

Журнал неорганической химии, 2019, T. 64, № 11, стр. 1198-1205
Синтез, строение и свойства координационных соединений бромида меди(II) с N-дизамещенными тиокарбамоил-N'-циклогексилсульфенамидами
Г. Н. Масановец 1, Н. В. Хитрич 1, *, И. И. Сейфуллина 1, Л. С. Скороход 1, Н. В. Шматкова 1, Н. Н. Ефимов 2, Е. А. Уголкова 2, В. Г. Власенко 3, А. Л. Тригуб 4, В. В. Минин 2
1 Одесский национальный университет им. И.И. Мечникова
65082 Одесса, ул. Дворянская, 2, Украина
2 Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
3 Научно-исследовательский институт физики, Южный федеральный университет
344090 Ростов-на-Дону, пр-т Стачки, 194, Россия
4 Национальный исследовательский центр “Курчатовский институт”
123182 Москва, пл. Академика Курчатова, 1, Россия
* E-mail: khitrich@ukr.net
Поступила в редакцию 10.04.2019
После доработки 24.04.2019
Принята к публикации 13.05.2019
Аннотация
Взаимодействием эквимолярных количеств CuBr2 в метаноле и RRN–C(=S)–S–NHC6H11 (L) в диэтиловом эфире получены комплексы [CuLBr2], где RR = (C2H5)2, (CH2)5, (CH2)2O(CH2)2. Соединения исследованы методами элементного анализа, ИК-, ЭПР, рентгеновской спектроскопии поглощения, электронной спектроскопии, кондуктометрии, магнетохимии и дериватографии. Методом ИК-спектроскопии установлено, что в комплексах лиганды координированы к меди(II) бидентатно через атомы тионной серы и сульфенамидного азота. Точные значения параметров структуры ближайшего окружения меди(II) определены из анализа EXAFS рентгеновских Cu и BrK-краев поглощения. Длины связей Cu–N, Cu–S и Cu–Br лежат в пределах 2.06–2.08, 2.24–2.49 и 2.33–2.38 Å соответственно. Спектры ЭПР комплексов в растворе ДМФА при 293 K описываются изотропным спиновым гамильтонианом со спином S = 1/2, включающим сверхтонкое взаимодействие с ядерным спином центрального атома меди и дополнительное сверхтонкое взаимодействие с ядерными спинами двух эквивалентных атомов брома и одного атома азота.
ВВЕДЕНИЕ
Высокая реакционная способность и склонность к хелатообразованию тиокарбамоилсульфенамидов RRNC(=S)S–NR’R’ (L) обусловлены наличием в их молекулах лабильной связи S–N, относительно легко расщепляющейся под действием как нуклеофильных, так и электрофильных реагентов, а также атомов тионной серы и сульфенамидного азота, обладающих донорными свойствами. Следует отметить, что среди различных производных дитиокарбамовой кислоты RRNC(=S)S–H тиокарбамоилсульфенамиды наименее изучены, вероятно, из-за их нестабильности, связанной с особенностью строения сульфенамидной группы. К настоящему времени на их основе нами впервые получено около 50 различных координационных соединений, состав и строение которых зависят от условий синтеза, природы металла-комплексообразователя, аниона его соли и заместителей в молекулах тиокарбамоилсульфенамидов [1]. Установлено, что в результате реакции между MX2 (M = Co, Zn, X = Cl, Br, I, NCS; M = Cu, X = Cl) и тиокарбамоилсульфенамидами (RR = R'R' = (CH3)2; RR = (CH3)2, (C2H5)2, (CH2)5, (CH2)6, (CH2)2O(CH2)2, R'R' = (CH2)5) в смешанных органических растворителях образуются координационные соединения [MLX2] [2–8] либо [CuL2]Cl2 [8], а в случае 1,4-пиперазин-бис(карботиосульфендиэтиламида) – биядерные [M2LX4] [8–10]. Во всех этих соединениях лиганды координированы к ионам металлов бидентатно через атомы тионной серы и сульфенамидного азота с образованием пятичленных хелатных металлоциклов. Были синтезированы также димеры [Zn2L2X4] (RR = R'R' = (CH3)2, X = = Cl, Br, I) с мостиковыми атомами галогенов и монодентатной координацией L через тионный атом серы [11]. Следует отметить, что полученные соединения представляют не только научный, но и практический интерес как полифункциональные химикаты-добавки для переработки высокомолекулярных соединений (ускорители вулканизации резиновых смесей, промоторы адгезии резин к металлокорду [4], первичные и вспомогательные сиккативы [2], активаторы полимеризации виниловых мономеров [12]), а также как модификаторы активности различных ферментов [13, 14]. Показано, что комплексы Co(II) с тиокарбамоилсульфенамидами проявляют высокую каталазную активность [9, 15].
Настоящая работа является продолжением этих исследований и посвящена синтезу, изучению состава, строения и свойств комплексов CuBr2 с N,N-диэтил-, N-пентаметилен- и N-оксидиэтилентиокарбамоил-N'-циклогексилсульфенамидами. Такой выбор комплексообразователя и лигандных систем сделан в связи с отсутствием в литературе сведений о комплексах CuBr2 с тиокарбамоилсульфенамидами, в которых атом сульфенамидного азота, обычно участвующий в координации, не полностью замещен, а также перспективой создания новых эффекторов ферментов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
N,N-диэтил- (L1), N-пентаметилен- (L2) и N-оксидиэтилентиокарбамоил-N'-циклогексилсуль-фенамиды (L3) получали окислительной конденсацией соответствующих дитиокарбаматов натрия и циклогексиламина с использованием в качестве окислителя иода [16].
Синтез комплексов [CuLBr2], L = L1 (I), L2 (II), L3 (III). К раствору, содержащему 0.005 моля соответствующего L в 25 мл диэтилового эфира, при интенсивном перемешивании добавляли по каплям раствор CuBr2 (0.005 моля) в 10 мл метанола. Образовавшиеся мелкокристаллические осадки комплексов I–III отфильтровывали, промывали метанолом, диэтиловым эфиром и высушивали в вакууме.
Комплекс I – красно-коричневые кристаллы. Выход 76%.
| Br | Cu | N | S | |
| Найдено, %: | 34.41; | 13.44; | 6.11; | 13.84. |
| Для C11H22Br2CuN2S2 | ||||
| вычислено, %: | 34.02; | 13.53; | 5.96; | 13.65. |
Электронный спектр поглощения (ЭСП) в CHCl3, λмакс, нм (lg ε): 276 (4.14), 344 (3.77), 373 (3.78), 541 (2.96).
Комплекс II – коричневые кристаллы. Выход 78%.
| Br | Cu | N | S | |
| Найдено, %: | 33.34; | 13.01; | 5.94; | 13.18. |
| Для C12H22Br2CuN2S2 | ||||
| вычислено, %: | 33.17; | 13.19; | 5.81; | 13.31. |
ЭСП (CHCl3), λмакс, нм (lg ε): 263 (4.03), 341 (3.89), 377 (4.20), 550 (3.17).
Комплекс III – темно-коричневые кристаллы. Выход 80%.
| Br | Cu | N | S | |
| Найдено, %: | 32.88; | 13.29; | 5.88; | 13.36. |
| Для C11H20Br2CuN2OS2 | ||||
| вычислено, %: | 33.03; | 13.14; | 5.79; | 13.25. |
ЭСП (CHCl3), λмакс, нм (lg ε): 273 (4.28), 343 (3.74), 377 (3.89), 541 (2.98).
Элементный анализ. Содержание меди в синтезированных соединениях определяли комплексонометрическим титрованием [17], брома и серы – колбовым методом Шенигера, азота – методом Дюма [18].
Удельную электропроводность 0.001 М растворов комплексов в органических растворителях измеряли на кондуктометре Эксперт-002 при 25°С.
ИК-спектры поглощения образцов (суспензии в вазелиновом масле и таблетки с KBr) записывали на ИК-Фурье-спектрометре Frontier FT-IR компании Perkin–Elmer в интервале волновых чисел 200–4000 см–1.
Рентгеновские Cu и BrK-края поглощения комплексов I–III в твердом состоянии получали в режиме пропускания на EXAFS-спектрометре станции Структурного материаловедения в Курчатовском синхротронном центре (Москва) [19]. Энергия электронного пучка, использованного в качестве источника рентгеновского синхротронного излучения, составляла 2.5 ГэВ при токе 100–120 мА. Для монохроматизации рентгеновского излучения использовали двухкристальный Si(111) монохроматор. Обработку полученных спектров осуществляли с помощью стандартных процедур выделения фона, нормирования на величину скачка K-края и выделения атомного поглощения μ0, затем проводили Фурье-преобразование полученных EXAFS (χ)-спектров в интервале волновых векторов фотоэлектронов k от 2.5 до 13.5 Å–1 с весовой функцией k3. Полученные модули Фурье-преобразования (МФТ) представляли собой псевдорадиальное распределение атомов ближайших координационных сфер (КС) вокруг поглощающих атомов со значениями радиусов КС, получаемых с точностью до фазовых поправок. Пороговую энергию ионизации E0 выбирали по значению максимума первой производной K-края и в дальнейшем варьировали при подгонке. Точные значения параметров структуры ближайшего окружения ионов меди(II) и брома в соединениях определяли путем нелинейной подгонки параметров соответствующих КС при сопоставлении рассчитанного EXAFS-сигнала и выделенного из полного EXAFS-спектра методом Фурье-фильтрации МФТ. Указанную нелинейную подгонку производили с использованием пакета программ IFFEFIT-1.2.11 [20]. Необходимые для построения модельного спектра фазы и амплитуды рассеяния фотоэлектронной волны рассчитывали с использованием программы FEFF7 [21] и атомных координат соединений с близкой атомной структурой [3, 5, 22]. Функцию качества подгонки $\Re ,$ минимизацию которой проводили при нахождении параметров структуры ближайшего окружения, рассчитывали по формуле:
ЭСП растворов соединений в CHCl3 регистрировали на спектрофотометре СФ-56 в области 240–1100 нм.
Магнитную восприимчивость поликристаллических образцов определяли методом Гуи в температурном интервале 100–293 K. В качестве эталона для калибровки использовали Co[Hg(CNS)4]. Диамагнитные поправки вводили по инкрементам, взятым из [23].
Спектры электронного парамагнитного резонанса (ЭПР) комплексов меди(II) как в поликристаллическом виде, так и в растворе ДМФА регистрировали на радиоспектрометре E-680X Elexsys фирмы Bruker в X-диапазоне при комнатной температуре. Параметры спектров ЭПР находили методом наилучшего приближения между экспериментальными и теоретическими спектрами путем минимизации функционала ошибки:
Термическую устойчивость соединений изучали в платиновых тиглях на Q-дериватографе системы F. Paulik, J. Paulik, L. Erdey (МОМ, Будапешт) в воздушной среде в интервале температур 20–1000°С. Управление работой дериватографа, усовершенствованного системой автоматического управления и регистрации явлений, и обработку дериватограмм осуществляли с помощью персонального компьютера. Нагревание проводили со скоростью 10 град/мин. Масса образцов составляла 200 мг. В качестве эталона использовали Al2O3.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Из результатов элементного анализа видно, что при взаимодействии эквимолярных количеств CuBr2 в метаноле с RRN–C(=S)–S–NHC6H11, где RR = (C2H5)2, (CH2)5, (CH2)2O(CH2)2, в диэтиловом эфире образуются комплексы I–III с мольным соотношением Cu : L = 1 : 1. Комплексы такого же состава, но с меньшим выходом были получены при использовании ацетона или смеси ацетон–диэтиловый эфир в качестве растворителей. Осуществить синтез указанных комплексов в метаноле, этаноле или ацетонитриле не удалось. Варьирование в широких пределах (от 6 : 1 до 1 : 6) соотношения CuBr2 : L не привело к образованию соединений другого состава.
Комплексы I–III хорошо растворимы в ДМФА, ДМСО, ацетонитриле, ацетоне, хуже – в метаноле, хлороформе. Величины молярной электропроводности 0.001 М растворов [CuLBr2] в ацетоне (15–23 Ом–1 см2 моль–1) ниже, чем для электролитов типа 1 : 1 [27], что позволяет отнести их к неэлектролитам. Следует отметить, что растворы комплексов I–III в ацетоне, хлороформе окрашены в коричневый цвет, такой же, как в твердом виде, а в ацетонитриле, метаноле – в зеленый. Изменение цвета растворов сопровождалось увеличением молярной электропроводности, которая намного превышала значения для электролитов типа 1 : 1.
Для определения центров локализации координационных связей был проведен сравнительный
анализ ИК-спектров L и [CuLBr2]. Поскольку ИК-спектры комплексов, запрессованных в таблетки с KBr, были искажены
из-за взаимодействия с KBr, их ИК-спектры изучали в виде суспензий в вазелиновом масле.
В ИК-спектрах L и [CuLBr2] достаточно сложно различить колебания отдельных связей C–N, C=S, C–C и др. из-за
их существенного смешения, характерного для соединений с тиоамидной группой 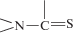 , поэтому для интерпретации ИК-спектров была использована концепция тиоамидных полос
I–V [28–30]. Полосы I (1480–1540 см–1), IV (960–980 см–1) и V (530–585 см–1) обусловлены в основном вкладами ν(C–N), ν(C=S) и δ(NCS) соответственно, а полосы
II (1230–1280 см–1) и III (1120–1160 и 1010–1050 см–1) следует рассматривать как скелетные колебания со значительным вкладом всех связей
тиоамидной системы и ее ближайшего окружения [30]. Отнесение основных колебательных частот в ИК-спектрах соединений приведено в табл. 1. Видно, что частоты тиоамидных полос II, III и V в спектрах тиокарбамоилсульфенамидов
и их комплексов мало различаются. Наиболее чувствительная к структурным изменениям
тиоамидная полоса I в ИК-спектрах L наблюдается при 1477–1488 см–1, т.е. занимает промежуточное положение между ν(C=N) (1640–1690 см–1) и ν(C–N) (1250–1360 см–1). Теоретически строение дитиокарбаматного иона можно представить в виде трех равновероятных
канонических структур, отличающихся длиной и кратностью связи C–N:
, поэтому для интерпретации ИК-спектров была использована концепция тиоамидных полос
I–V [28–30]. Полосы I (1480–1540 см–1), IV (960–980 см–1) и V (530–585 см–1) обусловлены в основном вкладами ν(C–N), ν(C=S) и δ(NCS) соответственно, а полосы
II (1230–1280 см–1) и III (1120–1160 и 1010–1050 см–1) следует рассматривать как скелетные колебания со значительным вкладом всех связей
тиоамидной системы и ее ближайшего окружения [30]. Отнесение основных колебательных частот в ИК-спектрах соединений приведено в табл. 1. Видно, что частоты тиоамидных полос II, III и V в спектрах тиокарбамоилсульфенамидов
и их комплексов мало различаются. Наиболее чувствительная к структурным изменениям
тиоамидная полоса I в ИК-спектрах L наблюдается при 1477–1488 см–1, т.е. занимает промежуточное положение между ν(C=N) (1640–1690 см–1) и ν(C–N) (1250–1360 см–1). Теоретически строение дитиокарбаматного иона можно представить в виде трех равновероятных
канонических структур, отличающихся длиной и кратностью связи C–N:
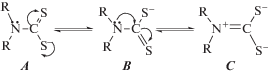
Таблица 1.
Основные колебательные частоты (см–1) в ИК-спектрах тиокарбамоилсульфенамидов L1–L3 и комплексов I–III (суспензии в вазелиновом масле)
| Соединение | Тиоамидные полосы | ν(NH) | ν(SN) | ν(CuN) | ν(CuS) | ν(CuBr) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | ||||||
| L1 | 1488 | 1266 | 1146, 1062 | 977 | 559 | 3216 | 645 | – | – | – |
| I | 1512 | 1277 | 1146, 1062 | 940 | 560 | 3086 | 686 | 496 | 354 | 230, 218, 215 |
| L2* | 1477 | 1239 | 1133, 1060 | 968 | 552 | 3229 | 645 | – | – | – |
| II | 1516 | 1246 | 1137, 1062 | 938 | 556 | 3101 | 685 | 500 | 347 | 230, 222, 214 |
| L3* | 1477 | 1266 | 1110, 1067 | 982 | 538 | 3263, 3253 | 618 | – | – | – |
| III | 1493 | 1273 | 1111, 1063 | 942 | 540 | 3103 | 686 | 494 | 338 | 230, 222, 214 |
В ИК-спектрах комплексов I–III тиоамидная полоса I смещается на 16–39 см–1 в высокочастотную область, что свидетельствует об увеличении кратности связи C–N и большом вкладе резонансной формы C в их структуру. Смещение в низкочастотную область на 30–40 см–1 тиоамидной полосы IV, ответственной в основном за валентные колебания связи C=S, указывает на участие атомов тионной серы в координации. Валентные колебания связей Cu–S проявляются в области 338–354 см–1. Полосы валентных колебаний связей N–H в ИК-спектрах [CuLBr2] существенно смещаются в низкочастотную, а связей S–N – в высокочастотную область по сравнению с их положением в ИК-спектрах L (табл. 1). Это происходит в результате вовлечения сульфенамидного азота в координацию. Полосы в области 494–500 см–1 отнесены к ν(Cu–N), а в области 214–230 см–1 – к ν(Cu–Br). Таким образом, методом ИК-спектроскопии установлено, что в [CuLBr2] лиганды координированы к меди(II) бидентатно через атомы тионной серы и сульфенамидного азота:
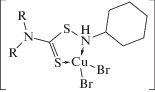
Анализ XANES и EXAFS Cu и BrK-краев поглощения комплексов I–III подтвердил такое окружение меди(II) и брома. Нормированные XANES CuK-краев поглощения и их первых производные (рис. 1) демонстрируют существенные различия, указывая, что геометрия ближайшего окружения меди(II) в этих комплексах меняется в зависимости от вида координированного тиокарбамоилсульфенамида RRN–C(=S)–S–NHC6H11. В то же время XANES BrK-краев и их первые производные поглощения близки по своей форме и энергетическим характеристикам для комплексов I–III, из этого следует, что атомы брома имеют схожую координацию к ионам меди(II). Количественные характеристики локального атомного окружения меди(II) и брома получены из анализа EXAFS рентгеновских Cu и BrK-краев поглощения комплексов I–III. Из рис. 2 видно, что все МФТ EXAFS Cu и BrK-краев поглощения состоят из основного пика при r = 1.90–2.00 Å, который соответствует рассеянию на ближайших КС, и нескольких пиков с меньшей амплитудой, соответствующих дальним КС. Результаты расчетов параметров локального атомного окружения меди(II) и брома в комплексах I–III при выборе соответствующих моделей атомного строения координационного узла (табл. 2) указывают на близкие структурные параметры ближайшего окружения во всех комплексах. Вокруг меди(II) на расстояниях 2.34–2.36 Å находятся два атома брома, на расстояниях 2.06–2.08 и 2.24–2.49 Å – один атом азота и один атом серы соответственно. Каждый атом брома координирован к одному иону меди(II) (длина связи 2.33–2.38 Å). Длины связей Cu–Br и Br–Cu, полученные из анализа EXAFS Cu и BrK-краев поглощения соответственно, равны с точностью до ошибки эксперимента. Связь Cu–S в комплексе I существенно длиннее (2.49 Å), чем в II, III (2.24 Å), что может быть причиной различия в XANES этих соединений. С целью получения дополнительных данных о структурно-геометрических параметрах комплексов I–III их модельные структуры были оптимизированы методом теории функционала плотности (DFT) с использованием гибридного функционала B3LYP [31, 32] с базисным набором 6-311G** [33]. Рассчитанные квантово-химически значения межатомных расстояний в комплексах I–III (табл. 3) хорошо согласуются с экспериментально установленными из анализа EXAFS рентгеновских Cu и BrK-краев поглощения (табл. 2), что подтверждает выводы о строении комплексов.
Рис. 1.
XANES-спектры Cu (а) и BrK-краев поглощения (б) комплексов I–III. На вставках показаны первые производные K-краев.
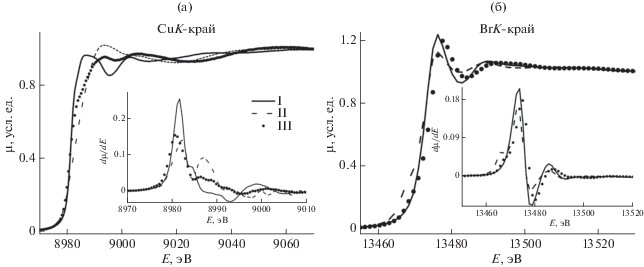
Рис. 2.
Модули Фурье-преобразования EXAFS-спектров Cu (а) и BrK-краев поглощения (б) комплексов I–III (сплошными линиями показаны экспериментальные данные, кружками – результаты расчетов).
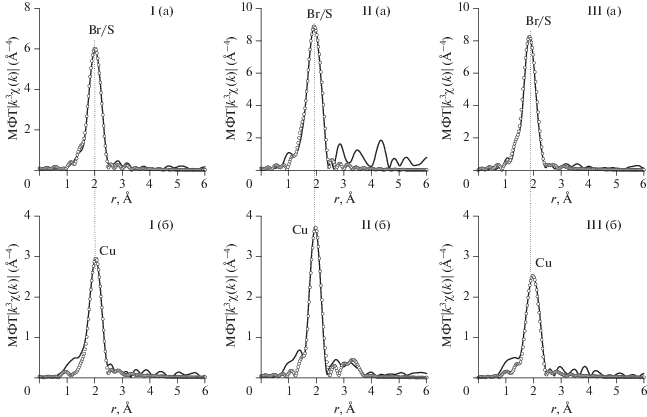
Таблица 2.
Параметры локального атомного окружения ионов меди(II) и брома в комплексах I–III, полученные из многосферной подгонки EXAFS Cu и BrK-краев поглощения (N – координационное число, R – межатомные расстояния, σ2 – фактор Дебая–Валлера, ℜ – функция качества подгонки)
| Комплекс | N | R, Å | σ2, Å2 | Атом | ℜ, % |
|---|---|---|---|---|---|
| I* | 1 | 2.08 | 0.0045 | N | 4.1 |
| 1 | 2.49 | 0.0056 | S | ||
| 2 | 2.36 | 0.0076 | Br | ||
| I** | 1 | 2.38 | 0.0069 | Cu | 5.0 |
| II* | 1 | 2.06 | 0.0045 | N | 4.5 |
| 1 | 2.24 | 0.0050 | S | ||
| 2 | 2.34 | 0.0063 | Br | ||
| II** | 1 | 2.33 | 0.0062 | Cu | 2.1 |
| III* | 1 | 2.06 | 0.0045 | N | 5.6 |
| 1 | 2.24 | 0.0060 | S | ||
| 2 | 2.35 | 0.0063 | Br | ||
| III** | 1 | 2.34 | 0.0080 | Cu | 3.3 |
Таблица 3.
Рассчитанные (DFT/B3LYP/6‑311G**) значения межатомных расстояний (R) в комплексах I–III
| Комплекс | R, Å | ||
|---|---|---|---|
| Cu–N | Cu–S | Cu–Br | |
| I | 2.09 | 2.52 | 2.31, 2.37 |
| II | 2.08 | 2.40 | 2.34, 2.40 |
| III | 2.08 | 2.41 | 2.33, 2.40 |
Эффективные магнитные моменты комплексов I, II и III при 293 K равны соответственно 1.82, 1.78 и 1.73 М.Б. и не изменяются при понижении температуры до 100 K, что указывает на их моноядерное строение.
Для установления геометрического и электронного строения комплексов I–III были изучены их спектры ЭПР как в поликристаллическом виде, так и в растворе ДМФА (рис. 3). Спектры ЭПР поликристаллических образцов малоинформативны, тогда как в спектрах комплексов в растворе наблюдается богатая СТС от двух эквивалентных атомов брома. Эти спектры описываются изотропным спиновым гамильтонианом (СГ) со спином S = 1/2, включающим сверхтонкое взаимодействие с ядерным спином центрального атома меди и дополнительное сверхтонкое взаимодействие с ядерными спинами двух эквивалентных атомов брома и одного азота:
где g – усредненная компонента g-тензора мономера; A – усредненная компонента СТС-тензора атома меди; a – усредненная компонента ДСТС-тензора атомов лигандов, выраженная в гауссах. Полученные параметры СГ приведены в табл. 4.Рис. 3.
Спектры ЭПР комплексов I–III в поликристаллическом виде (а) и в растворе ДМФА (б). T = 293 K. 1 – эксперимент, 2 – теория.
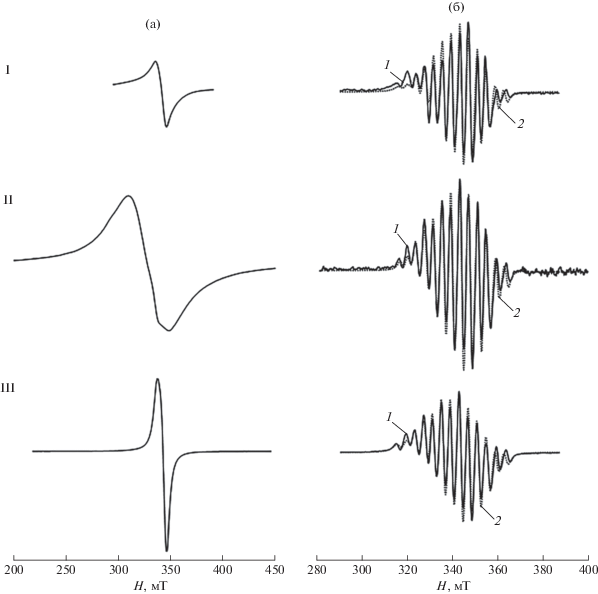
Таблица 4.
Параметры СГ спектров ЭПР комплексов I–III в растворе ДМФА и в поликристаллическом виде (в скобках). T = 293 K
| Комплекс | g | A × 103, см–1 | a(Br), Гс | a(N), Гс |
|---|---|---|---|---|
| I | 2.062 (2.056) | 7.44 | 38.38 | 4.85 |
| II | 2.061 (2.141) | 7.38 | 39.79 | 5.66 |
| III | 2.063 (2.047) | 7.24 | 40.66 | 5.28 |
ЭСП растворов комплексов I–III в хлороформе независимо от заместителей у атома тиокарбамоильного азота характеризуются подобными по форме, положению и разными по интенсивности полосами поглощения. Полосы при 541–550 нм отнесены к d–d-переходу, а при 373–377 и 341–344 нм – к d → π-переходам между центральным атомом меди и лигандами. В ЭСП комплексов [CuLBr2] трудно различить n → σ*- и π → π*-переходы, наблюдаемые в спектрах исходных L при 284–287 и 260–265 нм соответственно, поскольку они сливаются в одну широкую полосу с максимумом поглощения при 263–276 нм.
Исследование термической устойчивости комплексов I–III показало, что их термолиз протекает однотипно и имеет многоступенчатый характер (табл. 5). Термическая деструкция [CuLBr2] начинается при более низких температурах, чем исходных L. В узком температурном интервале (90–110°С) на кривых ДТА комплексов I–III наблюдаются четко выраженные экзоэффекты, сопровождаемые потерей незначительной части начальной массы образцов. Основная потеря массы органической составляющей соединений происходит в интервале 260–420°С и протекает с поглощением теплоты. Интересной особенностью термодеструкции комплексов I–III является образование при нагревании выше 550°С летучих медьсодержащих частиц и практически полное отсутствие остатков, тогда как обычно конечным продуктом термолиза координационных соединений меди(II) на воздухе является CuO.
Таблица 5.
Результаты термического анализа комплексов I–III
| Комплекс | Интервал температур по ТГ, °С | Tмакс экзо- (↑) и эндоэффектов (↓), °С | Суммарная потеря массы, % |
|---|---|---|---|
| I | 50–110 | 105 (↑) | 8.4 |
| 110–170 | 155 (↓) | 11.1 | |
| 170–260 | 200 (↓) | 14.6 | |
| 260–410 | 355 (↓) | 60.0 | |
| 410–800 | 455 (↑), 555 (↑) | 94.6 | |
| II | 50–105 | 100 (↑) | 7.0 |
| 105–200 | 165 (↑) | 12.1 | |
| 280–465 | 370 (↓), 390 (↓) | 57.0 | |
| 465–820 | 485 (↓), 590 (↑) | 98.6 | |
| III | 50–105 | 100 (↑) | 6.6 |
| 105–195 | 170 (↓) | 13.6 | |
| 195–270 | 235 (↑) | 19.3 | |
| 270–450 | 350 (↓), 400 (↓) | 54.4 | |
| 450–770 | 555 (↑) | 96.8 |
ЗАКЛЮЧЕНИЕ
В результате проведенного исследования синтезированы новые хелатные комплексы [CuLBr2], где L = N,N-диэтил-, N-пентаметилен- и N-оксидиэтилентиокарбамоил-N'-циклогексилсульфенамид, установлены их состав и строение, изучены спектральные, магнитные и термические свойства. Показано, что в отличие от ранее полученных N,N-диметил- и N,N-диэтилтиокарбамоил-N'-пентаметиленсульфенамидных комплексов меди(II) с мольным соотношением Cu : L = 1 : 1 и 1 : 2 [8] взаимодействие CuBr2 с N-дизамещенными тиокарбамоил-N'-циклогексилсульфенамидами независимо от условий синтеза приводит к образованию комплексов только эквимолярного состава.
Список литературы
Масановец Г., Сейфуллина И., Хитрич Н. Координационные соединения кобальта(II), меди(II) и цинка(II) с тиокарбамоилсульфенамидами. Saarbrücken: LAP, 2012. 130 с.
Хитрич Г.Н., Лебедева Е.Ю., Сейфуллина И.И., Пушкарев Ю.Н. // Вісн. Одеськ. нац. ун-ту. Хімія. 2009. Т. 14. № 3. С. 57.
Khitrich G.N., Seifullina I.I., Vologzhanina A.V. // Mendeleev Commun. 2010. V. 20. № 3. P. 180. https://doi.org/10.1016/j.mencom.2010.05.020
Сейфуллина И.И., Хитрич Г.Н., Овчаров В.И., Охтина О.В. // Вопр. химии и хим. технологии. 2010. № 3. С. 111.
Seifullina I.I., Khitrich G.N., Vologzhanina A.V. // Russ. J. Inorg. Chem. 2011. V. 56. № 2. P. 184. https://doi.org/10.1134/S0036023611020252
Khitrich G.N., Seifullina I.I., Khitrich N.V. // Russ. J. Gen. Chem. 2011. V. 81. № 5. P. 840. https://doi.org/10.1134/S1070363211050070
Масановец Г.Н., Сейфуллина И.И. // Укр. хим. журн. 2012. Т. 78. № 7. С. 50.
Хитрич Г.Н., Сейфуллина И.И., Зуб В.Я. // Укр. хим. журн. 2011. Т. 77. № 5. С. 12.
Chikhichin D.G., Kotseruba V.A., Levchenko O.A. et al. // Theor. Exp. Chem. 2011. V. 47. № 2. P. 115. https://doi.org/10.1007/s11237-011-9191-0
Масановец Г.Н., Сейфуллина И.И. // Вісн. Одеськ. нац. ун-ту. Хімія. 2012. Т. 17. № 2. С. 26.
Khitrich G.N., Seifullina I.I. // Theor. Exp. Chem. 2010. V. 46. № 5. P. 334. https://doi.org/10.1007/s11237-010-9162-x
Грекова А.В., Иванченко П.А., Сейфуллина И.И., Хитрич Г.Н. // Вісн. Одеськ. нац. ун-ту. Хімія. 2010. Т. 15. № 12–13. С. 97.
Варбанец Л.Д., Мацелюх Е.В., Гудзенко Е.В. и др. // Укр. біохім. журн. 2011. Т. 83. № 3. С. 25.
Варбанець Л.Д., Мацелюх О.В., Нідялкова Н.А. та інш. // Biotechnol. Acta. 2013. V. 6. № 1. P. 73. https://doi.org/10.15407/biotech6.01.073
Chihichin D.G., Kotseruba V.A., Levchenko O.A et al. // Russ. J. Gen. Chem. 2013. V. 83. № 5. P. 915. https://doi.org/10.1134/S107036321305006X
Smith G.E.P., Alliger G., Carr E.L., Young K.C. // J. Org. Chem. 1949. V. 14. № 6. P. 935. https://doi.org/10.1021/jo01158a002
Шварценбах Г., Флашка Г. Комплексонометрическое титрование. М.: Химия, 1970. 360 с.
Климова В.А. Основные микрометоды анализа органических соединений. М.: Химия, 1975. 224 с.
Chernyshov A.A., Veligzhanin A.A., Zubavichus Y.V. // Nucl. Instrum. Methods. Phys. Res., Sect. A. 2009. V. 603. № 1–2. P. 95. https://doi.org/10.1016/j.nima.2008.12.167
Newville M. // J. Synchrotron Rad. 2001. V. 8. № 2. P. 96. https://doi.org/10.1107/S0909049500016290
Zabinsky S.I., Rehr J.J., Ankudinov A. et al. // Phys. Rev. B. 1995. V. 52. № 4. P. 2995. https://doi.org/10.1103/PhysRevB.52.2995
van de Leemput P.J.H.A.M., Cras J.A., Willemse J. et al. // Recl. Trav. Chim. Pays-Bas. 1976. V. 95. № 7–8. P. 191. https://doi.org/10.1002/recl.19760950713
Калинников В.Т., Ракитин Ю.В. Введение в магнетохимию. Метод статической магнитной восприимчивости в химии. М.: Наука, 1980. 302 с.
Ракитин Ю.В., Ларин Г.М., Минин В.В. Интерпретация спектров ЭПР координационных соединений. М.: Наука, 1993. 399 с.
Лебедев Я.С., Муромцев В.И. ЭПР и релаксация стабилизированных радикалов. М.: Химия, 1972. С. 25.
Wilson R., Kivelson D. // J. Chem. Phys. 1966. V. 44. № 1. P. 154. https://doi.org/10.1063/1.1726439
Geary W.J. // Coord. Chem. Rev. 1971. V. 7. № 1. P. 81. https://doi.org/10.1016/s0010-8545(00)80009-0
Rao C.N.R., Venkataraghavan R. // Spectrochim. Acta. 1962. V. 18. № 4. P. 541. https://doi.org/10.1016/S0371-1951(62)80164-7
Jensen K.A., Nielsen P.H. // Acta Chem. Scand. 1966. V. 20. № 3. P. 597. https://doi.org/10.3891/acta.chem.scand.20-0597
D$\overset{\lower0.5em\hbox{$\smash{\scriptscriptstyle\smile}$}}{a} $escu C., Bacaloglu R., Ostrogovich G. // Bull. sti. si tehn. Inst. politehn. Timisoara. Ser. chim. 1973. V. 18. № 2. P. 121.
Becke A.D. // J. Chem. Phys. 1993. V. 98. № 7. P. 5648. https://doi.org/10.1063/1.464913
Lee C., Yang W., Parr R.G. // Phys. Rev. B. 1988. V. 37. № 2. P. 785. https://doi.org/10.1103/physrevb.37.785
Krishnan R., Binkley J.S., Seeger R., Pople J.A. // J. Chem. Phys. 1980. V. 72. № 1. P. 650. https://doi.org/10.1063/1.438955
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии