

Журнал неорганической химии, 2021, T. 66, № 2, стр. 282-290
Влияние строения производных перилена на их взаимодействие с монослоями восстановленного оксида графена
А. И. Звягина a, Е. А. Гусарова a, А. А. Аверин a, М. А. Калинина a, *
a Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Ленинский пр-т, 31, корп. 4, Россия
* E-mail: kalinina@phyche.ac.ru
Поступила в редакцию 26.03.2020
После доработки 23.04.2020
Принята к публикации 30.04.2020
Аннотация
Выявление факторов, влияющих на перенос энергии в гибридных системах на основе органических сенсибилизаторов и двумерных углеродных материалов, представляет собой одну из актуальных проблем физикохимии тонких пленок, решение которой позволит рационализировать дизайн оптоэлектронных устройств нового типа. С помощью спектроскопии комбинационного рассеяния (КР) установлена роль заместителей в молекуле перилена в переносе энергии между хромофором и монослоем восстановленного оксида графена (ОГ). Показано, что адсорбция производного перилена с плоской геометрией на восстановленном ОГ приводит к тушению флуоресценции и усилению КР хромофора. В то же время при иммобилизации “плоского” хромофора на чистом кремнии или перилена с изопентильными заместителями на монослое восстановленного ОГ не удается зарегистрировать КР-полосы перилена несмотря на присутствие его пиков флуоресценции в спектрах. Эффект усиленного поверхностью КР обусловлен сильными взаимодействиями между незамещенным хромофором и восстановленным ОГ за счет ароматического стекинга, обеспечивающими эффективный перенос энергии между компонентами системы. Взаимодействия между замещенным хромофором и углеродным каркасом ослаблены из-за стерических затруднений, что выражается в отсутствии эффекта усиления КР.
ВВЕДЕНИЕ
Оксид графена (ОГ) представляет собой окисленную форму графена, которая на сегодняшний день является одной из наиболее интенсивно изучаемых двумерных модификаций углерода. Интерес к оксиду графена обусловлен уникальной комбинацией физико-химических свойств и особой структурой латерально протяженного материала нанометровой толщины [1], при этом простота его получения из доступного сырья имеет большое значение с точки зрения практического применения материалов на основе ОГ. Как правило, оксид графена получают в виде гидрозолей частиц нанометровой толщины и с латеральными размерами от десятков нанометров до десятков микрон [2–4] путем расщепления графитовой пудры в водных растворах сильных окислителей [5–8]. Кислородсодержащие группы (гидроксильные, карбоксильные и эпоксидные), расположенные по обеим сторонам плоскости углеродного каркаса ОГ, предоставляют возможность для его дальнейшей функционализации путем их химической модификации и/или прививки к ним различных молекулярных фрагментов, ассоциатов или других неорганических частиц [9–11]. При необходимости кислородные группы могут быть удалены из структуры ОГ с помощью термического отжига или химической обработки с образованием восстановленного ОГ, обладающего выраженными полупроводниковыми свойствами [12–15]. Специфическое строение двумерной частицы “Арлекин”, сочетающей в своей структуре гидрофобные участки и гидрофильные группы, позволяет использовать ОГ в качестве мягкого поверхностно-активного вещества для получения адсорбционных монослоев на межфазных границах и стабилизаторов эмульсий Пикеринга [16]. Наконец, ОГ и материалы на его основе обладают сенсорными и (фото)каталитическими свойствами, что делает ОГ особенно ценным базовым компонентом различных композитных материалов и функциональных устройств [17, 18]. Двумерная структура ОГ позволяет легко интегрировать тонкие пленки и монослои на его основе в архитектуру слоистых систем различного назначения, таких как сенсоры, катализаторы, оптические переключатели, элементы оптических ячеек и т.д.
Благодаря сочетанию перечисленных качеств ОГ в настоящее время является одним из самых популярных наноразмерных материалов для сборки слоистых гибридных систем, в которых он выступает в качестве неорганической матрицы, объединенной с различными фотоактивными органическими соединениями, прежде всего полициклическими хромофорами, которые широко используются при создании фото- и электрофотокатализаторов, сенсибилизированных солнечных ячеек, фотовольтаических элементов и т.д. [19]. Одна из главных целей создания таких систем заключается в достижении синергетического эффекта за счет согласованного действия органического и неорганического компонентов, подразумевающего управление взаимодействиями между ними. Предполагается, что для систем на основе ОГ и его производных и органических хромофоров такие эффекты, как правило, реализуются при сильных взаимодействиях между углеродным каркасом и органическими молекулами за счет процессов эффективного переноса энергии и заряда, которые могут быть измерены различными оптическими, электрохимическими или электрофизическими методами [20–22]. В то же время по мере компактификации таких систем и перехода к ультратонким композитам, содержащим однослойные частицы и единичные молекулы или их ансамбли, прямая оценка силы этих взаимодействий связана со значительными экспериментальными трудностями в силу малого количества измеряемого материала. Поэтому особое значение приобретает поиск методов, которые могли бы прямо или косвенно определять силу взаимодействий и эффективность процессов переноса между ОГ и молекулами хромофора в тонкой пленке и тем самым обеспечивали бы возможность предварительного выбора оптимальной композиции слоистой структуры на основе ОГ.
В качестве одного из возможных подходов к решению этой задачи в настоящей работе предлагается использовать метод спектроскопии усиленного поверхностью комбинационного рассеяния (КР) [23–25], где ОГ может выступать в качестве субстрата за счет химического механизма усиления сигнала КР, основанного на переносе энергии и/или заряда между аналитом и субстратом [26–28]. Таким образом, спектры КР для ультратонкой системы ОГ–хромофор могут служить индикатором взаимодействий между углеродной матрицей и органическим компонентом.
Как и графен, ОГ способен эффективно тушить флуоресценцию хромофоров, адсорбированных на его поверхности. Это позволяет решить проблему фоновой люминесценции, мешающей регистрации сигнала КР в отсутствие тушителя [29–31]. Благодаря наличию функциональных групп на поверхности ОГ адсорбцией молекул на его поверхности можно управлять, подбирая оптимальное значение рН анализируемого раствора в зависимости от катионной или анионной природы хромофора [32]. Восстановление ОГ позволяет увеличить сопряжение в системе и ее гидрофобность и тем самым увеличить взаимодействие ОГ с поли- и макроциклическими соединениями. Существуют три основные стратегии получения тонких пленок на основе восстановленного ОГ для спектроскопии КР: а) химическое восстановление в объеме гидрозоля с последующим нанесением на предварительно силанизированную твердую подложку [26, 33]; б) химическое или термическое восстановление ОГ в уже сформированных на поверхности пленках [27]; в) соосаждение восстановленного ОГ с адсорбированными из раствора молекулами хромофора на подложку [33]. Как правило, эти методы приводят к образованию островковых или полислойных пленок значительной толщины (до 200 мкм) и не позволяют формировать тонкие однородные пленки, пригодные для интеграции в наноразмерные слоистые архитектуры. В настоящей работе для формирования ультратонких однородных пленок восстановленного ОГ был использован разработанный нами ранее метод переноса адсорбционных слоев ОГ непосредственно с межфазной границы вода/масло с последующим восстановлением с помощью микроволнового излучения [16, 34].
Для оценки влияния строения молекул-хромофоров на интенсивность их взаимодействия с монослоем ОГ были выбраны два структурно различных производных перилена с различными заместителями: диангидрид перилентетракарбоновой кислоты (PTCDA) и изопентилдиимид перилен-3,4,9,10-тетракарбоновой кислоты (EP-PDI) (рис. 1). Производные перилена широко используются в органических солнечных элементах в качестве красителей-сенсибилизаторов [35]. Благодаря ароматической системе периленового фрагмента оба хромофора склонны к π–π-стекингу. В то же время наличие в молекуле EP-PDI изопентильных заместителей, развернутых перпендикулярно к плоскости π-системы, должно затруднять взаимодействие такого хромофора с углеродным субстратом [36]. Напротив, плоская геометрия PTCDA должна обеспечивать максимально эффективный контакт между молекулами хромофора и двумерным углеродом [37].
Рис. 1.
Структурные формулы диангидрида перилентетракарбоновой кислоты (PTCDA) и изопентилдиимида перилен-3,4,9,10-тетракарбоновой кислоты (EP-PDI).
С помощью спектроскопии КР и флуоресцентной микроскопии были исследованы оптические свойства ультратонких композитов на основе этих соединений и монослоев восстановленного ОГ. На основе полученных данных проведена оценка влияния молекулярной структуры хромофора на эффективность его взаимодействия с однослойной пленкой восстановленного оксида графена как оптического субстрата.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Гидрозоль ОГ с концентрацией 1.5 мас. % синтезировали по модифицированному методу Хаммера, описанному в [16]. Диангидрид перилентетракарбоновой кислоты изопентилдиимид перилен-3,4,9,10-тетракарбоновой кислоты, н-гексан, ДМСО, хлороформ аналитической чистоты были приобретены у Sigma-Aldrich. Деионизированную воду с сопротивлением 16 MΩ, полученную с помощью системы “Водолей” (Химэлектроника), использовали для приготовления гидрозоля ОГ заданной концентрации. Кремниевые и кварцевые пластины очищали смесью Н2О2 и Н2SO4 в соотношении 1 : 1 в течение 20 мин, промывали деионизированной водой 20 раз и затем сушили в сушильном шкафу при температуре 170°С на воздухе. Чистые подложки хранили в закрытом контейнере на воздухе.
Для получения монослоев ОГ на твердых подложках 25 мл гидрозоля ОГ с концентрацией 0.4 г/л наливали в стеклянный бюкс диаметром 40 мм и высотой 30 мм. Затем с помощью автоматического диппера в гидрозоль вертикально опускали очищенную пластину на 2/3 высоты. Поверх гидрозоля ОГ наслаивали 5 мл н-гексана. Полученную двухфазную систему оставляли на 40 мин для формирования адсорбционного слоя ОГ на границе фаз гидрозоль ОГ/н-гексан. Затем пластинку поднимали со скоростью 1 мм/мин, сохраняя межфазную границу на протяжении всего процесса переноса. Подложку с перенесенным таким способом монослоем ОГ сушили на воздухе в течение суток [16, 34].
Для получения покрытий восстановленного ОГ монослои оксида графена восстанавливали с помощью нагрева в микроволновой печи Panasonic NN-DF383BZPE (1000 Вт) в атмосфере аргона. В случае кремниевой подложки время восстановления составляло 3 мин, для кварцевой подложки – 80 мин [16].
Для осаждения PTCDA на твердые подложки 25 мкл раствора PTCDA в ДМСО с концентрацией 1 × 10–6 М наносили на субстрат каплями по 5 мкл, затем сушили под вакуумом при 150°С в два этапа (40 мин и 12 ч) и промывали горячим ДМСО для удаления избытка красителя, после чего еще сушили под вакуумом 40 мин при 150°С.
Для иммобилизации EP-PDI подложки погружали в 1 мл раствора красителя в хлороформе с концентрацией 1 × 10–8 М, налитого в пенициллиновый флакон диаметром 20 мм, и оставляли неплотно прикрытым до полного испарения растворителя.
Спектры КР и флуоресценции монослоев восстановленного ОГ с иммобилизированными хромофорами регистрировали с помощью спектрометра КР Senterra фирмы Bruker. В качестве источника излучения использовали полупроводниковый Nd:YAG-лазер с длиной волны 532 нм. Фокусировку лазерного пучка проводили с использованием ×20 объектива (диаметр освещаемой площадки ~2 мкм). Мощность лазера составляла <0.2 мВт.
Оптические микрофотографии получены с помощью микроскопа Ломо Микмед-2 в видимом свете и в свете флуоресценции с использованием ×20 объектива.
Для определения углов смачивания использовали KSV Contact Angles Measurement System-101, объем капель составлял 5 мкл. Из-за маленького размера подложек (1 × 1 см) для одной подложки возможно только одно измерение. Для получения достоверных результатов монослой ОГ переносили на три кремниевые подложки, для каждой подложки углы смачивания измеряли до и после нагрева в микроволновой печи. Усредненный угол смачивания монослоев ОГ и восстановленного ОГ рассчитывали как среднее арифметическое полученных значений.
Электронные спектры поглощения исследуемых систем на кварцевых подложках регистрировали с помощью спектрофотометра Shimadzu UV 2450 PC в диапазоне длин волн 200−700 нм.
Электронные микрофотографии получали с помощью сканирующего электронного микроскопа Carl Zeiss NVision 40 с детектором вторичных электронов, ускоряющее напряжение составляло 1 кВ.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
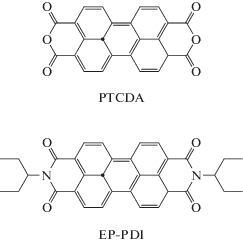
В качестве субстратов для спектроскопии КР использовали монослои восстановленного оксида графена. Во-первых, такая морфология субстрата исключает влияние контакта между слоями в пленке ОГ на взаимодействие между субстратом и органическими молекулами. Во-вторых, восстановление ОГ увеличивает долю сопряженных доменов на листе, что облегчает взаимодействие ароматического остова периленовых производных с электронной системой восстановленного ОГ. Монослои формировали на межфазной границе масло/вода, а затем переносили на твердые подложки по разработанной ранее методике [16, 34]. Для этого подложку вертикально погружали в гидрозоль ОГ на 2/3 высоты, поверх водной фазы наслаивали н-гексан и оставляли систему на некоторое время для формирования адсорбционного слоя ОГ. Затем подложку медленно поднимали, сохраняя межфазную границу жидкость/жидкость, и сушили на воздухе в течение суток. Для восстановления оксида графена подложки с монослоем ОГ нагревали в микроволновой печи в атмосфере аргона. Разогрев подложки при взаимодействии с микроволновым излучением приводит к быстрому восстановлению ОГ (время восстановления зависит от материала подложки и составляет от 4 до 80 мин). Данные СЭМ подтверждают формирование плотноупакованных монослоев с низкой долей перекрывания между листами (рис. 2а). По данным атомно-силовой микроскопии, толщина такого покрытия составляет ∼1.5 нм, что соответствует толщине одиночного листа ОГ [16, 34]. Исследование монослоев ОГ до и после обработки в микроволновой печи показало, что микроволновой нагрев приводит к увеличению угла смачивания от (23 ± 1)° до (49 ± 1)°. Кроме того, возрастает интенсивность основной полосы поглощения ОГ в области 230 нм в сочетании с батохромным сдвигом на 5 нм (рис. 2б). Полученные данные указывают на восстановление сопряжения в электронной системе ОГ. Несмотря на это в спектрах КР монослоев ОГ до и после восстановления нет достоверных различий, что может быть связано с дефектностью листов на площади, соизмеримой с площадью пятна возбуждающего лазера. Как видно из рис. 2в, в спектре присутствует выраженная полоса G, характеризующая колебания системы sp2-углеродных связей (~1580 см–1), и полоса D, отвечающая дефектам в двумерной структуре (1354 см–1), а также полосы второго порядка: 2D при 2700 см–1 и 3S в области 2900 см–1, представляющая собой сумму полос D и G.
Рис. 2.
СЭМ-фотография монослоя восстановленного ОГ на кремниевой подложке (а), оптические спектры поглощения монослоя ОГ до (1) и после (2) обработки в микроволновой печи (б), спектр КР монослоя ОГ на кремниевой подложке, звездочкой обозначена полоса кремния при 975 см–1 (в).
Процедура иммобилизации PTCDA на монослое восстановленного ОГ и чистой кремниевой подложке включала три стадии (рис. 3). На первой стадии раствор PTCDA в ДМСО раскапывали на подложке и сушили под вакуумом 40 мин, на второй – образцы подвергали длительной сушке (в течение 12 ч), на третьей стадии подложки промывали большим объемом растворителя для удаления не связанного с субстратом хромофора и снова сушили.

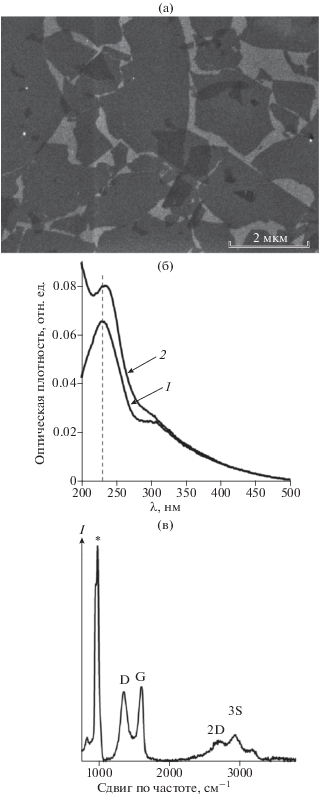
Как видно из рис. 4а, 4в, после первого этапа PTCDA формирует на поверхности восстановленного ОГ островковую пленку из люминесцирующих зеленым светом доменов. В спектре флуоресценции такой пленки присутствует основная полоса в области 575 нм с плечом при 670 нм (рис. 4д, кривая 1). Форма спектра повторяет флуоресценцию раствора PTCDA в мономерной форме (рис. S1), однако весь спектр смещен в длинноволновую область на 35 нм, что свидетельствует о сближении молекул в домене без формирования агрегатов. Регистрация КР-спектра такой пленки невозможна из-за перекрывания интенсивных полос флуоресценции и слабых сигналов от комбинационного рассеяния (рис. 4е, кривая 1). В то же время при адсорбции на чистом кремнии PTCDA образует сильно неоднородное покрытие из мелких агрегатов (рис. 4б, 4г). Максимум флуоресценции полученной пленки лежит в области 750 нм (рис. 4д, кривая 2), что отвечает формированию J-агрегатов. Из-за сильного смещения флуоресценции в длинноволновую область при образовании агрегатов перекрывание полос флуоресценции и комбинационного рассеяния уменьшается, благодаря чему на склоне пика флуоресценции проявляются слаборазрешенные полосы КР (рис. 4е, кривая 2). Следует отметить, что в случае адсорбции PTCDA на восстановленном ОГ скорость “выгорания” хромофора под пучком лазера значительно ниже, чем на поверхности чистого кремния. Эти результаты указывают на то, что при адсорбции PTCDA на монослое восстановленного ОГ протекают два конкурирующих процесса: взаимодействие молекул хромофора между собой с образованием агрегатов и их адсорбция на субстрате. Поскольку на поверхности восстановленного ОГ, в отличие от чистого кремния, хромофор не образует агрегаты, взаимодействие ароматической системы одиночной молекулы PTCDA с большой сопряженной π-системой восстановленного ОГ оказывается энергетически более выгодным, чем стекинг с молекулой такого же размера. При осаждении на чистый кремний молекулы PTCDA взаимодействуют только друг с другом и минимизируют контакт с гидрофильной поверхностью кремния.
Рис. 4.
Оптические микрофотографии PTCDA на монослое восстановленного ОГ (а, в) и PTCDA на чистой кремниевой подложке (б, г), полученные с помощью флуоресцентного микроскопа, в видимом свете (а, б) и в свете флуоресценции (в, г). Спектры флуоресценции (д) и комбинационного рассеяния (е) PTCDA на монослое восстановленного ОГ (1) и чистом кремнии (2). Спектры нормированы на максимальное значение интенсивности. Анализ образцов проведен после первой стадии иммобилизации: раствор PTCDA в ДМСО нанесли на подложки и сушили под вакуумом 40 мин при 150°С.
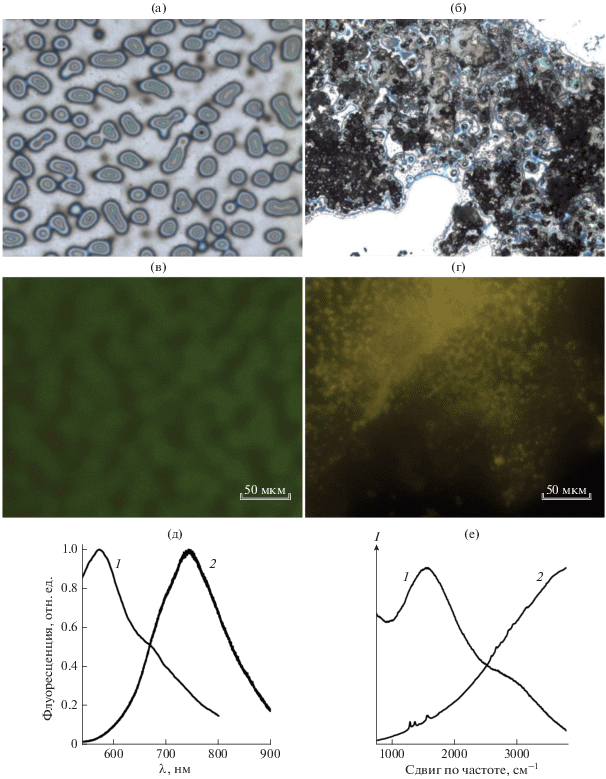
После длительной сушки образцов на второй стадии в спектре КР пленки PTCDA, адсорбированной на монослое восстановленного ОГ, проявляются хорошо разрешенные полосы хромофора (рис. 5, кривая 1). С одной стороны, это может быть следствием сильного тушения флуоресценции за счет переноса энергии между хромофором и субстратом. Анализируя спектры КР, невозможно судить об абсолютной интенсивности полос, поэтому при сравнении их нормируют относительно какой-либо референсной полосы. В данном случае была выбрана полоса кремния в области 975 см–1. Тот факт, что эта полоса видна в спектре образца после сушки, подтверждает низкую интенсивность флуоресценции системы. С другой стороны, удаление растворителя из пленки приводит к дополнительной агрегации не взаимодействующих с субстратом молекул PTCDA и смещению максимума флуоресценции в длинноволновую область, наблюдаемому также при адсорбции хромофора на чистом кремнии (рис. 4е, кривая 2). Удаление несвязанного PTCDA на третьей стадии с помощью промывки образца приводит к полному исчезновению фоновой люминесценции при сохранении интенсивности полос КР (рис. 5 кривая 2). Это означает, что вклад в комбинационное рассеяние вносят только молекулы, которые невозможно удалить механически из-за сильного взаимодействия π-систем PTCDA и восстановленного ОГ.
Рис. 5.
Спектры комбинационного рассеяния PTCDA на монослое восстановленного ОГ, записанные после сушки под вакуумом в течение 12 ч при 150°С (1) и последующей промывки горячим раствором ДМСО и сушки под вакуумом (2). Спектры нормализованы относительно полосы кремния при 975 см–1, отмеченной звездочкой.
Длительная сушка PTCDA на поверхности чистого кремния не влияет на спектральные характеристики агрегатов. После промывки подложки растворителем спектр флуоресценции смещается в коротковолновую область, что соответствует формированию тонкой неагрегированной пленки хромофора. Спектр флуоресценции перекрывает область комбинационного рассеяния PTCDA, в результате чего КР-полосы хромофора исчезают из спектра (рис. S2).
Таким образом, можно сделать вывод, что при адсорбции PTCDA на листах восстановленного ОГ между хромофором и субстратом возникает сильное взаимодействие за счет ароматического стекинга периленового фрагмента PTCDA c сопряженными фрагментами листа восстановленного ОГ и перенос энергии между органическими молекулами и субстратом. Это взаимодействие определяет структуру пленки в случае избытка хромофора на подложке, препятствуя агрегации PTCDA, и приводит к тушению флуоресценции и усилению комбинационного рассеяния хромофора после удаления не связанного с субстратом органического материала.
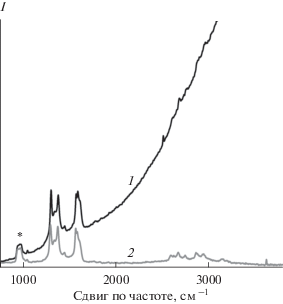
Чтобы определить, как влияет геометрия молекулы хромофора на взаимодействие с восстановленным ОГ, использовали производное перилена EP-PDI с двумя изопентильными заместителями, развернутыми перпендикулярно к плоскости периленового фрагмента. Для иммобилизации EP-PDI на углеродном субстрате кремниевую подложку с монослоем восстановленного ОГ горизонтально погружали в раствор EP-PDI в хлороформе и оставляли в темном месте до полного испарения растворителя. Аналогичным образом получали контрольную пленку EP-PDI на чистом кремнии. Как видно из рис. 6, общий вид КР-спектра EP-PDI на монослое восстановленного ОГ (кривая 1) соответствует спектру флуоресценции раствора мономеров EP-PDI (рис. S3). В спектре также присутствуют все характеристические полосы восстановленного ОГ и линия кремния, но не разрешены полосы EP-PDI. Форма КР-спектра EP-PDI на чистом кремнии отвечает спектру флуоресценции J-агрегатов (рис. 6, кривая 2). При этом на склоне пика флуоресценции видны слабо разрешенные КР-полосы хромофора. После промывки подложек избытком растворителя из КР-спектров систем исчезает флуоресценция хромофора, что говорит о полном удалении EP-PDI с подложки.
Рис. 6.
Спектры комбинационного рассеяния EP-PDI, адсорбированного на монослое восстановленного ОГ (1) и чистом кремнии (2) при раскапывании раствора EP-PDI в хлороформе. Интенсивность кривой 1 увеличена в 10 раз по сравнению с кривой 2 относительно полосы кремния при 975 см–1, отмеченной звездочкой.
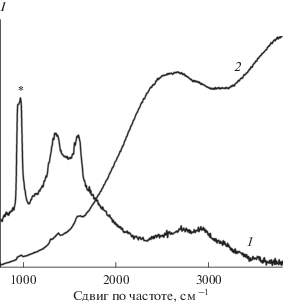
Таким образом, на поверхности чистого кремния молекулы EP-PDI ведут себя аналогично молекулам PTCDA, вступая между собой в стекинговые взаимодействия с образованием J-агрегатов, КР-спектр которых плохо разрешен из-за сильной фоновой люминесценции. При адсорбции на монослое восстановленного ОГ взаимодействие с субстратом подавляет агрегацию EP-PDI и приводит к практически полному тушению флуоресценции, однако этого взаимодействия недостаточно для усиления спектра КР. Снижение эффективности переноса заряда между органическими молекулами и субстратом связано со стерическими затруднениями, вызванными наличием разветвленных заместителей в молекуле EP-PDI.
ЗАКЛЮЧЕНИЕ
На основании анализа спектров КР периленовых производных, адсорбированных на монослоях восстановленного ОГ, установлена зависимость переноса энергии в системе от геометрии контакта между хромофором и углеродным субстратом. Продемонстрирована возможность использования метода спектроскопии КР для оценки взаимодействия между органическими соединениями и графеноподобными структурами. Показано, что производные перилена с планарной структурой вступают в сильные π–π-взаимодействия с восстановленным ОГ за счет полного перекрывания электронных систем периленого фрагмента хромофора и сопряженных доменов на листах восстановленного ОГ. Это взаимодействие приводит к эффективному переносу энергии, что проявляется в резком тушении флуоресценции и усилении КР хромофора. В случае периленовых производных с разветвленными заместителями стерические затруднения, вызванные разворотом заместителей относительно плоскости периленового фрагмента, уменьшают перекрывание электронных систем хромофора и субстрата, тем самым затрудняя перенос энергии между ними. Такие системы характеризуются лишь частичным тушением флуоресценции и слабым сигналом хромофора в спектрах КР.
Полученные результаты имеют принципиальное значение для разработки новых оптоэлектронных и фотовольтаических устройств, основанных на переносе энергии между производными перилена и двумерными углеродными материалами.
Список литературы
Gao W. // The Chemistry of Graphene Oxide. Graphene Oxide. Cham: Springer Int. Publ., 2015. P. 61. https://doi.org/10.1007/978-3-319-15500-5_3
Dong L., Yang J., Chhowalla M. et al. // Chem. Soc. Rev. 2017. V. 46. № 23. P. 7306. https://doi.org/10.1039/C7CS00485K
Kim D.W., Kim D., Min B.H. et al. // Carbon. 2015. V. 88. P. 126. https://doi.org/10.1016/j.carbon.2015.02.076
Kumar P., Shahzad F., Yu S. et al. // Carbon. 2015. V. 94. P. 494. https://doi.org/10.1016/j.carbon.2015.07.032
Hummers W.S., Offeman R.E. // JACS. 1958. V. 80. № 6. P. 1339. https://doi.org/10.1021/ja01539a017
Xu Y., Bai H., Lu G. et al. // JACS. 2008. V. 130. № 18. P. 5856. https://doi.org/10.1021/ja800745y
Alam S.N., Sharma N., Kumar L. // Graphene. 2017. V. 6. № 1. P. 1. https://doi.org/10.4236/graphene.2017.61001
Zaaba N., Foo K., Hashim U. et al. // Procedia Eng. 2017. V. 184. P. 469. https://doi.org/10.1016/j.proeng.2017.04.118
Huang X., Yin Z., Wu S. et al. // Small. 2011. V. 7. № 14. P. 1876. https://doi.org/10.1002/smll.201002009
Jiang X., Ruan G., Huang Y. et al. // J. Sep. Sci. 2020. P. jssc.201900694. https://doi.org/10.1002/jssc.201900694
Buslaeva E.Yu., Kraevskii S.V., Groshkova Yu.A. et al. // Russ. J. Inorg. Chem. 2020. V. 65. № 1. P. 5. https://doi.org/10.1134/S0036023620010040
Mattevi C., Eda G., Agnoli S. et al. // Adv. Funct. Mater. 2009. V. 19. № 16. P. 2577. https://doi.org/10.1002/adfm.200900166
Renteria J.D., Ramirez S., Malekpour H. et al. // Adv. Funct. Mater. 2015. V. 25. № 29. P. 4664. https://doi.org/10.1002/adfm.201501429
Bocharov G.S., Eletskii A.V. // J. Struct. Chem. 2018. V. 59. № 4. P. 806. https://doi.org/10.1134/S0022476618040091
Ziatdinov A.M., Saenko N.S., Skrylnik P.G. // Russ. J. Inorg. Chem. 2020. V. 65. № 1. P. 133. https://doi.org/10.1134/S0036023620010210
Zvyagina A.I., Melnikova E.K., Averin A.A. et al. // Carbon. 2018. V. 134. P. 62. https://doi.org/10.1016/j.carbon.2018.03.075
Rowley-Neale S.J., Randviir E.P., Abo Dena A.S. et al. // Appl. Mater. Today. 2018. V. 10. P. 218. https://doi.org/10.1016/j.apmt.2017.11.010
Toda K., Furue R., Hayami S. // Anal. Chim. Acta. 2015. V. 878. P. 43. https://doi.org/10.1016/j.aca.2015.02.002
Georgakilas V., Tiwari J.N., Kemp K.C. et al. // Chem. Rev. 2016. V. 116. № 9. P. 5464. https://doi.org/10.1021/acs.chemrev.5b00620
Yang L., Wang P., Yin J. et al. // Appl. Catal., B. Environ. 2019. V. 250. P. 42. https://doi.org/10.1016/j.apcatb.2019.02.076
Li X. // J. Chem. 2018. V. 2018. P. 1. https://doi.org/10.1155/2018/8050524
Stylianakis M., Viskadouros G., Polyzoidis C. et al. // Nanomaterials. 2019. V. 9. № 2. P. 137. https://doi.org/10.3390/nano9020137
Lai H., Xu F., Zhang Y. et al. // J. Mater. Chem B. 2018. V. 6. № 24. P. 4008. https://doi.org/10.1039/C8TB00902C
Lin J., Zhang N., Tong L. et al. // ACS Symposium Series. 2016. V. 1246. P. 97. https://doi.org/10.1021/bk-2016-1246.ch005
Hu Y., López-Lorente Á.I., Mizaikoff B. // ACS Photonics. 2019. V. 6. № 9. P. 2182. https://doi.org/10.1021/acsphotonics.9b00645
Yin F., Wu S., Wang Y. et al. // J. Solid State Chem. 2016. V. 237. P. 57. https://doi.org/10.1016/j.jssc.2016.01.015
Yu X., Cai H., Zhang W. et al. // ACS Nano. 2011. V. 5. № 2. P. 952. https://doi.org/10.1021/nn102291j
Yan T., Zhang L., Jiang T. et al. // Appl. Surf. Sci. 2017. V. 419. P. 373. https://doi.org/10.1016/j.apsusc.2017.05.052
Xie L., Ling X., Fang Y. et al. // JACS 2009. V. 131. № 29. P. 9890. https://doi.org/10.1021/ja9037593
Thrall E.S., Crowther A.C., Yu Z. et al. // Nano Lett. 2012. V. 12. № 3. P. 1571. https://doi.org/10.1021/nl204446h
Kozhemyakina N.V., Englert J.M., Yang G. et al. // Adv. Mater. 2010. V. 22. № 48. P. 5483. https://doi.org/10.1021/nl204446h
Liu Z., Li S., Hu C. et al. // J. Raman Spectrosc. 2013. V. 44. № 1. P. 75. https://doi.org/10.1002/jrs.4142
Yu S., Wang X., Yao W. et al. // Environ. Sci. Technol. 2017. V. 51. № 6. P. 3278. https://doi.org/10.1021/acs.est.6b06259
Zvyagina A.I., Gusarova E.A., Baranchikov A.E. et al. // Surf. Innov. 2019. V. 7. № 3–4. P. 210. https://doi.org/10.1680/jsuin.19.00003
Macedo A.G., Christopholi L.P., Gavim A.E.X. et al. // J. Mater. Sci. Mater. Electron. 2019. V. 30. № 17. P. 15 803. https://doi.org/10.1007/s10854-019-02019-z
Wiatrowski M., Dobruchowska E., Maniukiewicz W. et al. // Thin Solid Films. 2010. V. 518. № 8. P. 2266. https://doi.org/10.1016/j.tsf.2009.08.037
Wang Q.H., Hersam M.C. // Nat. Chem. 2009. V. 1. № 3. P. 206. https://doi.org/10.1038/nchem.212
Дополнительные материалы
- скачать ESM.docx
- Рис. S1. Нормированные спектры (1) поглощения и (2) флуоресценции раствора PTCDA в ДМСО.
Рис. S2. Спектр комбинационного рассеяния PTCDA, адсорбированного на чистой кремниевой пластинке, записанный после промывки подложки ДМСО для удаления несвязанных с субстратом молекул.
Рис. S3. Нормированные спектры (1) поглощения и (2) флуоресценции раствора EP-PDI в хлороформе.
Инструменты
Журнал неорганической химии