

Журнал неорганической химии, 2021, T. 66, № 2, стр. 160-167
Роль условий соосаждения гидроксидов в формировании нанокристаллического BiFeO3
О. В. Проскурина a, *, А. Н. Соколова a, А. А. Сироткин a, Р. Ш. Абиев a, В. В. Гусаров a
a Физико-технический институт им. А.Ф. Иоффе РАН
194021 Санкт-Петербург, Политехническая ул., 26, Россия
* E-mail: proskurinaov@mail.ru
Поступила в редакцию 18.07.2020
После доработки 14.08.2020
Принята к публикации 27.08.2020
Аннотация
Методами мягкой химии получен нанокристаллический ортоферрит висмута (BiFeO3). Показано влияние способов соосаждения гидроксидов на формирование нанокристаллического ортоферрита висмута. Определены условия синтеза нанокристаллического BiFeO3 без примесей других фаз системы Bi2O3–Fe2O3 с узким распределением кристаллитов по размерам с их минимальным значением ~6–7 нм.
ВВЕДЕНИЕ
Значительный интерес исследователей к материалам на основе ортоферрита висмута (BiFeO3) связан с его уникальными свойствами. Ортоферрит висмута является мультиферроиком при комнатной температуре, обладая высокой термической стабильностью магнитоупорядоченного и сегнетоэлектрического состояний, его температура Нееля ТN = 370°С, температура Кюри ТC = 830°С [1]. Ортоферрит висмута является полупроводником с шириной запрещенной зоны, лежащей в области видимого света от 2.1 до 2.8 эВ, что позволяет использовать его в качестве фотокатализатора [2, 3]. Материалы на основе BiFeO3 также было предложено использовать в устройствах энергонезависимой памяти, пьезоэлектрических устройствах, сенсорах, спинтронике и др. [4–7].
Однако для практического использования такого перспективного многофункционального материала есть два основных препятствия: во-первых, это сложность синтеза фазы BiFeO3 без примесей других фаз системы Bi2O3–Fe2O3 [8–10], а во-вторых, наличие циклоидной магнитной структуры с периодом ~62 нм [11]. Магнитная циклоида вызывает значительную компенсацию магнитных моментов, что приводит к ослаблению ферромагнетизма и снижению магнитоэлектрической связи. В качестве методов управления магнитными свойствами кристаллических материалов на основе феррита висмута используют введение примесных компонентов в структуру, формирование механически напряженных эпитаксиальных слоев, получение нанопорошков с определенным размером кристаллитов [12–16].
Появление примесных фаз объяснялось разными причинами: высокой летучестью висмута, нестехиометричностью и изменением области гомогенности при высоких температурах, большей термодинамической устойчивостью сосуществующих в системе Bi2O3–Fe2O3 фаз по сравнению с BiFeO3, наличием области эвтектоидного распада BiFeO3 [8–10, 17].
Сложность получения однофазного феррита висмута с определенным размером кристаллитов инициировала изучение механизмов формирования ортоферрита висмута в различных условиях синтеза [8, 18–23].
Перечисленные выше проблемы получения материалов на основе ортоферрита висмута привели к поиску новых методов синтеза феррита висмута и усовершенствованию уже разработанных методов. Для его синтеза используют методы твердофазных реакций [8, 18, 24], горения растворов [21, 25–27], различные методы мягкой химии [19, 20, 22, 28–31], ультразвуковой распылительный пиролиз [32]. Твердофазный синтез с термообработкой реагентов при высоких температурах часто приводит к появлению примесных фаз, таких как Bi25FeO39, Bi2Fe4O9 [8–10]. Кроме того, твердофазный синтез не позволяет получить наноразмерные частицы BiFeO3, в то время как в большинстве случаев именно нанокристаллические материалы демонстрируют значительно лучшие свойства по сравнению с порошками микрометрового размера [2, 15, 16, 33].
Хорошо известным преимуществом методов мягкой химии является потенциальная возможность получения нанокристаллического вещества [34]. Вместе с тем, как правило, синтез однофазного продукта даже при использовании методов мягкой химии требует повышения температуры и продолжительности процесса синтеза, в то время как возможность получения нанокристаллических частиц базируется на понижении температуры и уменьшении времени их образования [35]. Указанная противоречивость в условиях синтеза ставит задачу поиска методов интенсификации фазообразования для оптимизации процесса формирования нанокристаллического BiFeO3.
Для интенсификации химических реакций образования ортоферрита висмута без значительного повышения температуры используются такие методы, как ультразвуковая обработка [36], микроволновой нагрев при гидротермальном синтезе [20, 30], механохимическое воздействие [37]. В последнее время для получения нанокристаллического BiFeO3 использовался метод термообработки гидроксидов, соосажденных в микрореакторах со свободно сталкивающимися струями [23, 31].
Цель настоящей работы – определение роли условий соосаждения реагентов и термообработки соосажденных гидроксидов для получения однофазного нанокристаллического феррита висмута с минимальными размерами кристаллитов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез BiFeO3
Нанокристаллический BiFeO3 синтезировали путем соосаждения гидроксидов с последующей их термообработкой. Соосаждение гидроксидов проводили четырьмя способами: 1) с использованием микрореакторной техники в режиме “затопленных струй” [38], 2) в режиме смешения растворов реагентов в микрореакторе со свободно сталкивающимися струями с образованием так называемой “пелены” в соответствии с процедурой, описанной в [23, 31], 3) в режиме обратного осаждения при подаче смеси растворов солей в виде микроструи в раствор осадителя через его свободную поверхность; 4) в режиме обратного осаждения с одновременной ультразвуковой обработкой. Схематическое изображение установок описанных способов представлено на рис. 1.
В качестве исходных реагентов использовали Bi(NO3)3 ⋅ 5H2O, Fe(NO3)3 · 9H2O, 6 М раствор HNO3 и 4 М раствор NaOH. Все реактивы были аналитической чистоты. Первоначально навеску Bi(NO3)3 ⋅ 5H2O растворяли в 6 М растворе HNO3 при перемешивании магнитной мешалкой и нагревании до 70°С в течение 10 мин, затем в полученный раствор добавляли Fe(NO3)3 ⋅ 9H2O, перемешивали еще 10 мин, после чего добавляли дистиллированную воду и перемешивали еще 20 мин. Отдельно готовили 4 М раствор NaOH. Приготовленные таким образом растворы использовали для получения смеси гидроксидов висмута и железа.
Для приготовления образца 1 реактор заполняли раствором NaOH до указанного на схеме уровня (рис. 1а), который удерживали постоянным при помощи гидравлического затвора. После этого растворы нитратов и щелочи подавали с помощью перистальтических насосов Heidolph Pumpdrive 5201 одновременно через два сопла, расположенных на противоположных стенках реактора под углом 180° друг к другу. Через одно сопло диаметром 515 мкм подавали раствор нитратов висмута и железа, через другое сопло диаметром 470 мкм – раствор NaOH. Расход обоих растворов равен 150 мл/мин. Скорость струй на выходе из сопла составляла 15.3 м/с для раствора смеси солей и 16.7 м/с для раствора щелочи. При запуске насосов происходило “впрыскивание” раствора нитратов в реактор, заполненный NaOH, при этом сразу же начиналось осаждение гидроксидов металлов (рис. 2). Образовавшиеся в ходе химической реакции частицы соосажденных гидроксидов удаляли через шланг гидрозатвора в приемную емкость.
Рис. 1.
Схема установок для соосаждения гидроксидов по методу 1 (а), 2 (б), 3 (в), 4 (г). 1 – смесь растворов солей; 2 – раствор осадителя (NaOH).
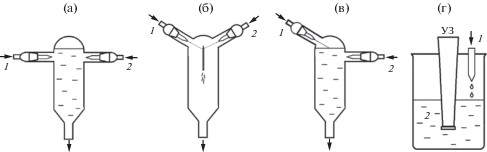
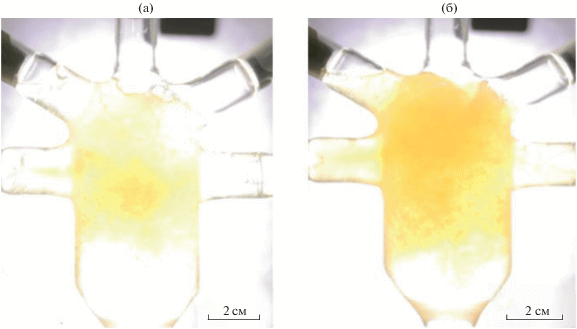
Рис. 2.
Фотографии микрореактора через 0.2 (а) и 0.4 с (б) от момента начала смешения “затопленных струй” при синтезе образца 1.
Для приготовления образца 2 растворы солей висмута и железа, а также раствор NaOH подавали с расходом обоих растворов 150 мл/мин через сопла, расположенные друг относительно друга под углом 150°, в виде свободно сталкивающихся струй, образующих жидкостную пелену, как показано на рис. 3. Оптимальные режимы подачи реагентов в микрореакторе со свободно сталкивающимися струями (скорость подачи и угол между сталкивающимися струями) были определены в работах [23, 31, 39].
Рис. 3.
Фотография жидкостной пелены при приготовлении образца 2. Снимок показывает пелену в плоскости, параллельной плоскости пелены.

Для приготовления образца 3 осуществляли обратное осаждение в соответствии со схемой, приведенной на рис. 1в. Раствор нитратов подавался под углом 30° к поверхности раствора щелочи. При этом для поддержания постоянного рН смеси непрерывно подавался раствор щелочи с расходом, равным расходу раствора солей висмута и железа (150 мл/мин).
Для приготовления образца 4 методом обратного осаждения смешивали растворы нитратов и щелочи при одновременном действии ультразвука (ультразвуковой диспергатор И100-6/1 (ИНЛАБ, Россия), частота 23 кГц). Смешение проходило в течение 1 мин (рис. 1г).
Соосажденные четырьмя способами гидроксиды промывали дистиллированной водой методом декантации до нейтральной среды и высушивали при температуре 65°С в течение 12 ч.
Термообработку образцов проводили следующим образом. Образец массой ~30 мг в течение 4–6 с помещали в предварительно нагретый в печи платиновый тигель, после чего тигель помещали в нагретую до определенной температуры печь на 30 с, затем тигель вынимали и образец высыпали на холодный металлический лист.
Исследование продуктов синтеза
Рентгеновские дифрактограммы снимали на дифрактометре Rigaku SmartLab 3 (CoKα-излучение) в диапазоне углов 2θ от 15° до 72° с шагом 0.01° и скоростью съемки 0.4 град/мин. Фазовый анализ образцов определяли по базе данных ICSD PDF-2. Распределение кристаллитов по размерам и параметры распределения определяли по методу фундаментальных параметров в приближении кристаллитов сферической формы и логнормальной модели распределения с помощью программного пакета SmartLab Studio II для рефлекса (012).
Исследование образцов методом растровой электронной микроскопии, а также рентгеноспектральный микроанализ проводили с помощью электронного микроскопа Tescan Vega 3 SBH с использованием детектора Oxford Instruments X-MAX.
Исследования методом просвечивающей электронной микроскопии (ПЭМ) выполнены с использованием микроскопа JEOL JEM-2100F при ускоряющем напряжении 200 кВ. Образцы для исследований подготавливали предварительной диспергацией исходного порошка в этиловом спирте в ультразвуковой ванне в течение 15 мин с последующей высадкой на поддерживающую пленку.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Результаты растровой электронной микроскопии (рис. 4) показали, что после соосаждения смесь гидроксидов висмута и железа представлена во всех случаях агломератами частиц. Размер частиц всех образцов варьируется в основном в диапазоне 1020 нм. Это хорошо видно также на примере данных ПЭМ (рис. 5). Причем результаты электронной дифракции и рентгенодифракционного анализа показывают, что все образцы после осаждения представлены аморфной фазой.
Рис. 4.
Микрофотографии растровой электронной микроскопии до термообработки образцов 1 (а), 2 (б), 3 (в), 4 (г).

Атомное отношение Bi : Fe во всех образцах до и после термообработки, по данным элементного анализа, остается на уровне Bi : Fe = (51 ± ± 2) : (49 ± 2) ат. %, что в пределах погрешности метода отвечает заданному при синтезе отношению, соответствующему стехиометрии BiFeO3.
Данные рентгеновской дифракции образцов после их последующего нагрева при температуре 490°С в течение 30 с (образцы 1– 4) приведены на рис. 6. Практически все рефлексы на рентгеновских дифрактограммах относятся к фазе ортоферрита висмута (рис. 6). Температура термообработки 490°С выбрана по той причине, что при этой температуре образование ортоферрита висмута было зафиксировано во всех образцах. Образцы соосажденных гидроксидов до начала формирования BiFeO3 (при более низких температурах) и при уменьшении времени термообработки гидроксидов до 20 с остаются рентгеноаморфными, и их дифрактограммы не приведены.
Рис. 6.
Рентгеновские дифрактограммы образцов 1–4, термообработанных при температуре 490°С в течение 30 с.
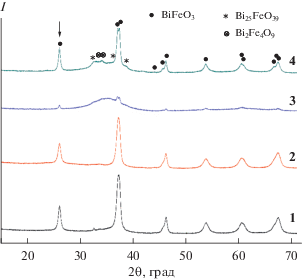
Следует отметить, что температура начала кристаллизации BiFeO3 хорошо коррелирует с температурой завершения процесса дегидратации соосажденных гидроксидов, как можно заключить на основании сравнения полученных результатов с данными синхронного термического анализа [23].
Таким образом, процесс формирования ортоферрита висмута в данном случае представляется как последовательность следующих превращений, реализующихся с повышением температуры:
(1)
$\begin{gathered} am{\text{ - }}\left( {{\text{B}}{{{\text{i}}}_{2}}{{{\text{O}}}_{3}},{\text{F}}{{{\text{e}}}_{2}}{{{\text{O}}}_{3}}} \right) \cdot n{{{\text{H}}}_{2}}{\text{O\;}}\xrightarrow{{ - {{{\text{H}}}_{2}}{\text{O}},T\sim {\text{\;}}400 - 450^\circ {\text{C}},{{{\tau }}_{1}}}} \\ \to am{\text{ - }}\left( {{\text{B}}{{{\text{i}}}_{2}}{{{\text{O}}}_{3}},{\text{F}}{{{\text{e}}}_{2}}{{{\text{O}}}_{3}}} \right){\text{\;}}\xrightarrow{{{\text{\;}}T\sim {\text{\;}}490^\circ {\text{C}},~{{{\tau }}_{2}}}}{\text{BiFe}}{{{\text{O}}}_{3}}, \\ \end{gathered} $Как можно заключить из приведенной цепочки превращений, часто наблюдаемая возможность формирования побочных продуктов – фазы со структурой силленита (Bi25FeO39), обогащенной оксидом висмута, и фазы со структурой муллита (Bi2Fe4O9), обогащенной оксидом железа по сравнению с BiFeO3, может реализоваться вследствие локальной неоднородности состава соосажденных гидроксидов. Пространственная неоднородность состава смеси гидроксидов может привести к еще большей неоднородности состава смеси оксидов после дегидратации, так как гидроксид железа дегидратирует с образованием оксидной фазы при более низкой температуре, т.е. раньше, чем начинается дегидратация гидроксида висмута [23], что приведет к еще большей сегрегации фаз, обогащенных висмутом и железом.
Наличие пространственной неоднородности в составе аморфной смеси оксидов висмута и железа может, в свою очередь, привести как к формированию примесных фаз, в данном случае фаз со структурами силленита (Bi25FeO39) и муллита (Bi2Fe4O9), так и к замедлению процесса кристаллизации BiFeO3 вследствие увеличения длины массопереноса компонентов к формирующимся кристаллическим частицам BiFeO3. В таком случае процесс дегидратации смеси аморфных гидроксидов может оказаться существенно растянутым по температуре и времени и привести к промежуточному формированию смеси различных фаз, соотношение между которыми будет меняться в зависимости от температуры и продолжительности процесса:
(2)
$\begin{gathered} am{\text{ - }}\left( {{\text{B}}{{{\text{i}}}_{2}}{{{\text{O}}}_{3}},{\text{F}}{{{\text{e}}}_{2}}{{{\text{O}}}_{3}}} \right) \cdot n{{{\text{H}}}_{2}}{\text{O}}\xrightarrow{{ - {{{\text{H}}}_{2}}{\text{O}},T{\text{\;}}}} \\ \to A~am{\text{ - }}\left( {{\text{B}}{{{\text{i}}}_{2}}{{{\text{O}}}_{3}},{\text{F}}{{{\text{e}}}_{2}}{{{\text{O}}}_{3}}} \right) \cdot n{\kern 1pt} '{{{\text{H}}}_{2}}{\text{O}} + \\ + \,\,~B~am{\text{ - }}\left( {{\text{B}}{{{\text{i}}}_{2}}{{{\text{O}}}_{3}},{\text{F}}{{{\text{e}}}_{2}}{{{\text{O}}}_{3}}} \right) + C~{\text{B}}{{{\text{i}}}_{{25}}}{\text{Fe}}{{{\text{O}}}_{{39}}} + \\ + \,\,D{\text{\;B}}{{{\text{i}}}_{2}}{\text{F}}{{{\text{e}}}_{4}}{{{\text{O}}}_{9}} + E{\text{\;BiFe}}{{{\text{O}}}_{3}}\xrightarrow{{T,{\tau }}}~{\text{BiFe}}{{{\text{O}}}_{3}}, \\ \end{gathered} $По-видимому, отражением некоторой пространственной неоднородности является заметная доля аморфной фазы и следовые количества примесей кристаллических фаз (Bi2Fe4O9 и Bi25FeO39) у образцов 3 и 4 по данным рентгеновской дифрактометрии (рис. 6). Таким образом, можно сделать вывод, что для быстрой дегидратации смеси гидроксидов и их трансформации в кристаллический ортоферрит висмута формирование смеси гидроксидов висмута и железа необходимо проводить в микрореакторах с “затопленными” и свободно сталкивающимися струями. Использование этих аппаратов интенсифицирует процесс микромасштабного смешения реагентов [23, 31, 38, 39], что позволяет в процессе гидролиза солей висмута и железа формировать смесь гидроксидов без их заметного пространственного разделения.
Объемное распределение кристаллитов BiFeO3 по размеру для образцов 1–4, определенное по рефлексу (012), отмеченному на рис. 6 стрелкой, приведено на рис. 7. Для образцов 1 и 2, полученных с помощью микрореактора с “затопленными” и свободно сталкивающимися струями, наблюдается более узкое распределение кристаллитов по размеру, чем для образцов 3 и 4, полученных при обратном осаждении без УЗ-обработки или с ней.
Рис. 7.
Объемное распределение кристаллитов по размеру для образцов 1–4, определенное по рефлексу (012). Пунктирная линия показывает область значений минимальных размеров нанокристаллитов BiFeO3.
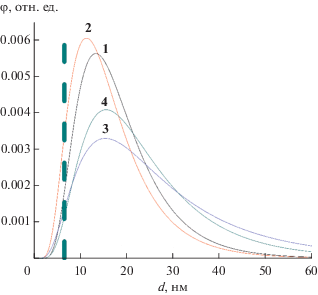
Средневзвешенные значения размеров кристаллитов BiFeO3 и их среднеквадратичные отклонения, по данным распределения кристаллитов по размерам (рис. 7), определенные по рефлексу (012), составляют для образцов: 1 – 19 ± 9, 2 – 17 ± 9, 3 – 28 ± 21, 4 – 24 ± 14 нм.
Следует отметить, что более широкое распределение кристаллитов по размерам и наличие значительной доли аморфной фазы и следовых количеств примесной кристаллической фазы после термообработки у образцов 3 и 4, полученных с использованием менее интенсивных способов смешения реагентов, свидетельствуют о том, что в этом случае фазообразование пошло по схеме (2) , в отличие от синтеза образцов 1 и 2, когда реализуется схема (1) быстрого формирования нанокристаллитов BiFeO3.
Существенную роль в формировании нанокристаллитов BiFeO3 с узким распределением по размерам при использовании для соосаждения смеси гидроксидов висмута и железа играет, по-видимому, и пространственная обособленность образующихся частиц смеси гидроксидов, размер которых варьируется в достаточно узких пределах –~8–20 нм (рис. 5). Возможность пространственного обособления частиц, образующихся при взаимодействии растворов реагентов в условиях микрореакторного смешения, на примере синтеза сложных оксидных фаз различного состава была продемонстрирована в работах [23, 31, 40] и связана с особенностями гидродинамики процесса столкновения жидких сред, приводящими к формированию вихрей масштаба Колмогорова, проявляющих себя как самоорганизующиеся нанореакторы. Дегидратация частиц гидроксидов висмута и железа указанных выше размеров будет приводить к образованию оксидных нанокристаллитов размером ~7–18 нм, если за рассматриваемый период термообработки (~30 с) не произойдет быстрый рост кристаллитов, например, по механизму ориентированного срастания [41]. Данное заключение хорошо коррелирует с экспериментальными результатами, приведенными на рис. 7.
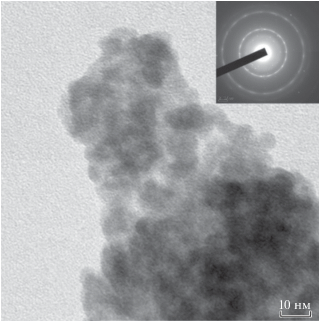
Просвечивающая электронная микроскопия (рис. 8) показала образование в образце 4 единичных монокристаллов феррита висмута размером около 6–7 нм, окруженных аморфной фазой. Расстояния между атомными плоскостями в кристаллите составляют 0.282 нм (рис. 8), что соответствует плоскости (104) для BiFeO3 (0.2818 нм по данным PDF 01-070-5668).

Следует отметить, что минимальные размеры кристаллитов BiFeO3, по данным ПЭМ, составляют 6–7 нм, как у кристаллитов, полученных с помощью методов 1 и 2, обеспечивающих интенсивное смешение реагентов [23, 31, 39, 40], так и в случае менее интенсивного смешения реагентов (рис. 7). Поскольку во всех указанных случаях образование кристаллического BiFeO3 происходило в очень короткие сроки путем кристаллизации аморфной фазы того же состава при одной и той же температуре, данные размеры кристаллитов можно, по-видимому, считать близкими к минимально возможным размерам кристаллической фазы на основе ортоферрита висмута. Это, в свою очередь, может быть связано как с предельным минимальным числом трансляций элементарной ячейки для формирования устойчивой кристаллической решетки [42], так и с формированием критического зародыша фазы ортоферрита висмута. Следует отметить, что полученные значения минимальных размеров кристаллитов (6–7 нм) находятся в хорошем соответствии с предложенным в работе [42] соотношением Dmin ≈ Lэл. яч.N (N = 10–12).
ЗАКЛЮЧЕНИЕ
Взаимодействие растворов реагентов – раствора эквимолярной смеси солей висмута и железа и раствора гидроксида натрия – при использовании микрореакторной техники в режиме “затопленных струй” и в условиях смешения растворов реагентов методом свободно сталкивающихся струй с образованием тонкопленочной реакционной зоны (так называемой “жидкостной пелены”) позволило кардинально повысить уровень смешения компонентов при образовании гидроксидов висмута и железа. Синтезированные таким образом гидроксиды при их термическом разложении при 490°C в течение 30 с позволили получить нанокристаллический ортоферрит висмута с узким распределением кристаллитов по размерам и с минимальными размерами кристаллитов (~6–7 нм) без примесей других фаз. Использование метода обратного осаждения даже с применением микрореакторной техники и ультразвуковой обработки не позволило достичь указанных результатов (узкого распределения нанокристаллитов BiFeO3 по размерам и отсутствия примесных фаз) при тех же режимах термообработки гидроксидов. Вместе с тем обнаруженные минимальные размеры кристаллических частиц BiFeO3 при использовании всех рассмотренных в работе вариантов формирования смеси гидроксидов висмута и железа были близки и составляли ~6–7 нм. Это позволяет считать, что данные значения приближаются к значениям предельных минимальных размеров кристаллитов BiFeO3, образующихся в рассматриваемых температурных условиях кристаллизации исходного аморфного вещества. Проведенное исследование показало решающую роль процесса смешения растворов реагентов при образовании смеси гидроксидов висмута и железа на возможность синтеза нанокристаллического ортоферрита висмута с узким распределением кристаллитов по размеру и без примеси других фаз, реализующихся в системе Bi2O3–Fe2O3.
Список литературы
Zvezdin A.K., Pyatakov A.P. // Phys. Usp. 2004. V. 47. P. 416. [Звездин А.К., Пятаков А.П. // Успехи физ. наук. 2004. Т. 174. № 4. С. 465. https://doi.org/10.3367/UFNr.0174.200404n.0465]https://doi.org/10.1070/PU2004v047n04ABEH001752
Bai X., Wei J., Tian B. et al. // J. Phys. Chem. C. 2016. V. 120. № 7. P. 3595. https://doi.org/10.1021/acs.jpcc.5b09945
Esmaili L., Gholizadeh A. // Iranan J. Crystallography and Mineralogy. 2019. V. 26. № 4. P. 1013. https://doi.org/10.29252/ijcm.26.4.1013
Catalan G., Scott J.F. // Adv. Mater. 2009. V. 21. № 24. P. 2463. https://doi.org/10.1002/adma.200802849
Kalinkin A.N., Skorikov V.M. // Russ. J. Inorg. Chem. 2010. V. 55. № 11. P. 1794. [Калинкин А.Н., Скориков В.М. // Журн. неорган. химии. 2020. Т. 55. № 11. С. 1903.]https://doi.org/10.1134/S0036023610110173
Pyatakov A.P., Zvezdin A.K. // Phys. Usp. 2012. V. 55. № 6. P. 557. [Пятаков А.П., Звездин А.К. // Успехи физ. наук. 2012. Т. 182. № 6. С. 593. https://doi.org/10.3367/UFNr.0182.201206b.0593]https://doi.org/10.3367/UFNe.0182.201206b.0593
Egorysheva A.V., Milenov T.I., Ellert O.G. et al. // Solid State Sci. 2015. V. 40. P. 31. https://doi.org/10.1016/j.solidstatesciences.2014.12.011
Morozov M.I., Lomanova N.A., Gusarov V.V. // Russ. J. Gen. Chem. 2003. V. 73. № 11. P. 1676. [Морозов М.И., Ломанова Н.А., Гусаров В.В. // Журн. общей химии. 2003. Т. 73. № 11. С. 1772.]
Silva J., Reyes A., Esparza H. et al. // Integr. Ferroelectr. 2011. V. 126. № 1. P. 47. https://doi.org/10.1080/10584587.2011.574986
Denisov V.M., Belousova N.V., Zhereb V.P. et al. // J. Siberian Federal University. 2012. V. 5. № 2. P. 146. [Денисов В.М., Белоусова Н.В., Жереб В.П. и др. // Журн. Сибирского федерального ун-та. Сер. Химия. 2012. Т. 5. № 2. С. 146. http://elib.sfu-kras.ru/handle/2311/3093]
Sosnowska I., Neumaier T.P., Streichele E. // J. Phys. C: Solid State Phys. 1982. V. 15. № 23. P. 4835. https://doi.org/10.1088/0022-3719/15/23/020
Lomanova N.A., Tomkovich M.V., Osipov A.V. et al. // Phys. Solid State. 2019. V. 61. P. 2535. [Ломанова Н.А., Томкович М.В., Осипов А.В. и др. // Физика твердого тела. 2019. Т. 61. № 12. С. 2503. https://doi.org/10.21883/FTТ.2019.12.48585.549]https://doi.org/10.1134/S1063783419120278
Patel R., Sawadh P. // Nanosyst.: Phys. Chem. Math. 2019. V. 10. № 3. P. 255. https://doi.org/10.17586/2220-8054-2019-10-3-255-265
Bai F., Wang J., Wuttig M. et al. // Appl. Phys. Lett. 2005. V. 86. P. 032511. https://doi.org/10.1063/1.1851612
Sheoran N., Saini M., Kumar A. et al. // MRS Adv. 2019. V. 4. № 28-29. P. 1659. https://doi.org/10.1557/adv.2019.167
Lomanova N.A., Panchuk V.V., Semenov V.G. et al. // Ferroelectrics. 2020. V. 569.
Selbach S.M., Einarsrud M.-A., Grande T. // Chem. Mater. 2009. V. 21. № 1. P. 169. https://doi.org/10.1021/cm802607p
Lomanova N.A., Gusarov V.V. // Nanosyst.: Phys. Chem. Math. 2013. V. 4. № 5. P. 696. http://nanojournal.ifmo.ru/en/wp-content/uploads/2013/10/NPCM_45P696.pdf
Lomanova N.A., Gusarov V.V. // Russ. J. Gen. Chem. 2013. V. 83. № 12. P. 2251. [Ломанова Н.А., Гусаров В.В. // Журн. общей химии. 2013. Т. 83. № 12. С. 1964.]https://doi.org/10.1134/S1070363213120049
Egorysheva A.V., Gaitko O.M., Rudnev P.O. et al. // Russ. J. Inorg. Chem. 2015. V. 60. № 11. P. 1304. [Егорышева А.В., Гайтко О.М., Руднев П.О. и др. // Журн. неорган. химии. 2015. Т. 60. № 11. С. 1425. https://doi.org/10.7868/S0044457X15110045]https://doi.org/10.1134/S0036023615110042
Proskurina O.V., Tomkovich M.V., Bachina A.K. et al. // Russ. J. Gen. Chem. 2017. V. 87. № 11. P. 2507. [Проскурина О.В., Томкович М.В., Бачина А.К. и др. // Журн. общей химии. 2017. Т. 87. № 11. С. 1761.]https://doi.org/10.1134/S1070363217110019
Ortiz-Quiñonez J.-L., Pal U., Villanueva M.S. // Inorg. Chem. 2018. V. 57. № 10. P. 6152. https://doi.org/10.1021/acs.inorgchem.8b00755
Proskurina O.V., Nogovitsin I.V., Il’ina T.S. et al. // Russ. J. Gen. Chem. 2018. V. 88. № 10. P. 2139. [Проскурина О.В., Ноговицин И.В., Ильина Т.С. и др. // Журн. общей химии. 2018. Т. 88. № 10. С. 1699.]https://doi.org/10.1134/S1070363218100183
Gil-González E., Perejón A., Sánchez-Jiménez P.E. et al. // J. Mater. Chem. A. 2018. V. 6. № 13. P. 5356. https://doi.org/10.1039/C7TA09239C
Lomanova N.A., Tomkovich M.V., Sokolov V.V., Gusarov V.V. // Russ. J. Gen. Chem. 2016. V. 86. № 10. P. 2256. [Ломанова Н.А., Томкович М.В., Соколов В.В., Гусаров В.В. // Журн. общей химии. 2016. Т. 86. № 10. С. 1605.]https://doi.org/10.1134/S1070363216100030
Peñalva J., Lazo A. // IOP Conf. Series: J. Phys.: Conf. Ser. 2018. V. 1143. P. 012025. https://doi.org/10.1088/1742-6596/1143/1/012025
Lomanova N.A., Tomkovich M.V., Sokolov V.V. et al. // J. Nanopart. Res. 2018. V. 20. P. 17. https://doi.org/10.1007/s11051-018-4125-6
Ghosh S., Dasgupta S., Sen A., Maiti H.S. // J. Am. Ceram. Soc. 2005. V. 88. P. 1349. https://doi.org/10.1111/j.1551-2916.2005.00306.x
Egorysheva A.V., Kuvshinova T.B., Volodin V.D. et al. // Inorg. Mater. 2013. V. 49. P. 310. [Егорышева А.В., Кувшинова Т.Б., Володин В.Д. и др. // Неорган. матер. 2013. Т. 49. С. 316. https://doi.org/10.7868/s0002337x13030032]https://doi.org/10.1134/S0020168513030035
Sun X., Liu Z., Yu H. et al. // Mat. Lett. 2018. V. 219. P. 225. https://doi.org/10.1016/j.matlet.2018.02.052
Proskurina O.V., Abiev R.S., Danilovich D.P. et al. // Chem. Eng. Process. 2019. V. 143. P. 107598. https://doi.org/10.1016/j.cep.2019.107598
Vladimirova E.V., Dmitriev A.V., Kandaurov M.V. // Russ. J. Inorg. Chem. 2019. V. 64. № 6. P. 689. [Владимирова Е.В., Дмитриев А.В., Кандауров М.В. // Журн. неорган. химии. 2019. Т. 64. № 6. С. 565. https://doi.org/10.1134/S0044457X19060163]https://doi.org/10.1134/S0036023619060160
Selbach S.M., Tybell T., Einarsrud M.A., Grande T. // Chem. Mater. 2007. V. 19. № 26. P. 6478. https://doi.org/10.1021/cm071827w
Popkov V.I., Almjasheva O.V., Semenova A.S. et al. // J. Mater. Sci.: Materials in Electronics. 2017. V. 28. № 10. P. 7163. https://doi.org/10.1007/s10854-017-6676-1
Gusarov V.V. // Russ. J. Gen. Chem. 1997. V. 67. № 12. P. 1846. [Гусаров В.В. Быстропротекающие твердофазные химические реакции // Журн. общей химии. 1997. Т. 67. № 12. С. 1959.]
Missaoui M., Coste S., Barré M. et al. // Nanomater. 2020. V. 10. № 1. P. 26. https://doi.org/10.3390/nano10010026
Egorysheva A.V., Volodin V.D., Ellert O.G. et al. // Inorg. Mater. 2013. V. 49. № 3. P. 303. [Егорышева А.В., Володин В.Д., Эллерт О.Г. и др. // Неорган. материалы. 2013. Т. 49. № 3. С. 308. https://doi.org/10.7868/S0002337X13030020]https://doi.org/10.1134/S0020168513030023
Fonte C.P., Sultan M.A., Santos R.J. et al. // Chem. Eng. J. 2015. V. 260. P. 316. https://doi.org/10.1016/j.cej.2014.08.090
Abiev R.S. // Theoret. Foundations Chem. Engineer. 2020. V. 54. № 6. [Абиев Р.Ш. // ТОХТ. 2020. Т. 54. № 6.]
Proskurina O.V., Sivtsov E.V., Enikeeva M.O. et al. // Nanosyst.: Phys. Chem. Math. 2019. V. 10. № 2. P. 206. https://doi.org/10.17586/2220-8054-2019-10-2-206-214
Ivanov V.K., Fedorov P.P., Baranchikov A.Y., Osiko V.V. // Russ. Chem. Rev. 2014. V. 83. № 12. P. 1204. [Иванов В.К., Федоров П.П., Баранчиков А.Е., Осико В.В. // Успехи химии. 2014. Т. 83. № 12. С. 1204.]https://doi.org/10.1070/RCR4453
Almyasheva O.V., Lomanova N.A., Popkov V.I. et al. // Nanosyst.: Phys. Chem. Math. 2019. V. 10. № 4. P. 428. https://doi.org/10.17586/2220-8054-2019-10-4-428-437
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии