

Журнал неорганической химии, 2021, T. 66, № 11, стр. 1523-1531
О термическом разложении гидроортофосфата церия(IV) Ce(PO4)(HPO4)0.5(H2O)0.5
Т. О. Козлова a, А. Е. Баранчиков a, *, К. В. Биричевская a, Д. А. Козлов a, b, Н. П. Симоненко a, А. В. Гавриков a, М. А. Теплоногова a, b, В. К. Иванов a
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
b Московский государственный университет им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
* E-mail: a.baranchikov@yandex.ru
Поступила в редакцию 21.05.2021
После доработки 28.05.2021
Принята к публикации 31.05.2021
Аннотация
Методом гидротермальной обработки гелей, полученных гидролизом церийсодержащих фосфорнокислых растворов, синтезирован ортофосфат-гидроортофосфат церия(IV) состава Ce(PO4)(HPO4)0.5(H2O)0.5. Полученное соединение и продукты его нагрева в широком температурном диапазоне проанализированы методами рентгенофазового анализа, ИК-спектроскопии, масс-спектрометрического анализа газообразных продуктов термолиза и растровой электронной микроскопии. Уточнены особенности термического разложения указанного соединения при температурах до 1200°С. В частности, показано, что формирование кристаллического ортофосфата церия(III) (CePO4) происходит при нагреве Ce(PO4)(HPO4)0.5(H2O)0.5 на воздухе уже при 600°С.
ВВЕДЕНИЕ
Ортофосфаты редкоземельных элементов (РЗЭ) в трехвалентном состоянии находят широкое применение в различных высокотехнологичных отраслях, в первую очередь для создания люминесцентных материалов, сцинтилляторов для детектирования рентгеновского и гамма-излучения, биоматериалов и др. [1–5]. Ортофосфаты РЗЭ в других степенях окисления (+4) описаны только для церия, но даже они малоизучены несмотря на то, что церий является одним из самых распространенных редкоземельных элементов. Церий в четырехвалентном состоянии, по-видимому, не образует простых средних ортофосфатов [6, 7] (в то же время отметим, что Машином и Долежалем [8] была предложена методика гравиметрического определения содержания церия(IV) в растворах путем его осаждения в виде среднего ортофосфата). Известны примеры достоверно охарактеризованных кристаллических двойных и смешанных ортофосфатов церия(IV) состава (NH4)2Ce(PO4)2 · H2O [9], K2Ce(PO4)2 [10], NH4Ce2(PO4)3 [11], Na10Ce2P6O24 [12], K4CeZr(PO4)4 [13], (NH4)[CeF2(PO4)] [14], а также основного фосфата CeOHPO4 и оксофосфата Ce2O(PO4)2 [15]. Кислые ортофосфаты Ce(IV) склонны к существованию в виде аморфных соединений переменного состава, представляющих собой гелеобразные вещества. Синтез и свойства таких соединений описаны в ряде работ [16–22]. Установлено, что аморфные ортофосфаты Ce(IV) могут обменивать находящиеся в их составе ионы гидроксония на ионы щелочных металлов (Li+, Na+ и K+). Определение химического состава таких соединений оказалось сложной задачей, им был приписан ряд возможных формул, в том числе Ce(HPO4)2 ⋅ H2O [18], Ce(OH)x(PO4)x(HPO4)2 – 2x ⋅ · yH2O, Ce(HPO4)2хН2О, Ce(OH)0.7(PO4)1.1 [19, 20] и даже CePO4 ⋅ H2O [21]. В целом, состав и структура аморфных гидроортофосфатов церия на настоящий момент надежно не установлены.
Структура кристаллических гидроортофосфатов церия(IV) была впервые определена Назарали и др. [23–25] и Бранделом и др. [26]. Так, кристаллический гидроортофосфат церия состава Ce(PO4)(HPO4)0.5(H2O)0.5 (или Ce2(PO4)2HPO4 · ⋅ H2O) был получен при гидротермальной обработке раствора диоксида церия в ортофосфорной кислоте (мольное соотношение P : Ce ~ 2.4) [23]. Данное соединение кристаллизуется в моноклинной сингонии и имеет слоистую структуру (пространственная группа С2/с, параметры элементарной ячейки: а = 21.0142(3) Å, b = 6.55082(7) Å, с = 6.94382(6) Å, β = 91.983(1)°. Другой кристаллический гидроортофосфат церия(IV) состава Ce(H2O)(PO4)1.5(H3O)0.5(H2O)0.5, кристаллизующийся в моноклинной сингонии (пр. гр. С2/c, параметры элементарной ячейки а = 15.7058(17) Å, b = 9.6261(9) Å, с = 10.1632(4) Å, β = 121.623(7)°), также был получен и структурно охарактеризован Назарали и др. [27].
Вышеприведенный краткий обзор охватывает практически все экспериментальные работы, касающиеся аморфных и кристаллических ортофосфатов церия(IV), и многие свойства этих соединений остаются практически неизученными. Несмотря на то, что термолиз гидроортофосфатов церия(IV) изучали в ряде работ [24–26, 28, 29], имеющиеся данные остаются в значительной степени неполными из-за многостадийного характера термического разложения этих соединений.
В настоящей работе впервые с использованием рентгенофазового анализа, ИК-спектроскопии и масс-спектрометрического анализа газообразных продуктов термолиза проведен детальный анализ термического разложения гидроортофосфата церия(IV) состава Ce(PO4)(HPO4)0.5(H2O)0.5, полученного в условиях гидротермальной обработки.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исходных веществ использовали Ce(NO3)3 ⋅ 6H2O (ч. д. а.), H3PO4 (85%-ный водный раствор, ч. д. а.), водный раствор аммиака (~25%, ч. д. а.), изопропанол (ос. ч.), дистиллированную воду.
На первом этапе синтеза получали нанокристаллический диоксид церия (размер частиц, определенный по формуле Селякова–Шеррера, 4–5 нм) [30], для чего водно-изопропанольный раствор нитрата церия(III) добавляли в водный раствор аммиака, полученный CeO2 отделяли, промывали и сушили.
Навеску CeO2 (100 мг) помещали в 5 мл ортофосфорной кислоты и нагревали полученную суспензию при 100°C до получения бесцветного истинного раствора (концентрация по церию – 0.12 М). Полное растворение навески CeO2 происходило за ~15 мин. К полученному раствору добавляли 35 мл дистиллированной воды при интенсивном перемешивании, при этом по всему объему реакционной смеси наблюдали формирование геля. Расчетное мольное соотношение H3PO4 : H2O в полученной смеси составило 1 : 27. Полученный гель подвергали гидротермальной обработке при 180°C в течение 24 ч (степень заполнения автоклава ~40%). После завершения обработки автоклав извлекали и остужали на воздухе. Полученный осадок промывали многократной декантацией относительно дистиллированной воды, затем сушили при 60°C в течение 24 ч на воздухе. Термическую обработку полученных порошков проводили в муфельной печи СНОЛ 10/11 на воздухе путем линейного нагрева (5 град/мин) навесок массой ~50 мг и закалкой на воздух немедленно по достижении температур 300, 450, 500, 600, 650, 680, 710, 740, 770, 900, 950 и 1200°С.
Термический анализ образцов проводили с помощью совмещенного ТГА/ДСК/ДТА анализатора TA Instruments SDT Q-600 в режиме нагрева до 1200°C (10 град/мин) в токе воздуха (100 мл/мин). Дополнительно осуществляли масс-спектрометрический анализ газообразных продуктов, выделяющихся в ходе термического разложения образцов. Для этого использовали термовесы Netzsch TG 209 F1, снабженные масс-спектрометром Netzsch QMS 403 C Aëolos. Анализ проводили в атмосфере аргона при скорости нагрева до 850°C 5 град/мин.
Рентгенофазовый анализ образцов проводили на дифрактометре с вращающимся анодом Rigaku D/MAX 2500 (Rigaku, Япония) в режиме отражения (геометрия Брегга–Брентано) с использованием CuKα1,2-излучения и графитового монохроматора. Получение дифрактограмм для осуществления фазового анализа проводили в низкофоновых кюветах из ориентированного в направлении [510] кремния. Съемку осуществляли в режиме θ–2θ-сканирования с шагом 0.02° по шкале 2θ и временем накопления сигнала 0.5 с в интервале углов 2θ 5°–70°. Для полнопрофильного анализа дифрактограммы регистрировали в диапазоне углов 5°–120° и обрабатывали с использованием программного обеспечения JANA2006 [31]. Базовую линию описывали полиномами Чебышева 4 степени (используя дополнительный обратный член). Несимметричность пиков учитывали по фундаментальным параметрам геометрии съемки.
Морфологию и химический состав образцов анализировали с помощью растрового электронного микроскопа Carl Zeiss NVision 40, оснащенного микрозондовым анализатором Oxford Instruments X-MAX, в диапазоне ускоряющих напряжений 1–20 кВ.
Инфракрасные спектры пропускания образцов регистрировали на спектрометре Bruker ALPHA в диапазоне частот 400–4000 см–1 в режиме нарушенного полного внутреннего отражения.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
По данным рентгенофазового анализа, продуктом гидротермальной обработки церийсодержащего фосфорнокислого геля являлся однофазный гидроортофосфат церия(IV) Ce(PO4)(HPO4)0.5(H2O)0.5 (PDF2 01-075-5561). Уточненные в результате полнопрофильного анализа полученной дифрактограммы (рис. 1) параметры элементарной ячейки полученного соединения составили а = = 21.023(2) Å, b = 6.5586(8) Å, с = 6.9554(8) Å, β = = 91.98(1)°, что удовлетворительно согласуется с данными работы [23].
Рис. 1.
Полнопрофильный анализ дифрактограммы образца гидроортофосфата церия(IV) Ce(PO4)(HPO4)0.5(H2O)0.5.
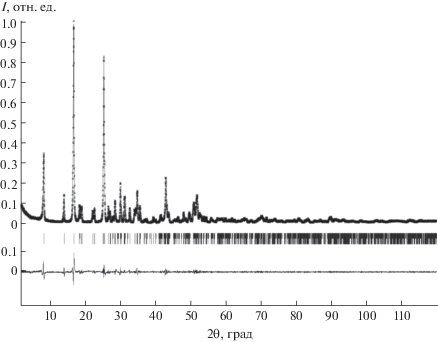
По результатам локального рентгеноспектрального анализа среднее мольное отношение Ce : P в полученном образце составило 1 : 1.5, что соответствует химической формуле Ce(PO4)(HPO4)0.5(H2O)0.5, приписываемой этому соединению.
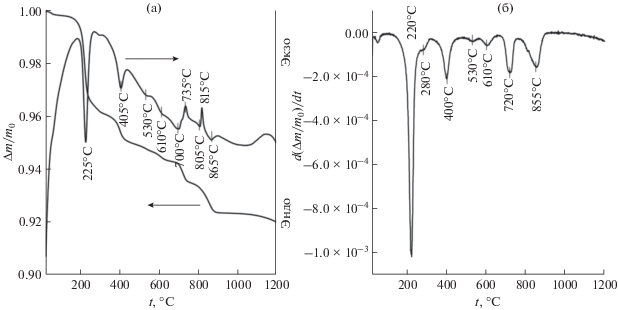
Рассмотрим результаты термического анализа образца гидроортофосфата церия(IV) (рис. 2). Из приведенных на рис. 2 данных следует, что термическое разложение указанного соединения протекает как минимум в 7 стадий (с максимумами скоростей термического разложения при 220, 280, 400, 530, 610, 720 и 855°С). Данные дифференциального термического анализа свидетельствуют о том, что в ходе нагрева образца протекает ряд эндотермических (при температурах 225, 405, 700 и 865°С) и экзотермических процессов (735, 815°С).
Рис. 2.
Результаты термогравиметрического и дифференциального термического анализа образца гидроортофосфата церия(IV) (а) и данные дифференциального термогравиметрического анализа (б).
Результаты масс-спектроскопического анализа газообразных продуктов термолиза Ce(PO4)(HPO4)0.5(H2O)0.5 приведены на рис. 3.
Рис. 3.
Результаты масс-спектроскопического анализа газообразных продуктов, выделяющихся в ходе термолиза гидроортофосфата церия(IV) Ce(PO4)(HPO4)0.5(H2O)0.5 для m/z = 18 (а), 16 (б), 32 (в).
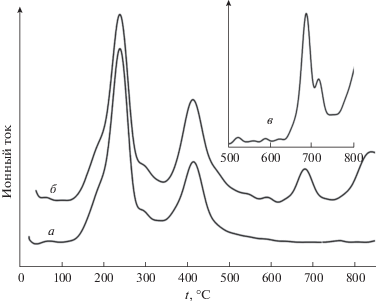
Сопоставление данных термического и масс-спектроскопического анализа (рис. 2 и 3) позволяет заключить, что низкотемпературные стадии потери массы (ниже 500°С) анализируемым соединением связаны с потерей воды, присутствующей в исходном соединении в молекулярной форме, а также в составе гидроортофосфат-ионов. Наиболее вероятным представляется, что при ~200–300°C происходит потеря молекулярной воды, а при ~400°C – дегидратация гидроортофосфат-анионов с образованием конденсированных фосфатов. При анализе данных, приведенных на рис. 3, обращает на себя внимание различное поведение зависимостей для m/z = 18 (H2O) и 16 (O) в области температур выше 600°C, что проявляется в наличии выраженных максимумов для сигнала кислорода (m/z = 16) в области температур ~700 и ~850°C и отсутствии аналогичных максимумов для сигнала воды (m/z = 18). Природа этих максимумов связана, по-видимому, с выделением в ходе термолиза элементарного кислорода, что подтверждается наличием аналогичных максимумов для сигнала двухатомного кислорода (рис. 3, кривая в, m/z = 32).
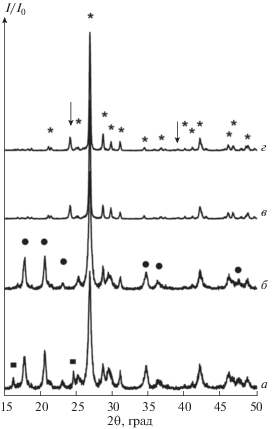
С целью уточнения последовательности фазовых превращений, протекающих в ходе термолиза гидроортофосфата церия(IV), произведен нагрев этого соединения до температур 300, 450, 500, 600, 650, 680°C, 710, 740, 770, 900, 950 и 1200°С с немедленной закалкой на воздух. Дифрактограммы продуктов нагрева приведены на рис. 4 и 6. Отметим, что ранее Брандел и др. [26] указали на то, что термолиз Ce(PO4)(HPO4)0.5(H2O)0.5 при температурах до 700°С сопровождается образованием соединений Ce(PO4)(HPO4)0.5 (образуется при ~300°С) и α-Ce(PO4)(P2O7)0.25 (существует в диапазоне 400–700°С), обладающих структурой, схожей с исходным гидроортофосфатом церия(IV). К сожалению, в этой работе не были приведены ни дифрактограммы продуктов отжига до температур 600°С, ни результаты определения параметров кристаллической решетки продуктов отжига, подтверждающие достоверность сделанных выводов. В свою очередь, полученные нами результаты рентгенофазового анализа указывают на то, что термическое разложение Ce(PO4)(HPO4)0.5(H2O)0.5 до ~700°С протекает через большее количество стадий. Согласно данным термического анализа, первая стадия потеря массы завершается примерно при 250°С, что соответствует удалению половины молекулы воды на формульную единицу Ce(PO4)(HPO4)0.5(H2O)0.5 и формированию соединения с брутто-формулой Ce(PO4)(HPO4)0.5. Дифрактограмма продукта нагрева Ce(PO4)(HPO4)0.5(H2O)0.5 до 300°С имеет существенные отличия от дифрактограммы исходного соединения. В частности, на ней отсутствуют рефлексы при 19.0°, 22.3°, 22.9°, 27.3°, 28.0°, 34.2°, 34.6°, 35.8°, 43.1° 2θ и присутствует ряд новых, и/или положение рефлексов, характерных для исходного соединения, сильно смещено в область больших углов. Такое смещение рефлексов может свидетельствовать об уменьшении расстояния между металл-анионными слоями в структуре гидроортофосфата церия(IV) за счет удаления молекул воды.
Рис. 4.
Дифрактограммы образцов, полученных в результате термической обработки Ce(PO4)(HPO4)0.5(H2O)0.5: исходный образец (а), нагрев до температур 300 (б), 450 (в), 500 (г), 600 (д), 650°C (е), 680 (ж) и 710°C (з). Помеченные звездочкой рефлексы отвечают монациту CePO4 (PDF2 00-032-199).
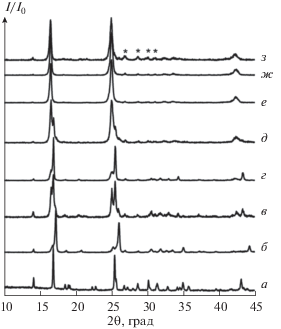
Рис. 5.
ИК-спектры образцов, полученных в результате термической обработки исходного гидроортофосфата церия(IV) (а) при температурах 300 (б), 450 (в), 500 (г), 650°С (д).
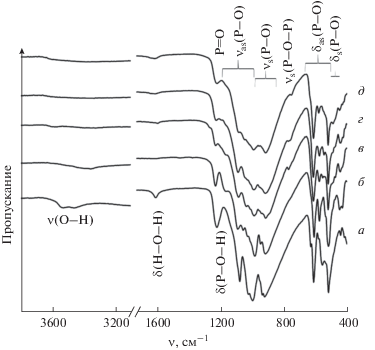
Рис. 6.
Дифрактограммы образцов, полученных в результате нагрева гидроортофосфата церия(IV) до температур 740 (а), 770 (б), 900 (в), 950°С (г). Помеченные рефлексы отвечают (◼) продукту нагрева до 710°С, (⚫) пирофосфату церия CeP2O7 (PDF2 00-016-0584), (↓) триполифосфату церия CeP3O9 (PDF2 00-033-0336), (*) монациту CePO4 (PDF2 00-032-199).
При более высоких температурах (300–450°С), согласно данным термического и рентгенофазового анализа, по всей видимости, реализуется превращение Ce(PO4)(HPO4)0.5 → α-Ce(PO4)(P2O7)0.25, которое завершается примерно при 450°С и носит двухстадийный характер с вероятным образованием промежуточного соединения неустановленного состава (о формировании этого соединения свидетельствует ступень на термогравиметрической кривой в районе 350°С). Отметим, что состав и структура соединений Ce(PO4)(HPO4)0.5 и α-Ce(PO4)(P2O7)0.25 на настоящий момент также надежно не установлены.
Анализ данных, приведенных на рис. 4, показывает, что дифрактограммы продуктов нагрева гидроортофосфата церия(IV) до температур 450 и 650°С содержат близкие по своему положению рефлексы, указывающие на формирование ряда промежуточных соединений с близким химическим составом, который может быть оценен по данным термогравиметрического анализа. Отметим также, что нагрев до 650 и 710°С приводит к формированию продуктов, имеющих почти идентичные дифрактограммы.
В свою очередь, потеря массы (рис. 2а) в диапазоне температур 450–710°С наиболее вероятно обусловлена выделением элементарного кислорода [32] и частичным переходом Ce(IV) –Ce(III), при этом могут образовываться промежуточные соединения близкого химического состава. По данным масс-спектроскопического анализа, выделение элементарного кислорода (рис. 3) при нагреве гидроортофосфата церия(IV) Ce(PO4)(HPO4)0.5(H2O)0.5 надежно зафиксировано в диапазоне свыше 600°С, и на дифрактограммах продуктов нагрева до 600 и 710°С уже можно наблюдать характеристические рефлексы примеси монацита в диапазоне 26°–31° 2θ (рис. 4).
Изменение структуры в ходе термолиза гидроортофосфата церия(IV) до температуры 650°С анализировали методом ИК-спектроскопии.
ИК-спектр исходного соединения полностью соответствует литературным данным [11, 25, 26]. Все представленные на рис. 5 ИК-спектры характеризуются наличием двух основных полос поглощения с разной степенью расщепления в области 1100–900 и 650–440 см–1, относящихся к валентным и деформационным колебаниям ортофосфат-аниона соответственно [33, 34]. В ИК-спектре Ce(PO4)(HPO4)0.5(H2O)0.5 также присутствуют полосы поглощения молекул воды с максимумами в области 3600–3300 и 1610 см–1, отвечающие валентным колебаниям О–H и деформационным колебаниям H–O–H, соответственно. Расщепление полосы ν(O–H) в ИК-спектре гидроортофосфата церия(IV) предположительно связано с присутствием в этом соединении двух типов гидроксо-групп – в молекулах H2O и анионах ${\text{HPO}}_{4}^{{2 - }}$ [35]. Полоса поглощения с максимумом при 1220 см–1 может быть отнесена к колебаниям P–O–H [36–38].
В ИК-спектрах продуктов нагрева до 300°С и выше полоса поглощения с максимумом при 1230 см–1, относится, по-видимому, к колебаниям P=O [39]. В ИК-спектрах продуктов нагрева до 450°С и выше присутствует также характерная для конденсированных фосфатов полоса поглощения в районе 750 см–1, отвечающая колебаниям P–O–P [39, 40].
ИК-спектры продуктов нагрева до 450 и 500°С практически идентичны, что согласуется с данными рентгенофазового анализа. При этом сравнение остальных ИК-спектров между собой позволяет выявить определенные различия, заключающиеся в изменении количества полос поглощения, отвечающих за валентные и деформационные колебания ${\text{PO}}_{4}^{{3 - }},$ и степени их разрешенности. В частности, в ИК-спектре продукта нагрева Ce(PO4)(HPO4)0.5(H2O)0.5 до 300°С присутствует большее количество полос поглощения (рис. 5 и табл. 1) по сравнению с исходным соединением, что может быть связано с искажениями геометрии ортофосфат-иона в поле большого многозарядного катиона (церия) [34]. С увеличением температуры нагрева, напротив, наблюдается уменьшение количества полос поглощения, а также степени их разрешенности, что может быть объяснено совместным присутствием в соответствующих образцах структурных единиц PO4 и P2O7 [41].
Таблица 1.
Отнесение частот максимумов полос поглощения (см–1) в ИК-спектрах исходного гидроортофосфата церия(IV) и продуктах его нагрева
| t, °С | δ(P–O–H) | P=O | νas(P–O) | νs(P–O) | νs(P–O–P) | δas(P–O) | δs(P–O) |
|---|---|---|---|---|---|---|---|
| До нагрева | 1220 | – | 1077, 1021, 999 | 935, 915 | – | 627, 610, 554, 515 | 441 |
| 300 | – | 1230 | 1160, 1087, 1053, 1025, 985 | 945, 915 | – | 610, 573, 515 | 441, 429 |
| 450 | – | 1230 | 1160, 1087, 1035, 985 | 915 | 770 | 610, 573, 554, 515 | 441 |
| 500 | – | 1230 | 1090, 1050, 985 | 915 | 760 | 610, 573, 547, 515 | 441 |
| 650 | – | 1230 | 1085, 1045, 985 | 915 | 750 | 610, 573, 518 | 486, 450 |
Рассмотрим фазовые превращения, происходящие с Ce(PO4)(HPO4)0.5(H2O)0.5 в ходе термолиза при более высоких температурах. В результате нагрева Ce(PO4)(HPO4)0.5(H2O)0.5 до 740 и 770°C (рис. 6) происходит разложение соединения, предположительно присутствующего в продукте нагрева до 710°С, с образованием двухфазной смеси пирофосфата церия(IV) (CeP2O7) и ортофосфата церия(III) (CePO4). Нагрев до 900 и 950°С приводит к исчезновению рефлексов, характерных для CeP2O7. Согласно данным рентгенофазового анализа (рис. 6), при этом происходит разложение пирофосфата церия(IV) по схеме:
(1)
${\text{Ce}}{{{\text{P}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{7}}}} \to {\text{Ce}}{{{\text{P}}}_{{\text{3}}}}{{{\text{O}}}_{{\text{9}}}} + {\text{CeP}}{{{\text{O}}}_{{\text{4}}}} + {\text{O}}_{2}^{ - } \uparrow $Нагрев гидроортофосфата церия(IV) до 1200°C приводит к формированию однофазного монацита (PDF2 00-032-199). Таким образом, триполифосфат церия, формирующийся в результате протекания реакции (1), разлагается с образованием ортофосфата церия(III) по реакции:
(2)
${\text{2Ce}}{{{\text{P}}}_{{\text{3}}}}{{{\text{O}}}_{{\text{9}}}} \to {\text{2CeP}}{{{\text{O}}}_{{\text{4}}}} + {{{\text{P}}}_{{\text{4}}}}{\text{O}}_{{10}}^{ - } \uparrow {\text{,}}$По данным растровой электронной микроскопии, фаза Ce(PO4)(HPO4)0.5(H2O)0.5 имеет пластинчатую морфологию (рис. 7), что согласуется с результатами [23, 26, 29]. В ходе нагрева Ce(PO4)(HPO4)0.5(H2O)0.5 до 300 и 450°C не наблюдается видимых изменений микроструктуры: полученные образцы состоят из частиц пластинчатой формы. При нагреве до 710°C однородность структуры пластинчатых кристаллов нарушается. При дальнейшем увеличении температуры нагрева до 900°С пластинки приобретают зернистую структуру с размером зерен около 100 нм. Термическая обработка при 1200°C приводит к существенному увеличению размера зерен (~500–1000 нм) и образованию ажурных пластинчатых агрегатов. Стоит особо отметить, что пластинчатая структура исходных кристаллов сохраняется для всех продуктов нагрева, что иллюстрирует эффект наследования морфологии в ходе протекания топохимических реакций.
Рис. 7.
Микрофотографии образцов, полученных нагревом Ce(PO4)(HPO4)0.5(H2O)0.5 (а) до температур 300 (б), 450 (в), 710 (г), 900 (д), 1200°С (е).

Отметим, что сохранение структурного мотива и наследование микроструктуры в ходе термолиза было отмечено и для других ортофосфатов церия(IV) [11, 15]. По всей видимости, структуры ортофосфатов четырехвалентного церия являются достаточно устойчивыми при нагреве до температур 500–600°С и не разрушаются при удалении воды. Коллапс структур и образование монацита происходят только при переходе Ce(IV)–Ce(III), сопровождающемся выделением газообразного кислорода.
ЗАКЛЮЧЕНИЕ
Проведен детальный анализ процесса термического разложения гидроортофосфата церия(IV) Ce(PO4)(HPO4)0.5(H2O)0.5 в диапазоне температур 300–1200°С с использованием комплекса взаимодополняющих методов физико-химического анализа. Установлено, что термолиз указанного соединения до температур около 700°С протекает как минимум в 5 стадий и сопровождается образованием ряда промежуточных соединений неустановленного состава, структура которых, вероятно, является искаженной структурой исходного соединения, Ce(PO4)(HPO4)0.5(H2O)0.5. Показано, что при температурах около 600°С происходит выделение кислорода и наблюдается частичный переход церия(IV) в церий(III). Высокотемпературные стадии термолиза Ce(PO4)(HPO4)0.5(H2O)0.5 сопровождаются формированием промежуточных фаз – пирофосфата церия(IV) и триполифосфата церия(III) – с образованием при 1200°С однофазного CePO4. Показан эффект наследования микроструктуры порошков в ходе термолиза Ce(PO4)(HPO4)0.5(H2O)0.5 до температур 1200°С.
Список литературы
The Rare Earth Elements: Fundamentals and Applications / Ed. Atwood D.A. Chichester: Wiley, 2012. 624 p.
Sharma S.K., Behm T., Köhler T. et al. // Crystals. 2020. V. 10. P. 593. https://doi.org/10.3390/cryst10070593
Prokop K., Guzik M., Guyot Y. et al. // Ceram. Int. 2020. V. 46. P. 26350. https://doi.org/10.1016/j.ceramint.2020.03.169
Gai S., Li C., Yang P. et al. // Chem. Rev. 2014. V. 114. P. 2343. https://doi.org/10.1021/cr4001594
Shen J., Suna L.-D., Yan C.-H. // Dalton Trans. 2008. V. 42. P. 5687. https://doi.org/10.1039/B805306E
Borhan A., Apetrăchioaei B., Popa K. // Rev. Roum. Chim. 2010. V. 55. P. 389. http://revroum.lew.ro/wp-content/uploads/2010/RRCh_7_2010/Art%2006.pdf
Tananaev I.V. // XXIVth International Congress of Pure and Applied Chemistry. 1974. P. 75. https://doi.org/10.1016/B978-0-408-70578-3.50007-5
Mašín V., Doležal J. // Analyt. Chim. Acta. 1978. V. 101. P. 413. https://doi.org/10.1016/S0003-2670(01)93377-X
Salvado M.A., Pertierra P., Trobajo C. et al. // J. Am. Chem. Soc. 2007. V. 129. P. 10970. https://doi.org/10.1021/ja0710297
Bevara S., Achary S.N., Patwe S.J. et al. // Dalton Trans. 2016. V. 45. P. 980. https://doi.org/10.1039/C5DT03288A
Shekunova T.O., Istomin S.Ya., Mironov A.V. et al. // Eur. J. Inorg. Chem. 2019. P. 3242. https://doi.org/10.1002/ejic.201801182
Lai Y., Chang Y., Wong T. et al. // Inorg. Chem. 2013. V. 52. P. 13639. https://doi.org/10.1021/ic402208s
Ogorodnyk I.V., Zatovsky I.V., Baumer V.N. et al. // Acta Crystallogr? Sect. C. Cryst. Struct. Commun. 2006. V. 62. P. 100. https://doi.org/10.1107/S0108270106044519
Yu R., Wang D., Takei T. et al. // J. Solid State Chem. 2001. V. 157. P. 180. https://doi.org/10.1006/jssc.2000.9072
Kozlova T.O., Mironov A.V., Istomin S.Y. et al. // Chem. Eur. J. 2020. V. 26. P. 12188. https://doi.org/10.1002/chem.202002527
Hartley W.N. // J. Chem. Soc. 1882. V. 41. P. 202. https://doi.org/10.1039/CT8824100202
Larsen E., Cilley W.A. // J. Inorg. Nucl. Chem. 1968. V. 30. P. 287. https://doi.org/10.1016/0022-1902(68)80092-2
Alberti G., Constantino U., Gregorio F.D. et al. // J. Inorg. Nucl. Chem. 1968. V. 30. P. 295. https://doi.org/10.1016/0022-1902(68)80093-4
Barboux P., Morineau R., Livage J. // Solid State Ionics. 1988. V. 27. P. 221. https://doi.org/10.1016/0167-2738(88)90213-5
Herman R.G., Clearfield A. // J. Inorg. Nucl. Chem. 1975. V. 37. P. 1697. https://doi.org/10.1016/0022-1902(75)80301-0
Rajesh K., Mukundan P., Pillai P.K. et al. // Chem. Mater. 2004. V. 16. P. 2700. https://doi.org/10.1021/cm0499139
König K.-H., Meyn E. // J. Inorg. Nucl. Chem. 1967. V. 29. P. 1153. https://doi.org/10.1016/0022-1902(67)80101-5
Nazaraly M., Wallez G., Chaneac C. et al. // Angew. Chem. Int. Ed. 2005. V. 44. P. 5691. https://doi.org/10.1002/anie.200501871
Nazaraly M., Chanéac C., Ribot F. et al. // J. Phys. Chem. Solids. 2007. V. 68. P. 795. https://doi.org/10.1016/j.jpcs.2007.03.010
Nazaraly M., Wallez G., Chaneac C. et al. // J. Phys. Chem. Solids. 2006. V. 67. P. 1075. https://doi.org/10.1016/j.jpcs.2006.01.028
Brandel V., Clavier N., Dacheux N. // J. Solid State Chem. 2005. V. 178. P. 1054. https://doi.org/10.1016/j.jssc.2005.01.005
Nazaraly M., Quarton M., Wallez G. et al. // Solid State Sci. 2007. V. 9. P. 672. https://doi.org/10.1016/j.solidstatesciences.2007.04.021
Shekunova T.O., Baranchikov A.E., Ivanova O.S. et al. // J. Non-Cryst. Solids. 2016. V. 447. P. 183. https://doi.org/10.1016/j.jnoncrysol.2016.06.012
Sato T., Li R., Sato C. et al. // Phosphorus Res. Bull., 2007. V. 21. P. 44. https://doi.org/10.3363/prb.21.44
Ivanov V.K., Polezhaeva O.S., Baranchikov A.E. et al. // Inorg. Mater. 2010. V. 46. P. 43. https://doi.org/10.1134/S0020168510010103
Petricek V., Dusek M., Palatinus L. // Z. Kristallogr. 2014. V. 229. P. 345. https://doi.org/10.1515/zkri-2014-1737
Bamberger C.E., Begun G.M., Brynestad J. et al. // Radiochim. Acta. 1982. V. 31. P. 57. https://doi.org/10.1524/ract.1982.31.12.57
Clavier N., Mesbah A., Szenknect S. et al. // Spectrochim. Acta, Part A: Mol. Biomol. Spectrosc. 2018. V. 205. P. 85. https://doi.org/10.1016/j.saa.2018.07.016
Skogareva L.S., Shekunova T.O., Baranchikov A.E. et al. // Russ. J. Inorg. Chem. 2016. V. 61. P. 1219. https://doi.org/10.1134/S0036023616100181
Hadrich A., Lautié A., Mhiri T. et al. // Vib. Spectrosc. 2001. V. 26. P. 51. https://doi.org/10.1016/S0924-2031(01)00100-X
Brandel V., Dacheux N., Genet M. et al. // J. Solid State Chem. 2001. V. 159. P. 139. https://doi.org/10.1006/jssc.2001.9143
Brandel V., Dacheux N., Pichot E. et al. // Chem. Mater. 1998. V. 10. P. 345. https://doi.org/10.1021/cm970513d
Pleshko N., Boskey A., Mendelsohn R. // Biophys. J. 1991. V. 60. P. 786. https://doi.org/10.1016/S0006-3495(91)82113-0
Garcia-lodeiro I., Irisawa K., Jin F. et al. // Cem. Concr. Res. 2018. V. 109. P. 243. https://doi.org/10.1016/j.cemconres.2018.04.019
Tomaz A.F., Sobral de Carvalho S.M., Barbosa R.C. et al. // Materials. 2018. V. 11. P. 2051. https://doi.org/10.3390/ma11102051
Dacheux N., Clavier N., Wallez G. et al. // Mater. Res. Bull. 2005. V. 40. P. 2225. https://doi.org/10.1016/j.materresbull.2005.06.011
Hirai H., Masui T., Imanaka N. et al. // J. Alloys Compd. 2004. V. 374. P. 84. https://doi.org/10.1016/j.jallcom.2003.11.069
Masui T., Hirai H., Imanaka N. et al. // Phys. Status Solidi A. 2003. V. 198. P. 364. https://doi.org/10.1002/pssa.200306623
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии