием в данных концентрациях стабильных фаз типа силленита–Bi12PbO19 (состав указан согласно фазовой диаграмме [3], далее по тексту Bi25MO40, где M = Fe, Pb) и твердого раствора на основе β-Bi8Pb5O17, который перспективен для использования в качестве ионного проводника [5]. Крайние точки разреза (PbO)0.6(Bi2O3)0.4–BiFeO3 были определены исходя из результатов исследования фотокаталитической активности первых трех составов (I–III) и литературных данных [13, 14] об ее усилении в висмутсодержащих композитах при формировании фазы BiFeO3.
Рис. 1.
Дифрактограммы образцов в разрезах: а – PbO–Bi2O3, б – (PbO)0.6(Bi2O3)0.4–BiFeO3, синтезированных твердофазным методом (нижние кривые) и пиролизом цитратно-нитратных композиций (верхние кривые). Состав композиций: І – (PbO)0.143(Bi2O3)0.857, ІІ – (PbO)0.55(Bi2O3)0.45, ІІІ – (PbO)0.6(Bi2O3)0.4, IV – (PbO)0.5(Bi2O3)0.42(Fe2O3)0.08, V – (PbO)0.4(Bi2O3)0.44(Fe2O3)0.16; 1 – Bi12PbO19, 2 – γ-Bi2O3, 3 – Bi8Pb5O17, 4 – Bi25MO40 (M = Fe; Pb), 5 – Bi2Fe4O9.
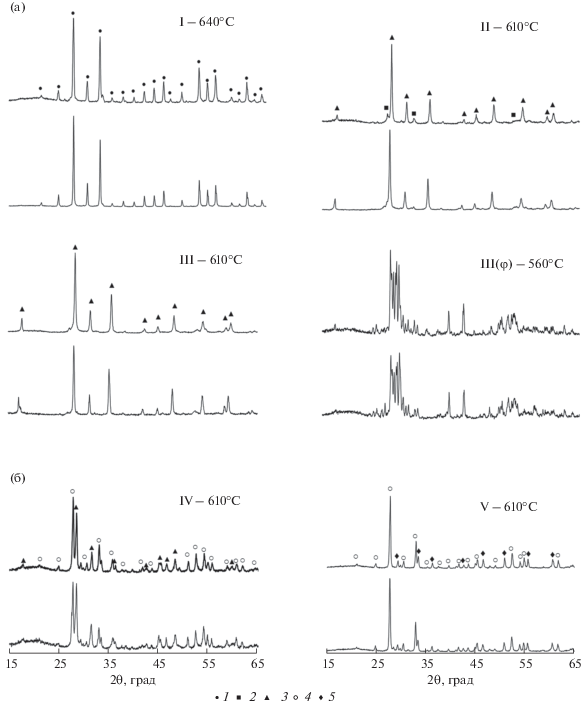
В системе PbO–Bi2O3 (рис. 1а) при первом стехиометрическом соотношении (состав I) обоими методами была получена чистая фаза со структурой типа силленита (Bi12PbO19 по данным PDF-2, карточка № 39-837). В случае двух других соотношений (составы II и III) основной фазой после обжига при 610°С был висмутат β-Bi8Pb5O17 (41-405). При меньшем содержании оксида свинца (состав II, синтезированный твердофазным методом) наблюдается присутствие небольшого количества примеси в виде кубического γ-Bi2O3 (2-542).
Следуя данным [25, 26], твердофазным методом синтеза с длительной термической обработкой состава ІІІ при 560°C в течение 72 ч была получена фаза φ-Bi8Pb5O17 (COD № 96-152-6076) [27]. Использование метода пиролиза цитратно-нитратных композиций при синтезе фазы φ-Bi8Pb5O17 не привело к снижению необходимой температуры обжига, однако позволило значительно уменьшить его длительность. Однофазный образец был получен при термообработке в течение 24 ч.
В разрезе (PbO)0.6(Bi2O3)0.4–BiFeO3 (рис. 1б) твердофазным и цитратно-нитратным методами синтеза были получены образцы двухфазной керамики в точках (PbO)0.5(Bi2O3)0.42(Fe2O3)0.08 (состав IV) и (PbO)0.4(Bi2O3)0.44(Fe2O3)0.16 (состав V). Образцы состава IV при применении обоих методов имели близкий фазовый состав, в котором основными фазами являлись Bi8Pb5O17 и твердый раствор со структурой силленита Bi25MO40 (M = = Fe, Pb). При увеличении содержания Fe2O3 (состав V) также была получена двухфазная керамика, состоящая из силленита Bi25MO40 (M = Fe, Pb) и феррита висмута Bi2Fe4O9 (20-836).
На рис. 2 показаны результаты сканирующей электронной микроскопии некоторых синтезированных материалов. Видно, что синтез цитратно-нитратным методом приводит к получению более плотно спеченной керамики. Для состава IV, содержащего железо, приведены микрофотографии с большим увеличением, где отмечены две фазы (рис. 2). По результатам микрозондового анализа, они имеют состав Bi6.35Pb4.29Fe2.06O17 (фаза 1) и Bi23.9Pb0.93Fe0.80O40 (фаза 2), что хорошо согласуется с данными РФА, показавшими наличие в указанных образцах твердых растворов на основе β-Bi8Pb5O17 и силленита Bi25MO40 (M = Fe, Pb). Химический состав образца III, синтезированного цитратно-нитратным методом, соответствует соотношению (Bi2O3)0.39(PbO)0.61, твердофазным методом – (Bi2O3)0.38(PbO)0.62, что близко к взятому по шихте.
Рис. 2.
Микрофотографии образцов IV – (PbO)0.5(Bi2O3)0.42(Fe2O3)0.08 (а, б) и ІІІ – (PbO)0.6(Bi2O3)0.4 (в, г), синтезированных цитратно-нитратным (слева) и твердофазным (справа) методами: 1 – Bi6.35Pb4.29Fe2.06O17, 2 – Bi23.9Pb0.93Fe0.80O40.

В связи с тем, что цитратно-нитратный метод синтеза не позволил уменьшить необходимую температуру конечного обжига, использованные для эксперимента по определению фотокаталитической активности образцы представляли собой порошки механически измельченной керамики с частицами субмикронного и микронного размера (по данным динамического светорассеяния, средний размер частиц составлял от 250 до 450 нм).
Удельная поверхность ряда синтезированных образцов была исследована с помощью многоточечного метода БЭТ и составила ~1 м2/г (табл. 1 ). Из полученных данных можно сделать вывод, что метод пиролиза цитратно-нитратных композиций позволяет увеличить площадь удельной поверхности образцов на ~20% по сравнению с классическим твердофазным методом синтеза.
Результаты исследования фотокаталитической активности представлены на рис. 3. Все исследованные образцы проявляют фотокаталитическую активность в реакции деградации органического красителя МО. Следует отметить, что метиловый оранжевый в отсутствие фотокатализатора не разлагается и спустя 3 ч облучения показывает тот же уровень светопропускания, что и в начале эксперимента.
Рис. 3.
Фотокаталитическая деградация красителя МО под воздействием облучения на образцах состава: І – (PbO)0.143(Bi2O3)0.857; ІІ – (PbO)0.55(Bi2O3)0.45; ІІІ – (PbO)0.6(Bi2O3)0.4; IV – (PbO)0.5(Bi2O3)0.42(Fe2O3)0.08, V – (PbO)0.4(Bi2O3)0.44(Fe2O3)0.16, синтезированных твердофазным (а) и цитратно-нитратным (б) методами. Для сравнения приведена гистограмма деградации красителя под действием коммерческого образца катализатора Aeroxide® TiO2 P25, также известного как Degussa P25 – VI (в). Длительность облучения: 1 – 60, 2 – 120, 3 – 180 мин.
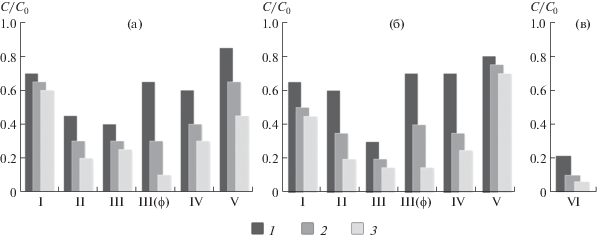
Применение метода пиролиза цитратно-нитратных композиций в сравнении с классическим твердофазным синтезом положительно влияет на скорость разложения красителя и приводит к увеличению значения ΔС/С0 за 3 ч экспонирования: на 36% для (PbO)0.143(Bi2O3)0.857 (І) и (PbO)0.6(Bi2O3)0.4 (ІІІ) и на 13% для (PbO)0.5(Bi2O3)0.42(Fe2O3)0.08 (IV).
Образец на основе фазы φ-Bi8Pb5O17 (состав ІІІφ), синтезированный как твердофазным методом, так и пиролизом цитратно-нитратных композиций, проявляет высокую фотокаталитическую активность с показателями разложения красителя МО ≥ 85%. Минимальный уровень деградации красителя у него составляет С/С0 = 0.1 (в случае применения твердофазного метода).
В ходе эксперимента ряд образцов (составы ІІ–IV) изменили цвет. По результатам РФА, в них происходит частичное разложение твердых растворов на основе Bi8Pb5O17 с образованием моноклинного α-Bi2O3, что свидетельствует о переходе части свинца в раствор. Таким образом, несмотря на высокий уровень фотокаталитической активности данные образцы не могут быть рекомендованы для очистки водных растворов. Однако они представляют интерес для изучения разложения на них органических загрязнителей в воздушной среде, что требует дополнительных исследований. Композиции на основе силленитовой фазы (І, V) оставались стабильными на протяжении всего эксперимента. Среди них наилучшие характеристики имел порошок состава (PbO)0.4(Bi2O3)0.44(Fe2O3)0.16, синтезированный твердофазным методом. Коэффициент фотодеградации на нем составил С/С0 = 0.45.
Помимо исследования синтезированных образцов был проведен эксперимент на коммерческом образце катализатора фирмы Aeroxide®. Условия проведения опыта совпадали с применяемыми к рассматриваемым составам. Коэффициент фотодеградации красителя после облучения в течение 3 ч составил С/С0 = 0.05. При этом невозможно сказать, разложился ли краситель полностью, так как в растворе присутствует взвесь катализатора, которую не удалось до конца отцентрифугировать, что не позволяет добиться полной светопропускаемости раствора.
Анализ спектров оптического поглощения с использованием построения Тауца в предположении прямых разрешенных электронных переходов (n = 1/2) представлен на рис. 4. По ней графическим методом были определены края полос поглощения. Данные значения можно принять близкими по величине к энергии перехода электронов из валентной зоны в зону проводимости, т.е. к ширине запрещенной зоны (Eg).
Рис. 4.
Аппроксимация спектров оптического поглощения в предположении модели прямых разрешенных переходов для образцов в разрезах PbO–Bi2O3 (а) и (PbO)0.6(Bi2O3)0.4–BiFeO3 (б): І – (PbO)0.143(Bi2O3)0.857; ІІ – (PbO)0.55(Bi2O3)0.45; ІІІ – (PbO)0.6(Bi2O3)0.4; IV – (PbO)0.5(Bi2O3)0.42(Fe2O3)0.08; V – (PbO)0.4(Bi2O3)0.44(Fe2O3)0.16, синтезированных твердофазным (сплошная линия) и цитратно-нитратным (пунктирная линия) методами.
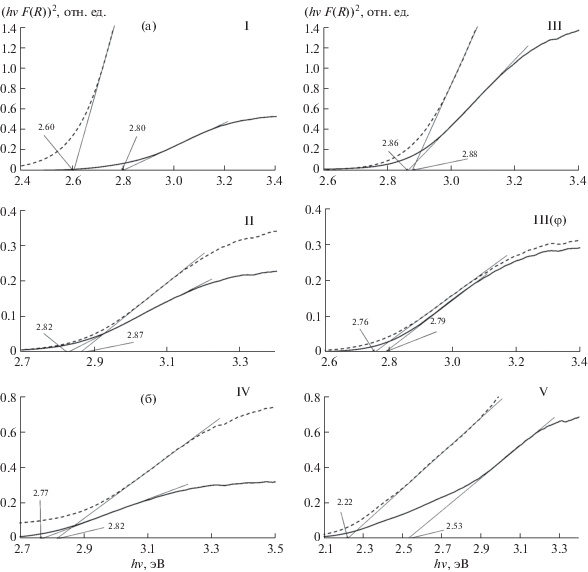
Eg рассмотренных образцов находится в диапазоне от 2.22 до 2.88 эВ, что соответствует видимому диапазону длин волн от 430 до 558 нм. Это является одним из необходимых условий возможного использования материалов в качестве фотокатализаторов, активируемых видимым светом. Таким образом, рассмотренные составы могут быть перспективны для данного применения.
Следует отметить, что для всех порошков, полученных цитратно-нитратным методом, наблюдается большее значение F(R). Данное явление обычно связывают с меньшим размером частиц и, как следствие, с большей величиной рассеивания падающего на образец излучения. Особенно отчетливо это видно на составе (PbO)0.143(Bi2O3)0.857 (І), при синтезе которого цитратно-нитратным методом не проводился дополнительный высокотемпературный обжиг.
Таблица 1.
Полная удельная поверхность образцов, полученных двумя методами синтеза
| № образца | Состав образца | Удельная поверхность (м2/г) для составов, полученных | |
|---|---|---|---|
| твердофазным методом | цитратно-нитратным методом | ||
| ІІІ | (PbO)0.6(Bi2O3)0.4 | 0.95 ± 0.04 | 1.16 ± 0.01 |
| ІІІφ | φ-Bi8Pb5O17 | 0.43 ± 0.01 | 1.11 ± 0.08 |
| IV | (PbO)0.5(Bi2O3)0.42(Fe2O3)0.08 | 0.98 ± 0.04 | 1.27 ± 0.08 |
| V | (PbO)0.4(Bi2O3)0.44(Fe2O3)0.16 | 0.91 ± 0.01 | 1.01 ± 0.02 |
Зависимость общей электропроводности полученных твердофазных методом керамических материалов в координатах Аррениуса близка к линейной (рис. 5).
Рис. 5.
Температурная зависимость электропроводности образцов в системе PbO–Bi2O3–Fe2O3, синтезированных твердофазным методом: 1 – (PbO)0.143(Bi2O3)0.857; 2 – (PbO)0.55(Bi2O3)0.45; 3 – (PbO)0.5(Bi2O3)0.42(Fe2O3)0.08; 4 – (PbO)0.4(Bi2O3)0.44(Fe2O3)0.16; 5 – β-Bi8Pb5O17 [6].
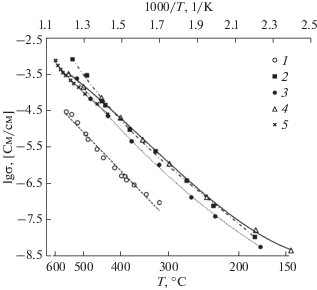
Как видно из приведенных графиков, значения удельной проводимости всех рассмотренных образцов, несмотря на различия в фазовом составе, очень близки, за исключением (PbO)0.143(Bi2O3)0.857 (I). Электропроводность состава I примерно на порядок ниже. Следует отметить, что результаты, полученные для образца (PbO)0.55(Bi2O3)0.45 (ІІ), в основе которого лежит фаза β-Bi8Pb5O17, в значительной степени согласуются с ранее опубликованными в работе [6]. Высокая проводимость в керамике, содержащей железо (составы IV, V), может быть обусловлена как наличием β-Bi8Pb5O17, так и присутствием фаз на основе силленита Bi25MO40 (M = = Fe, Pb).
По температурным зависимостям проводимости для каждого из составов в диапазоне от 150 до 500°C была рассчитана энергия активации электропроводности Ea, значения которой находятся в диапазоне от 0.87 эВ для образца (PbO)0.4(Bi2O3)0.44(Fe2O3)0.16 (V) до 1.01 эВ для (PbO)0.143(Bi2O3)0.857 (I).
Ввиду механической хрупкости состава (PbO)0.6(Bi2O3)0.4 (ІІІ) не удалось измерить температурную зависимость электропроводности для данного образца.
ЗАКЛЮЧЕНИЕ
Установлено преимущество пиролиза цитратно-нитратных композиций по сравнению с твердофазным методом при синтезе образцов, формирующихся в тройной системе PbO–Bi2O3–Fe2O3. Данный метод позволяет снизить длительность термической обработки в 2–3 раза в зависимости от конкретного состава. С его помощью также удалось впервые получить однофазный образец φ-Bi8Pb5O17 и керамику на основе (PbO)0.6(Bi2O3)0.4–BiFeO3.
Все полученные составы проявляют фотокаталитическую активность под воздействием видимого света, что предполагает их использование в качестве фотокатализаторов. Однако в ходе эксперимента было выявлено разложение некоторых образцов при длительном нахождении в растворе, и, несмотря на хороший уровень фотодеградации красителя, это ставит под сомнение безопасность использования данных материалов для очистки воды от загрязнителей. Тем не менее полученные образцы могут быть интересны для изучения на них разложения органических загрязнителей в воздушной среде.
Впервые определена общая электропроводность керамики, формирующейся в разрезе (PbO)0.6(Bi2O3)0.4–BiFeO3. Значения удельной проводимости изученных образцов оказались близки, и при 500°C σ составила ~(2.0–2.6) × 10–4 См/см.
Список литературы
Biefeld R.M., White S.S. // J. Am. Ceram. Soc. 1981. V. 64. № 3. P. 182. https://doi.org/10.1111/j.1151-2916.1981.tb10253.x
Braileanu A., Zaharescu M., Crişan D. et al. // J. Therm. Anal. 1997. V. 49. P. 1197. https://doi.org/10.1007/BF01983675
Diop I., David N., Fiorani J.M. et al. // J. Chem. Thermodyn. 2009. V. 41. P. 420. https://doi.org/10.1016/j.jct.2008.10.012
Dapčević A., Poleti D., Karanović L. et al. // J. Serb. Chem. Soc. 2017. V. 82. № 12. P. 1433. https://doi.org/10.2298/JSC170711111D
Sammes N.M., Tompsett G., Phillips R. et al. // Solid State Ionics. 1996. V. 86–88. P. 125. https://doi.org/10.1016/0167-2738(96)00106-3
Demonchy P., Boivin J.-C., Thomas D. et al. // C.R. Acad. Sci. Paris. 1980. V. 290. Série C. P. 279.
Fee M.G., Sammes N.M., Tompsett G. et al. // Solid State Ionics. 1997. V. 95. P. 183. https://doi.org/10.1016/S0167-2738(96)00546-2
Honnart F., Boivin J.C., Thomas D. et al. // Solid State Ionics. 1983. V. 9–10. P. 921. https://doi.org/10.1016/0167-2738(83)90111-X
Бордовский Г.А., Анисимова Н.И. // Неорган. материалы. 1982. Т. 18. № 7. С. 1206.
Liu X. // For the Degree of Doctor of Philosophy. School of Biological and Chemical Sciences Queen Mary, University of London, United Kingdom. 2009. 295 p.
Ma Y., Yang A., Zhu H. et al. // Materials. 2019. V. 12. P. 1948. https://doi.org/10.3390/ma12121948
Sayama K., Sugita G., Sugihara H. et al. // National Institute of Advanced Industrial Science and Technology, Nissan Motor Co., Ltd. № 2007-073618. March 22, 2007.
Wang B., Wang S., Gong L. et al. // Ceram. Int. 2012. V. 38. P. 6643. https://doi.org/10.1016/j.ceramint.2012.05.051
Xiong Z., Cao L. // J. Alloys Compd. 2019. V. 773. P. 828. https://doi.org/10.1016/j.jallcom.2018.09.344
Ершов Д.С., Беспрозванных Н.В., Синельщикова О.Ю. // Физика и химия стекла. 2020. Т. 46. № 4. С. 416. [Ershov D.S., Besprozvannykh N.V., Sinel’shchikova O.Yu. // Glass Phys. Chem. 2020. V. 46. № 4. P. 329. https://doi.org/10.1134/S1087659620040057]https://doi.org/10.31857/S0132665120040058
Wang Y., He Y., Li T. et al. // Chem. Eng. J. 2012. V. 189–190. P. 473. https://doi.org/10.1016/j.cej.2012.02.079
Li Y., Zhao J., Zhang G. et al. // Water Res. 2019. V. 161. P. 251. https://doi.org/10.1016/j.watres.2019.06.011
Prakash C., Husain Sh., Singh R.J. et al. // J. Alloys Compd. 2001. V. 326. P. 47. https://doi.org/10.1016/S0925-8388(01)01224-5
Qiu H., Sakata H., Hirayama T. // J. Ceram. Soc. Jpn. 1996. V. 104. P. 1004. https://doi.org/10.2109/jcersj.104.1004
Кувшинова Т.Б., Егорышева А.В., Гайтко О.М. и др. // Журн. неорган. химии. 2015. Т. 60. № 10. С. 1294. https://doi.org/10.7868/S0044457X15100116
Владимирова Е.В., Дмитриев А.В., Кандауров М.В. // Журн. неорган. химии. 2019. Т. 64. № 6. С. 565. https://doi.org/10.1134/S0044457X19060163
Deganello F., Marcì G., Deganello G. // J. Eur. Ceram. Soc. 2009. V. 29. P. 439. https://doi.org/10.1016/j.jeurceramsoc.2008.06.012
Mukasyan A.S., Costello C., Sherlock K.P. et al. // Sep. Purif. Technol. 2001. V. 25. P. 117. https://doi.org/10.1016/S1383-5866(01)00096-X
Tauc J., Grigorovici R., Vancu A. // Phys. Status Solidi B. 1966. V. 15. P. 627. https://doi.org/10.1002/pssb.19660150224
Righi L., Calestani G., Gemmi M. et al. // Acta Crystallogr. 2001. V. 57. P. 237. https://doi.org/10.1107/S0108768101001628
Shyamkumar S., Reshmi P.R., Muthuambika T. et al. // J. Chem. Thermodynamics. 2021. V. 155. P. 106351. https://doi.org/10.1016/j.jct.2020.106351
Gemmi M., Righi L., Calestani G. et al. // Ultramicroscopy. 2000. V. 84. P. 133. https://doi.org/10.1016/S0304-3991(00)00007-3
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии