

Журнал неорганической химии, 2021, T. 66, № 5, стр. 682-694
Влияние допантов на функциональные свойства катодных материалов с высоким содержанием лития для литий-ионных аккумуляторов
Л. С. Печень a, *, Е. В. Махонина a, А. Е. Медведева a, А. М. Румянцев b, Ю. М. Коштял c, Ю. А. Политов a, А. С. Головешкин d, И. Л. Еременко a
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
b Физико-технический институт им. А.Ф. Иоффе РАН
194021 Санкт-Петербург, ул. Политехническая, 26, Россия
c Санкт-Петербургский политехнический университет Петра Великого
195251 Санкт-Петербург, ул. Политехническая, 29, Россия
d Институт элементоорганических соединений им. А.Н. Несмеянова РАН
119334 Москва, ул. Вавилова, 28, Россия
* E-mail: lidia.s.maslennikova@gmail.com
Поступила в редакцию 02.12.2020
После доработки 07.12.2020
Принята к публикации 08.12.2020
Аннотация
Проведено сравнительное исследование влияния природы допанта на электрохимические характеристики оксидов с высоким содержанием лития общего состава 0.5Li2MnO3 ∙ 0.5LiMn0.33Ni0.33Co0.31M0.02O2 (M = Mg, Cr, Zr). Полученные материалы испытаны в качестве катода в дисковых макетах относительно лития. Результаты проведенных исследований свидетельствуют в пользу того, что основную роль в деградации материала играет миграция переходных металлов, которая зависит от энергии связи допанта с кислородом. Введение магния стабилизирует структуру оксида, замедляя процесс фазовой перестройки. К 110-му циклу в диапазоне напряжений 2.5–4.8 В при токе 100 мА/г образец с магнием сохраняет на 10% больше удельной энергии, чем исходный оксид.
ВВЕДЕНИЕ
Создание высокоэнергоемких аккумуляторов – одна из наиболее актуальных задач на сегодняшний день в связи с большим распространением электротранспорта, необходимостью иметь мощные резервные источники питания, развитием электросетей с накопителями энергии. Основной ресурс повышения удельной энергии литий-ионных аккумуляторов (ЛИА) – использование более энергоемких материалов положительного электрода (катода). Используемые в настоящее время коммерческие материалы, такие как LiCoO2, LiMn2O4, LiFePO4, LiNixCo(1 –x)O2, LiNi(x – y)Co(1 –x–y)AlyO2 и LiNixMnyCozO2, практически достигли пределов своих возможностей по удельной энергии и мощности. Недавно возникшее новое направление развития катодных материалов – сложные оксиды с избытком лития, в которых проявляет редокс-активность не только металл, но и кислород. Их использование позволяет существенно (практически двукратно) повысить удельную емкость ЛИА по сравнению с такими катодными материалами, как LiCoO2, LiMn2O4 и LiFePO4. Общая формула оксидов с высоким содержанием лития (Li-rich, Li-excess оксиды) может быть записана как Li(1 +y)M(1 –y)O2, где M – металлы с изменяющейся степенью окисления или их сочетание. Структура сложных оксидов также может быть различна: слоистая [1, 2], структура шпинели [3, 4], интегрированная слоисто-шпинельная [5, 6], разупорядоченная структура галита (каменной соли) [7, 8]. С точки зрения высокой удельной емкости наиболее привлекательны слоистые структуры. Обогащенные литием оксиды переходных металлов (Mn, Ni, Co и др.) обладают удельной разрядной емкостью 250 мА ч/г и более [9–12], что при среднем разрядном напряжении 3.6–3.7 В обеспечивает удельную энергию материала порядка 750–900 Вт ч/кг. Широко используемый материал катода LiCoO2 имеет удельную энергию ~500 Вт ч/кг.
Структура любого катодного материала вообще, и рассматриваемых сложных оксидов в особенности, оказывает большое влияние на их электрохимические свойства [11, 13]. К настоящему времени проведено большое число исследований оксидов на основе никеля, кобальта и марганца, которые были предложены первыми из обогащенных литием материалов [14–16]. Структуру таких оксидов можно рассматривать как состоящую из моноклинной (C2/m, Li2MnO3) и тригональной ($R\bar {3}m,$ LiMO2) фаз, а состав оксида может быть записан как хLi2MnO3 ∙ (1 – х)LiMO2, где M = Mn, Ni, Co и т.д. Моноклинная фаза манганата лития Li2MnO3 и слоистая тригональная фаза LiMO2 содержат слои с плотной гранецентрированной кубической упаковкой с межслоевым расстоянием ~4.7 Å между плоскостями (001) в моноклинной и (003) в тригональной фазах. Благодаря этому они могут легко интегрироваться одна в другую. Моноклинная фаза в составе таких структур стабилизирует тригональную фазу в процессе интеркаляции/деинтеркаляции лития до напряжения не выше 4.4 В [16, 17]. При заряде до более высокого напряжения (4.5–4.8 В) моноклинная структура Li2MnO3 электрохимически активируется [18, 19]. Глубокая деинтеркаляция лития по реакции Li2MnO3 = Li2O + MnO2 ведет к высоким значениям емкости и формированию при разряде (обратном внедрении лития) электрохимически активной фазы LiMnO2. Компенсация заряда происходит за счет вовлечения в обратимый редокс-процесс аниона кислорода, при этом может происходить удаление молекулярного кислорода [16, 20], формирование соединений типа перекисных (О2––O2n–) [10, 21–23] или образование кислородных вакансий [16, 19]. Процесс более глубокого окисления-восстановления катионов переходного металла (ПМ), как правило, исключают, поскольку существование Mn5+ (наиболее вероятного кандидата на дальнейшее окисление) маловероятно в октаэдрических позициях.
Несмотря на большое число исследований структур оксидов с высоким содержанием лития в последнее десятилетие, для них остается ряд нерешенных проблем. Гистерезис напряжений заряда/разряда и падение емкости и напряжения при циклировании, необратимая емкость в первых циклах заряда/разряда за счет активации моноклинной фазы, сопровождающейся фазовой перестройкой с возможным выделением кислорода, что небезопасно при эксплуатации, являются основными препятствиями на пути коммерциализации данного типа катодных материалов. Наиболее дискутируемыми вопросами являются механизм анионного окисления, его связь с миграцией катионов и причины структурной перестройки сложных оксидов в процессе циклирования.
Ранее нами было показано, что на стабильность кристаллической структуры и сохранение емкости в процессе длительного циклирования оксидов состава xLi2MnO3 ∙ (1 – x)LiMn1/3Ni1/3Co1/3O2 значительно влияют как метод синтеза и морфология материала, в том числе наличие дефектов [24, 25], так и фазовый состав обогащенных литием оксидов [26]. Введение допирующих добавок в позиции переходных металлов может изменять электронную структуру оксидов, что, в свою очередь, будет влиять на функциональные свойства катодов на их основе.
В настоящей работе исследовано влияние на свойства оксида состава xLi2MnO3 ∙ (1 – ‒ x)LiMn1/3Ni1/3Co1/3O2 допантов магния, хрома и циркония (2 мол. %), введенных в позиции катионов ПМ. Синтезированные оксиды охарактеризованы методами сканирующей электронной микроскопии (СЭМ), масс-спектрометрии с индуктивно-связанной плазмой (ICP-MS), рентгенофазового анализа (РФА), рентгеновской фотоэлектронной спектроскопии (РФЭС) и гранулометрического анализа с использованием статического рассеяния света. Исследованы стабильность циклирования и скоростные характеристики катодов на основе синтезированных оксидов в дисковых макетах относительно лития. Обнаружено существенное различие допированных образцов в отношении фазового перехода слоистой в шпинелеподобную фазу и циклировании высокими токами.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Оксиды с высоким содержанием лития получали методом соосаждения с последующей твердофазной реакцией по методике, подробно описанной ранее [24]. В качестве исходных реагентов при синтезе использовали нитраты переходных металлов Ni(NO3)2 · 6H2O (99.9%, ABCR), Co(NO3)2 · 6H2O (99%, Acros Organics) и Mn(NO3)2 · 4H2O (98%, Alfa Aesar), гидроксид лития LiOH · H2O (99+%, Sigma-Aldrich). В качестве осадителя применяли карбонат натрия Na2CO3 (х. ч.), ХИММЕД с добавлением гидроксида аммония NH4OH (ос. ч.), ХИММЕД. При допировании основного состава другими катионами дополнительно использовали нитрат магния Mg(NO3)2 · 6H2O (ч. д. а.), ХИММЕД, нитрат хрома Cr(NO3)2 · 9H2O (99%, Acros Organics) и нитрат цирконила ZrO(NO3)2 · 2H2O (ч. д. а.), ХИММЕД.
При синтезе исходного оксида сравнения (обозначение LR) и допированных образцов (обозначения LR-Mg, LR-Cr, LR-Zr) методом соосаждения карбонатных прекурсоров предварительно готовили смешанный водный раствор нитратов никеля, кобальта, марганца (при допировании добавляли соль допирующего элемента) с общей концентрацией 2 моль/л и раствор осадителя – Na2CO3 (2 моль/л) с добавкой NH4OH (0.3 моль/л). Концентрация вводимого допанта составляла 2 мол. % по отношению к сумме ПМ за счет уменьшения концентрации кобальта. Растворы с помощью перистальтических насосов автоматически подавали в стеклянный реактор с использованием специально разработанной программы для поддержания рН, температуры синтеза и скорости перемешивания. Осаждение карбонатного прекурсора проводили при подобранных ранее условиях в атмосфере углекислого газа [24, 25].
При расчете добавляемого лития на стадии твердофазной реакции образца LR-Cr учитывали, что карбонат хрома подвергается быстрому гидролизу до гидроксида хрома, а при синтезе образца LR-Zr – тот факт, что использование нитрата цирконила ведет к образованию ZrO2.
Полученные осадки промывали большим количеством (5–7 л) деионизированной воды, ацетоном и диэтиловым эфиром и сушили в токе аргона при температуре 110°С в течение 16–20 ч. Высушенные прекурсоры смешивали с гидроксидом лития и тщательно гомогенизировали, перетирая в агатовой ступке в спиртовой среде, после чего отжигали в муфельной печи на воздухе при температуре 480°С в течение 6 ч. Затем образец снова тщательно перетирали и отжигали при температуре 900°С в течение 12 ч, в процессе высокотемпературного отжига смеси неоднократно гомогенизировали.
МЕТОДЫ ИССЛЕДОВАНИЯ
Морфологию и микроструктуру, а также равномерность распределения элементов по составу полученных оксидов изучали с помощью СЭМ и рентгеноспектрального микроанализа (РСМА) на приборе NVision-40 (Carl Zeiss).
Измерение распределения частиц по размерам проводили с использованием лазерного анализатора Analysette 22 MicroTec Plus в соответствии с процедурой, описанной в работах [24, 25]. Содержание металлов в полученных образцах определяли методом ICP-MS на приборе Agilent 7500ce (Agilent Technologies Inc.).
Химическое состояние металлов определяли методом РФЭС на приборе PH I5500 Versa Probe II. Источник возбуждения – монохроматизированное AlKα-излучение (hν = 1486.6 эВ), мощность – 50 Вт, диаметр – 200 мкм. Область анализа составляла 800 × 400 мкм2. Энергию связи (Есв) фотоэлектронных линий определяли по спектрам высокого разрешения C1s, O1s, Mn3s, снятым при энергии пропускания анализатора 23.5 эВ и плотности сбора данных 0.2 эВ/шаг, и спектрам Mn2p, Co2p3 и Ni2p3, Zr3d, а также Cr2p, Mg1s, снятым при 29.35 и 0.25 эВ/шаг. Аппроксимацию спектров выполняли нелинейным методом наименьших квадратов с использованием функции Гаусса–Лоренца с добавлением (или без добавления) асимметрии. Калибровка шкалы энергии связи проведена по Au4f – 83.96 эВ и Cu2p3 – 932.6 эВ. Шкалу энергии связи корректировали по энергии связи спектра С1s адсорбированных углеводородов (284.9 эВ).
Дифрактометрические исследования проводили при комнатной температуре на рентгеновском дифрактометре Bruker D8 Advance (CuKα-излучение, λ = 0.15418 нм, 40 кВт/40 мА) в режиме θ/2θ-сканирования в интервале углов 2θ 5°–90° с шагом 0.02°. Съемку осуществляли в геометрии Брэгга–Брентано, образец наносили на кремниевый прободержатель (без фона) в виде суспензии в гексане. Для повышения точности измерения параметров элементарной ячейки съемку образцов осуществляли в широком угловом диапазоне с добавлением внутреннего стандарта (высококристаллический кремний). РФА проводили также на порошковом рентгеновском дифрактометре Bruker D8 Advance Vario (CuKα1-излучение) с Ge-монохроматором и позиционно-чувствительным детектором LynxEye (θ/2θ-геометрия) с вращением.
Сбор данных осуществляли с помощью программного комплекса Bruker DIFFRACplus, анализ – с помощью программ EVA, TOPAS. Методом Ритвельда проводили уточнение дифрактограмм, что позволило оценить соотношение фаз в обогащенных литием оксидах. Уширение линий было описано методом Вильямсона–Холла. Анизотропное уширение пиков моноклинной формы проводилось с помощью модели Стефенса.
Определение электрохимической активности полученных катодных материалов осуществляли в дисковых макетах габарита CR2032 с использованием зарядно-разрядных стендов Neware CT3008W-5V10mA. Электродный слой состоял из исследуемого катодного материала (92 мас. %), электропроводящей добавки (сажа Super C65 (Timcal) – 5 мас. %) и связующего (поливинилиденфторид Solef 5130 (Solvay) – 3 мас. %). Для изготовления электрода в сухую смесь катодного вещества добавляли связующее, растворенное в н-метилпирролидоне (СТП ТУ КОМП 2-207-10, х. ч., Компонент реактив). Полученную активную массу тщательно перемешивали и наносили на алюминиевую фольгу с применением намазочной машины (AFA 1, MTI) и приспособления Dr Blade (зазор 100 мкм). Затем фольгу с нанесенным активным слоем сушили на воздухе при температуре 60°С, вырубали диски электродов и сушили их под вакуумом при температуре 110°С, затем без контакта с атмосферой переносили в камеру перчаточного бокса Omni-Lab (VAC) для последующей сборки дисковых макетов CR 2032 в атмосфере сухого аргона (ГОСТ 10157-20160, ЗАО “НУЗ”). В качестве отрицательного электрода, сепаратора и электролита применяли литиевую фольгу, два слоя Celgard 2325 и TC-E918 (Tinci) соответственно.
После сборки дисковых макетов при малых токах ~20 мА/г (<С/10) проводили циклы формировки: два цикла заряда/разряда в диапазоне напряжений 2.5–4.2 В, два цикла в диапазоне 2.5–4.4 В, за которыми следовала электрохимическая активация моноклинной фазы в диапазоне напряжений 2.5–4.8 В. Величина тока при гальваностатическом циклическом заряде/разряде составляла 80–100 мА/г. Работоспособность материалов оценивали также при высоких токах разряда в диапазоне 10–500 мА/г, ток заряда был постоянным – 80–100 мА/г.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
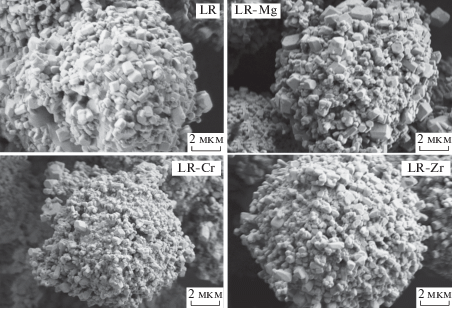
Полученные катодные материалы обладают схожей микроструктурой и представляют собой шарообразные агломераты, состоящие из первичных частиц, размер которых находится в основном в диапазоне 250–750 нм, но может достигать 1.5–3 мкм (рис. 1).
По результатам гранулометрического анализа с использованием статического рассеяния света, все синтезированные образцы имеют узкое одномодальное распределение агломератов по размерам (рис. 2).
Рис. 2.
Дифференциальные (а) и интегральные (б) кривые распределения агломератов по размерам для оксидов.
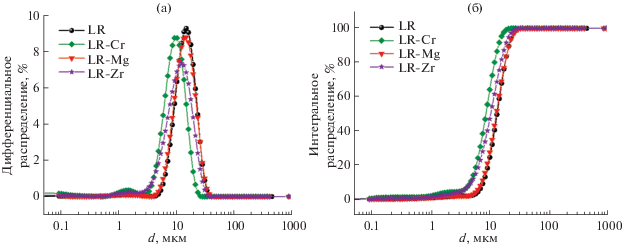
Образец сравнения LR (D50 = 13.1 мкм, D90 = = 20.9 мкм) и образец, допированный магнием, LR-Mg (D50 = 12.8 мкм, D90 = 20.9 мкм) имеют близкие параметры распределения. Допирование оксида хромом (D50 = 8.5 мкм, D90 = 13.9 мкм) и цирконием (D50 = 10.1 мкм, D90 = 18.0 мкм) приводит к смещению распределения агломератов в сторону меньших значений.
По данным РСМА, переходные металлы и допанты равномерно распределены по объему агломератов (рис. 3).
Рис. 3.
Карты распределения элементов, полученные с использованием РСМА образцов LR-Mg, LR-Cr и LR-Zr.

В соответствии с результатами химического анализа методом ICP-MS, концентрации элементов в синтезированных оксидах близки к заложенным при синтезе соотношениям (табл. 1).
Таблица 1.
Состав полученных оксидов по данным ICP-MS анализа в сравнении с заложенным составом
| Образец | Состав по данным ICP-MS | Заложенный состав |
|---|---|---|
| LR | Li1.22Mn0.529Ni0.125Co0.126O2 | Li1.200Mn0.540Ni0.130Co0.130O2 |
| LR-Mg | Li1.20Mn0.537Ni0.124Co0.119Mg0.02O2 | Li1.200Mn0.540Ni0.130Co0.114Mg0.016O2 |
| LR-Cr | Li1.22Mn0.527Ni0.127Co0.110Cr0.014O2 | Li1.200Mn0.540Ni0.130Co0.114Cr0.016O2 |
| LR-Zr | Li1.21Mn0.542Ni0.130Co0.107Zr0.011O2 | Li1.200Mn0.540Ni0.130Co0.114Zr0.016O2 |
Таким образом, в процессе синтеза получены допированные образцы катодных материалов 0.5Li2MnO3∙ 0.5LiMn1/3Ni1/3Co1/3 –zMzO2 заданного состава с равномерным распределением всех элементов по объему частиц.
Дифрактограммы полученных оксидных материалов схожи, сильных различий в уширении пиков и их положении не наблюдается (рис. 4). Наиболее интенсивные пики отвечают тригональной структуре с пр. гр. $R\bar {3}m.$ Пики низкой интенсивности в интервале углов 20°–30° 2θ (приведены для каждого образца во вставке) характеризуют наличие моноклинной структуры с пр. гр. C2/m в образцах катодных материалов. Уточнение дифрактограмм методом Ритвельда позволило установить соотношение фаз в образцах литированных оксидов. По результатам этой оценки, содержание моноклинной фазы в образцах составляет ~25–35% (табл. 2).
Рис. 4.
Теоретические (красные линии) и экспериментальные (синие линии) дифрактограммы образцов и их разность (серые линии). Синие штрихи обозначают расчетные положения пиков моноклинной фазы, черные – тригональной.
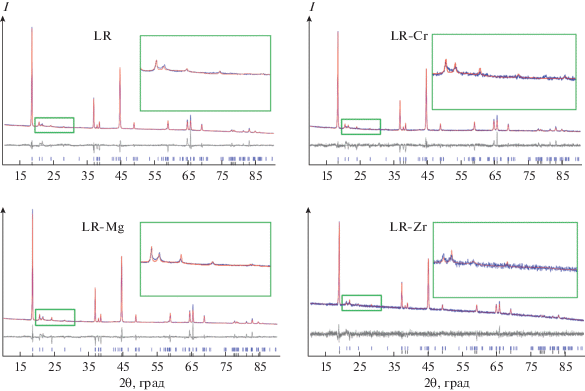
Таблица 2.
Параметры элементарной ячейки и процентное содержание моноклинной фазы в образцах
| Параметр | LR | LR-Mg | LR-Cr | LR-Zr |
|---|---|---|---|---|
| a, Å | 2.8477(3) | 2.8491(4) | 2.8483(4) | 2.8504(7) |
| c, Å | 14.223(4) | 14.227(5) | 14.222(5) | 14.234(9) |
| V, Å3 | 99.888(4) | 100.011(4) | 99.927(5) | 100.154(8) |
| фаза C2/m, % | 29 | 32 | 26 | 34 |
По зависимости уширения пиков от дифракционного угла для тригональной формы можно заключить, что наибольший вклад в рамках метода Вильямсона–Холла на уширение пиков вносит наличие микронапряжений в образце, тогда как размер кристаллитов вносит меньший вклад и составляет >200 нм.
Пики моноклинной фазы имеют сильное анизотропное уширение и перекрываются с пиками тригональной модификации, причем вклад в интенсивность перекрывающихся пиков гораздо выше у тригональной модификации. Поэтому определение размера кристаллитов и параметров ячейки моноклинной фазы может содержать систематические погрешности, исходя из этого, сопоставлять эти параметры между разными образцами достаточно проблематично. Достоверно удалось установить только параметры ячейки тригональной модификации во всех образцах, их значения приведены в табл. 2.
Для всех допированных образцов параметры ячейки больше, чем для образца LR, что подтверждает включение допирующих катионов с большими ионными радиусами в состав тригональной фазы.
Методом РФЭС определена энергия связей фотоэлектронных линий элементов по спектрам высокого разрешения для всех оксидов, данные приведены в табл. 3.
Таблица 3.
Энергия связи в синтезированных катодных материалах
| Образец | Энергии связи, эВ | ΔMn3s, эВ/q ±0.05 эВ |
|||||
|---|---|---|---|---|---|---|---|
| C1s | Mn2p3 ±0.30 эВ |
Co2p3 ± 0.2 эВ/ ПШПВ | Ni2p3 ± 0.2 эВ/ ПШПВ | O1s | |||
| пик 1 ±0.1 эВ |
пик 1 ±0.2 эВ |
пик 2 ±0.2 эВ |
|||||
| LR | 284.9 | 642.0 | 779.8/1.2 | 854.6/1.7 | 529.1 | 531.0 | 4.54; q = +4.0 |
| LR-Mg | 284.9 | 642.0 | 779.9/1.3 | 854.8/1.9 | 529.1 | 531.3 | 4.25; q = +4.0 |
| LR-Cr | 284.9 | 642.0 | 779.9/1.3 | 854.8/2.0 | 529.1 | 531.3 | 4.32; q = +4.0 |
| LR-Zr | 284.9 | 642.0 | 779.9/1.3 | 854.8/2.0 | 529.1 | 531.3 | 4.50; q = +4.0 |
Энергия связи Mn2p3 для всех образцов одинакова и составляет 642.0 эВ, что типично для иона Mn4+. Однако величина энергетического расстояния между высокоспиновым и низкоспиновым компонентами в Mn3s в фотоэлектронных спектрах ΔMn3s различается для образцов (рис. 5). Зависимость ΔMn3s от числа неспаренных 3d-электронов (степени окисления) на атоме марганца носит линейный характер [27] и дает возможность определения формальной валентности марганца в исследуемых оксидах.
В случае недопированного образца LR и образца с цирконием LR-Zr величины ΔMn3s составляют 4.54 и 4.50 эВ, что соответствует заряду иона марганца q = +4 [27], а в образцах LR-Mg и LR-Cr эти значения равны соответственно 4.25 и 4.32 эВ. Вместе с тем линейную зависимость ΔMn3s от числа неспаренных 3d-электронов нельзя использовать напрямую для определения степени окисления в сложных системах [28], а наблюдаемые отклонения невелики, что позволяет предполагать в совокупности с данными энергии связи Mn2p3 в этих образцах четырехвалентное состояние марганца. Энергия связи Co2p3 для всех образцов находится в диапазоне значений 779.8–779.9 эВ. Все спектры Co2p3 имеют одинаковую форму – достаточно узкий асимметричный пик и сателлит, сдвинутый на +9.7 эВ с интенсивностью 7–9%, что характерно для Co3+ [29, 30]. Спектры Ni2p для всех образцов подобны по форме – широкий пик и сильный сателлит, отстоящий на 6.5 эВ. Полученные значения энергии связи Ni2p3 соответствуют заряду иона 2+ [29, 30].
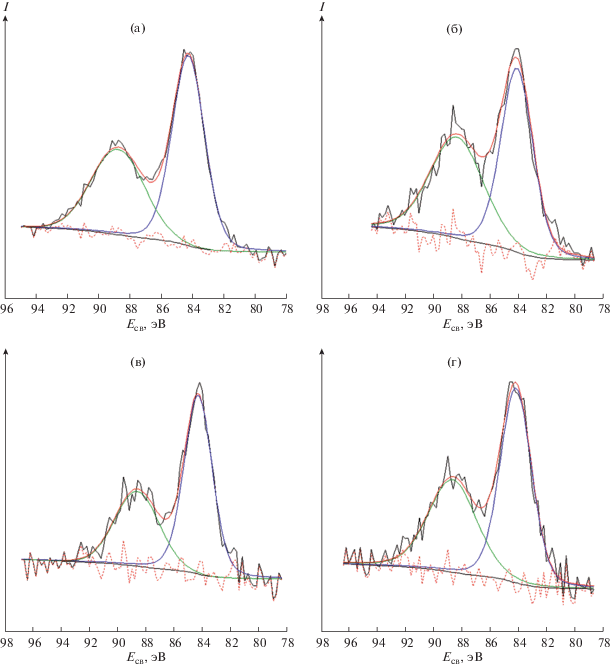
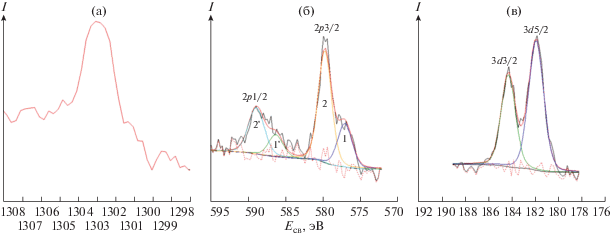
Энергия связи Mg1s в образце LR-Mg составила 1302.9 эВ (рис. 6а), эта величина характерна для зарядового состояния ионов магния 2+ [31]. Энергия связи Zr3d, равная 182.0 эВ (рис. 6б), соответствует Zr4+ в образце LR-Zr. В спектре Cr2p3 образца LR-Cr обнаружено два дублета (рис. 6в): дублет 1-1' с Есв(Cr2p3/2) = 577.0 эВ (30%) и дублет 2-2' с Есв(Cr2p3/2) = 579.7 эВ (70%). Последний приписывают Cr6+ [30, 31]. По данным [32], медленное окисление гидроксида хрома до шестивалентного состояния возможно при температурном отжиге в анаэробной атмосфере, а также в присутствии MnO2 в водной среде как при наличии воздуха, так и без него. Данные РФЭС свидетельствуют в пользу такого сценария.
Рис. 6.
Фотоэлектронные спектры Mg1s образца LR-Mg (а), Cr2p образца LR-Cr – пики 1 и 2 – 2p3/2, пики 1' и 2' – 2p1/2 (б), и Zr3d образца LR-Zr (в).
Зарядно-разрядные кривые первого цикла гальваностатического циклирования до высокого напряжения 4.8 В при токе 20 мА/г (циклы формировки) представлены на рис. 7а, соответствующие им дифференциальные кривые емкости по напряжению – на рис. 7б.
Рис. 7.
Зарядно-разрядные кривые на первом цикле циклирования до высокого напряжения (а) и первая производная емкости по напряжению от напряжения на том же цикле (б) (диапазон напряжений 2.5–4.8 В, удельный ток 20 мА/г; пик при 4.5 В не показан).
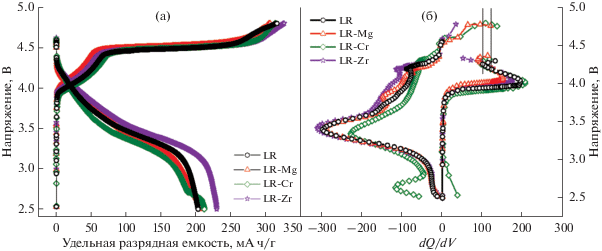
На зарядных кривых наблюдается два плато, одно из которых соответствует деинтеркаляции лития из тригональной фазы, сопровождающейся окислением никеля и кобальта; ему соответствует пик при 4.0–4.05 В на дифференциальной кривой. Положение пика несколько смещается в зависимости от образца. Второе длинное плато при 4.5 В соответствует дальнейшему удалению лития, связанному с редокс-процессом с участием кислорода [12] (на рисунке пик на дифференциальной кривой не показан). Разрядные кривые сглажены, а дифференциальные разрядные кривые характеризуются присутствием обратного катодного пика при 3.4 В, соответствующего восстановлению никеля и кобальта.
Значительно отличается поведение образца, допированного хромом. Уже в первом цикле на разрядной кривой для него наблюдается небольшое плато в районе 2.6 В, необычное для структур данного типа. На дифференциальной кривой этому плато соответствует пик, который не может быть отнесен к появлению в системе шпинели или шпинелеподобной структуры, для которых характерен пик при 2.7–2.9 В [33, 34]. Авторы работ [35, 36] также наблюдали наличие катионов хрома в степени окисления +6 в подобных оксидах. В работе [36] его наличие трактуют как результат процесса окисления первоначально присутствующего трехвалентного хрома или как возможное наличие обратимо работающей фазы Li2CrO4. Однако в нашем случае Cr6+ присутствует в исходном соединении, как и в работе [35], авторами которой этот факт не комментируется. Возможно, в данном диапазоне напряжений при разряде происходит восстановление Cr6+ до более низких степеней окисления [37]. Следует отметить, что обратного пика окисления в нашем случае мы также не видим, а пик при 2.6 В постепенно исчезает в процессе циклирования. Такое поведение образца, допированного хромом, несмотря на первоначально большие значения зарядной и разрядной емкости (табл. 4), должно приводить к постепенному ухудшению циклируемости образца.
Таблица 4.
Сравнение удельной емкости, величин необратимой емкости в первом цикле до 4.8 В и теоретических значений разрядной емкости оксидов
| Образец | Зарядная емкость | Разрядная емкость | Необратимая емкость | Теоретическая емкость (на 1 ион лития) |
|---|---|---|---|---|
| 1 цикл 2.5–4.8 В | ||||
| LR | 314 | 203 | 111 | 250 |
| LR-Mg | 307 | 203 | 104 | 254 |
| LR-Cr | 312 | 212 | 100 | 250 |
| LR-Zr | 323 | 230 | 93 | 251 |
Приведенные значения (табл. 4) близки для всех оксидов; наибольшей емкостью и меньшей необратимостью первого цикла обладает образец, допированный цирконием.
В зависимости от условий заряда/разряда снижается не только разрядная емкость, но и среднее разрядное напряжение, в связи с этим для оценки работоспособности целесообразно использовать изменение удельной энергии, включающей оба параметра. Поэтому результаты гальваностатического циклирования образцов в диапазоне напряжений 2.5–4.8 В (рис. 8) приведены в единицах отдаваемой при разряде энергии.
Рис. 8.
Удельная энергия, отдаваемая материалом при разряде в диапазоне 2.5–4.8 В, в зависимости от номера цикла (а) и удельного тока разряда (б).
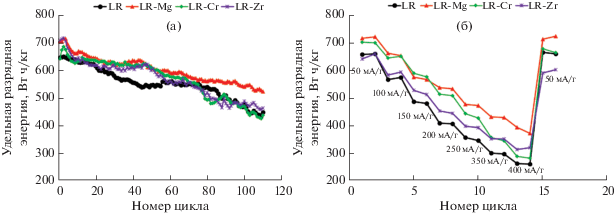
О ресурсе работы можно судить по величине сохранения энергии в результате проведения фиксированного числа циклов. Введение хрома и циркония практически не влияет на работоспособность катодных материалов. С другой стороны, площадь под кривой зависимости удельной энергии от числа циклов для этих образцов выше, чем для исходного образца. Это свидетельствует о большей величине запасаемой и отдаваемой энергии за фиксированное число циклов заряд/разряд. Вместе с тем отметим, что образец LR-Cr имеет самый большой процент падения по сравнению с остальными образцами, что согласуется с вышеприведенным предположением о необратимости восстановления хрома в процессе интеркаляции лития.
Образец, допированный магнием, демонстрирует более высокие значения удельной энергии, чем все остальные (рис. 8а); для него к 110-му циклу сохраняется почти 80% первоначальной энергии, и зависимость удельной энергии от числа циклов располагается выше, чем у других образцов. Этот же образец показывает лучшие характеристики при разряде высокими токами (рис. 8б). По сравнению с исходным образцом лучшую циклируемость при высоких скоростях показывают все допированные образцы, что, скорее всего, связано с большим ионным радиусом допантов.
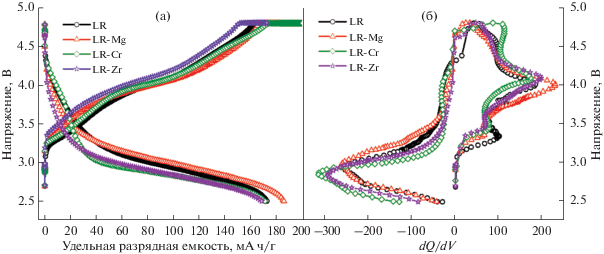
Кривые заряда-разряда на 80-м цикле в том же диапазоне напряжений для всех образцов приведены на рис. 9а. Видно, что прогиб разрядной кривой имеется в районе 3.0 В, который соответствует переходу в шпинельную фазу, больше всего для образцов, допированных хромом и цирконием; средние разрядные напряжения для этих образцов составляют 2.98 и 2.96 В соответственно. Образцы LR и LR-Mg показывают большие значения, около 3.10 В. При этом образец, допированный магнием, сохраняет значительно большую разрядную емкость. Кривые первой производной разрядной емкости по напряжению, соответствующие окислительно-восстановительным потенциалам в процессе электрохимической работы ячейки, приведены на рис. 9б. Катодный пик в районе 2.7 В подтверждает образование шпинельной фазы [33, 34] во всех оксидах, однако смещение в сторону низких напряжений для образцов с хромом и цирконием больше, чем для образца с магнием.
Рис. 9.
Зарядно-разрядные кривые на 80-м цикле (а) и первая производная емкости по напряжению от напряжения на 80-м цикле заряда-разряда (б) (диапазон напряжений 2.5–4.8 В, удельный ток 100 мА/г).
При выборе допантов для исследования мы руководствовались тем, что процесс миграции ПМ рассматривается как основной механизм, ведущий к формированию шпинели или шпинелеподобной структуры [11, 12], что приводит к снижению среднего разрядного напряжения и разрядной емкости материала. Для подтверждения этого предположения выбраны элементы, энергия связи с кислородом у которых больше (хром, цирконий) и меньше (магний) по сравнению с Mn, Ni, Co (табл. 5) при сравнимых ионных радиусах. Большая энергия связи должна ослаблять связь ПМ с кислородом и способствовать их миграции в тетраэдрические позиции лития, тем самым ускоряя фазовый переход материала катода. Действительно, введение магния ведет к замедлению фазового перехода и стабилизации структуры образца LR-Mg.
Таблица 5.
Характеристики выбранных допантов, входящих в состав синтезируемых оксидов, по сравнению с Mn, Ni, Co и Li
| Ион | Ионный радиус, Å | КЧ | Энергия связи с кислородом, кДж/моль |
|---|---|---|---|
| Co3+ | 0.545 | 6 | 397.4 ± 8.7 |
| Ni2+ | 0.69 | 6 | 366.0 ± 30.0 |
| Mn4+ | 0.530 | 6 | 362.0 ± 25.0 |
| Zr4+ | 0.720 | 6 | 766.1 ± 10.6 |
| Mg2+ | 0.720 | 6 | 358.2 ± 7.2 |
| Cr3+ | 0.615 | 6 | 461.0 ± 8.7 |
| Li+ | 0.760 | 6 | 340.5 ± 6.3 |
Таким образом, различия в поведении допированных образцов обусловлены разной величиной их энергии связи с кислородом. В случае магния не исключена также роль вакансий кислорода, которые могут возникать при введении двухзарядного катиона Mg и, по данным [38, 39], улучшать электрохимические показатели обогащенных литием оксидов.
ЗАКЛЮЧЕНИЕ
Проведено сравнительное исследование влияния магния, хрома и циркония на электрохимические характеристики оксида состава 0.5Li2MnO3 ∙ 0.5LiMn0.33Ni0.33Co0.31M0.02O2 (М – допант) с высоким содержанием лития. Оксиды синтезированы методом соосаждения в виде шарообразных агломератов с размером первичных частиц 250–750 нм. Составы полученных материалов близки к заложенным при синтезе и характеризуются равномерным распределением элементов. Дифрактограммы оксидов идентифицированы на основе двух фаз: тригональной (пр. гр. $R\bar {3}m$) и моноклинной (пр. гр. C2/m). По данным РФА, доля моноклинной фазы варьируется от 25 до 35%. Все допированные образцы имеют лучшие скоростные характеристики, чем исходный образец, что, по-видимому, связано с большим ионным радиусом элементов по сравнению с радиусами ПМ. Среди допированных материалов образец с магнием показал лучшие результаты как при высоких токах, так и в ресурсных испытаниях в диапазоне напряжений 2.5–4.8 В. Анализ электрохимических данных показал, что введение магния замедляет процесс фазового перехода слоистой структуры оксида в шпинельную, появление которой ведет к снижению удельной емкости, напряжения и в итоге удельной энергии материала. Различие в поведении допированных образцов обусловлено разной величиной энергии связи допанта с кислородом.
Список литературы
Ates M.N., Mukerjee S., Abraham K.M. // J. Electrochem. Soc. 2014. V. 161. № 3. P. A355. https://doi.org/10.1149/2.070403jes
Robert R., Villevieille C., Novák P. // J. Mater. Chem. A. 2014. V. 2. № 23. P. 8589. https://doi.org/10.1039/c3ta12643a
Liddle B.J., Collins S.M., Bartlett B.M. // Energy Environ. Sci. 2010. V. 3. № 9. P. 1339. https://doi.org/10.1039/c0ee00059k
Buzanov G.A., Simonenko N.P., Zhizhin K.Y. et al. // Russ. J. Inorg. Chem. 2019. V. 64. № 12. P. 1482. [Бузанов А.Г., Симоненко Н.П., Жижин К.Ю. и др. // Журн. неорган. химии. 2019. Т. 64. № 12. С. 1246.]https://doi.org/10.1134/S0036023619120040
Feng X., Yang Z., Tang D. et al. // Phys. Chem. Chem. Phys. 2015. V. 17. № 2. P. 1257. https://doi.org/10.1039/C4CP04087B
Long B.R., Croy J.R., Park J.S. et al. // J. Electrochem. Soc. 2014. V. 161. № 14. P. A2160. https://doi.org/10.1149/2.0681414jes
Wang R., Li X., Liu L. et al. // Electrochem. Commun. 2015. V. 60. P. 70. https://doi.org/10.1016/j.elecom.2015.08.003
Yu Z., Qu X., Dou A. et al. // ACS Appl. Mater. Interfaces. 2019. V. 11. № 39. P. 35777. https://doi.org/10.1021/acsami.9b12822
Ohzuku T., Nagayama M., Tsuji K. et al. // J. Mater. Chem. 2011. V. 21. № 27. P. 10179. https://doi.org/10.1039/c0jm04325g
Rozier P., Tarascon J.M. // J. Electrochem. Soc. 2015. V. 162. № 14. P. A2490. https://doi.org/10.1149/2.0111514jes
Manthiram A., Knight J.C., Myung S.-T. et al. // Adv. Energy Mater. 2016. V. 6. № 1. P. 1501010. https://doi.org/10.1002/aenm.201501010
Wang J., He X., Paillard E. et al. // Adv. Energy Mater. 2016. V. 6. № 21. P. 1600906. https://doi.org/10.1002/aenm.201600906
Nipan G.D., Smirnova M.N., Kornilov D.Y. et al. // Russ. J. Inorg. Chem. 2020. V. 65. № 4. P. 573. [Нипан Г.Д., Смирнова М.Н., Корнилов Д.Ю. и др. // Журн. неорган. химии. 2020. Т. 65. № 4. С. 534.]https://doi.org/10.1134/S0036023620040130
Lu Z., MacNeil D.D., Dahn J.R. // Electrochem. Solid-State Lett. 2001. V. 4. № 11. P. A191. https://doi.org/10.1149/1.1407994
Lu Z., Beaulieu L.Y., Donaberger R.A. et al. // J. Electrochem. Soc. 2002. V. 149. № 6. P. A778. https://doi.org/10.1149/1.1471541
Thackeray M.M., Kang S.-H., Johnson C.S. et al. // J. Mater. Chem. 2007. V. 17. № 30. P. 3112. https://doi.org/10.1039/b702425h
Johnson C.S., Li N., Lefief C. et al. // Electrochem. Commun. 2007. V. 9. № 4. P. 787. https://doi.org/10.1016/j.elecom.2006.11.006
Robertson A.D., Bruce P.G. // Chem. Mater. 2003. V. 15. № 10. P. 1984. https://doi.org/10.1021/cm030047u
Armstrong A.R., Holzapfel M., Nová P. et al. // JACS 2006. V. 128. № 26. P. 8694. https://doi.org/10.1021/ja062027
Fell C.R., Qian D., Carroll K.J. et al. // Chem. Mater. 2013. V. 25. № 9. P. 1621. https://doi.org/10.1021/cm4000119
Sathiya M., Rousse G., Ramesha K. et al. // Nat. Mater. 2013. V. 12. № 9. P. 827. https://doi.org/10.1038/nmat3699
Assat G., Iadecola A., Delacourt C. et al. // Chem. Mater. 2017. V. 29. № 22. P. 9714. https://doi.org/10.1021/acs.chemmater.7b03434
McCalla E., Abakumov A.M., Saubanere M. et al. // Science. 2015. V. 350. № 6267. P. 1516. https://doi.org/10.1126/science.aac8260
Pechen L.S., Makhonina E.V., Rumyantsev A.M. et al. // Russ. J. Inorg. Chem. 2018. V. 63. № 12. P. 1534. [Печень Л.С., Махонина Е.В., Румянцев А.М. и др. // Журн. неорган. химии. 2018. Т. 63. № 12. С. 1522.]https://doi.org/10.1134/S0036023618120173
Makhonina Е.V., Pechen L.S., Volkov V.V. et al. // Russ. Chem. Bull. 2019. V. 68. № 2. P. 301. [Махонина Е.В., Печень Л.С., Волков В.В. и др. // Изв. АН. Сер. химическая. 2019. С. 301.]https://doi.org/10.1007/s11172-019-2386-6
Pechen L.S., Makhonina E.V., Rumyantsev A.M. et al. // Russ. Chem. Bull. 2019. V. 68. № 2. P. 293. [Печень Л.С., Махонина Е.В., Румянцев А.М. и др. // Изв. АН. Сер. химическая. 2019. № 2. С. 293.]https://doi.org/10.1007/s11172-019-2385-7
Qi-Hui W., Thissen A., Jaegermann W. // Chin. Phys. Lett. 2006. V. 23. № 8. P. 2202. https://doi.org/10.1088/0256-307X/23/8/066
Kochur A.G., Ivanova T.M., Shchukarev A.V. et al. // Bull. Russ. Acad. Sci. Phys. 2010. V. 74. № 5. P. 625. https://doi.org/10.3103/S1062873810050126
Moses A.W., Flores H.G.G., Kim J.-G. et al. // Appl. Surf. Sci. 2007. V. 253. № 10. P. 4782. https://doi.org/10.1016/j.apsusc.2006.10.044
Biesinger M.C., Payne B.P., Grosvenor A.P. et al. // Appl. Surf. Sci. 2011. V. 257. № 7. P. 2717. https://doi.org/10.1016/j.apsusc.2010.10.051
https://srdata.nist.gov/xps/main_search_menu.aspx
Apte A., Tare V., Bose P. // J. Hazard. Mater. 2006. V. 128. № 2–3. P. 164. https://doi.org/10.1016/j.jhazmat.2005.07.057
Armstrong A.R., Dupre N., Paterson A.J. et al. // Chem. Mater. 2004. V. 16. № 16. P. 3106. https://doi.org/10.1021/cm034964b
Thackeray M.M. // Prog. Solid State Chem. 1997. V. 25. № 1–2. P. 1. https://doi.org/10.1016/S0079-6786(97)81003-5
Nisar U., Amin R., Shakoor A. et al. // Emergent Mater. 2018. V. 1. № 3–4. P. 155. https://doi.org/10.1007/s42247-018-0014-0
Mangani I.R., Park C.W., Yoon Y.K. et al. // J. Electrochem. Soc. 2007. V. 154. № 4. P. A359. https://doi.org/10.1149/1.2509096
Wang W., Meng J., Yue X. et al. // Chem. Commun. 2018. V. 54. № 98. P. 13809. https://doi.org/10.1039/C8CC07660J
Sun Y., Cong H., Zan L. et al. // ACS Appl. Mater. Interfaces 2017. V. 9. № 44. P. 38545. https://doi.org/10.1021/acsami.7b12080
Huang Z., Xiong T., Lin X. et al. // J. Power Sources. 2019. V. 432. P. 8. https://doi.org/10.1016/j.jpowsour.2019.05.069
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии