

Микроэлектроника, 2021, T. 50, № 4, стр. 299-313
Моделирование нуклеации многокомпонентного 2DGaSxSe1 – x с применением эволюционного уравнения
С. М. Асадов *
Институт катализа и неорганической химии Национальной академии наук Азербайджана
AZ-1143 Баку, Пр. Г. Джавида, 113, Азербайджан
* E-mail: salim7777@gmail.com
Поступила в редакцию 03.07.2020
После доработки 25.08.2020
Принята к публикации 21.09.2020
Аннотация
Проведено моделирование процессов нуклеации и кристаллизации многокомпонентных частицна примере формирования двумерных (2D) твердых растворов GaSxSe1 –x(0 ≤ х ≤ 1) в термодинамически закрытых системах. Разработана нелинейная физико-химическая модель формирования двумерных 2D термодинамических фаз с использованием эволюционного уравнения типа Фоккера–Планка (Ф–П) в пространстве размеров. Аппроксимация эволюции функции, распределения частиц проведена по размерам во времени. Модель процессов формирования фаз, содержащую систему нелинейных дифференциальных уравнений Ф–П, исследовали с учетом теорий гомогенной и гетерогенной кристаллизации, и уравнения модели решали методом конечных разностей. Численные аппроксимации проведены для формирования 2D-нанокристаллов систем GaSxSe1 –x. В описание модели выбранной функции распределения учитывали влияния химического потенциала компонентов, температуры, а также примесей на образование новой фазы. Рассмотрены также влияние коэффициента активности, распределения примесей, температурно-зависящих факторов и диффузионного эффекта на формирование 2D фаз.
1. ВВЕДЕНИЕ
Оптимизация методов получения и моделирование свойств наноматериалов, обладающих низкой дисперсностью, является актуальной задачей микро- и наноэлектроники [1]. Формирование таких материалов включает в себя процессы зарождения и роста наночастиц. Эти процессы образования новых фаз и наночастиц взаимосвязаны и их аналитическое описание затруднительно. Поэтому понимание и описание термодинамики и кинетики зарождения и роста являются неполными. Указанные затрудняют оптимизацию получения наночастиц с необходимыми характеристиками. Для повышения эффективности описания этих процессов значение приобретает численный анализ с использованием нелинейных кинетических уравнений.
Двумерные (2D) Ван-дер-Ваальсовы полупроводники, в частности,на основе системы GaS–GaSe привлекают внимание в основном благодаря их превосходным электронным и оптоэлектронным свойствам. Формирование качественных материалов на основе монохалькогенидов для нанотехнологий часто происходит с неоднородным распределением структурных дефектов по объему [2]. Например, плотность дислокаций приводит к механическим напряжениям и образованию собственных точечных дефектов. В частности, материалы GaSxSe1 –x с дефектной структурой имеют невоспроизводимые электрические, оптические, фотоэлектрические, люминесцентные и другие физические характеристики [3–5].
Менее изучены двумерные полупроводниковые твердые растворы монохалькогенидов, такие как на основе GaS [6–10], GaSe [11, 12] и InSe [13, 14]. Эти 2D материалы продемонстрировали новые физические свойства [15], которые значительно отличаются от свойств других слоистых материалов. Они имеют перестраиваемые запрещенные зоны и высокую стабильность, что делает их идеальным слоистым материалом для изготовления транзисторов и фотоприемников.
Кристаллы GaS и GaSe принадлежат к гексагональной сингонии, характеризуется слоистой структурой и пространственной группой $D_{{3h}}^{1} - P\bar {6}m2$. Они имеют несколько полиморфных модификаций. Так, например, GaSe имеет четыре модификации (β-, ε-, γ- и δ-GaSe). При комнатной температуре термодинамически более стабильными модификациями являются β-GaS и ε-GaSe. Изменение состава от GaS до GaSe сделало возможным непрерывно изменять природу электронной структуры и запрещенные зоны системы GaS–GaSe [12]. Образцы на основе неограниченных твердых растворов GaSxSe1 –x (х = 0–1), исследованные в их объемных и пленочных состояниях, показывают на непрерывное изменение свойств во всем диапазоне концентраций компонентов (х = 0–1), но подобных исследований на нанопластинах GaSxSe1 –х еще не проводилось.
Когда GaSe ограничен несколькими слоями, в нем возможно обнаружить переход из прямого к непрямому переходу через запрещенную зону [16, 17]. Он имеет очень близкие прямые и непрямые края полосы (~25 мэВ), что обеспечивает легкий путь для эффективного разделения электронно-дырочных пар [18]. Каждый слой GaSe состоит из четырех ковалентно связанных атомных плоскостей Se–Ga–Ga–Se толщиной 0.798 нм. GaSe является 2D-полупроводником p-типа и может быть объединен с другим 2D-полупроводником n-типа с образованием p-n-переходов, которые являются необходимыми строительными блоками для будущей наноэлектроники и оптоэлектроники.
Причины, вызывающие неоднородное распределение дефектов по объему формированных 2D материалов, определяются следующими процессами: нуклеация, кристаллизация, посткристаллизационное охлаждение и термообработка. В указанных процессах возникают градиенты концентрации, химического потенциала и температуры. Однако, основные причины и факторы, влияющие на формирование энергетической, электронной и кристаллической структуры, до сих пор мало изучены. Влияния нелинейных процессов на физические свойства 2D материалов, в частности, на основе системы GaS–GaSe также изучены недостаточно и не рассмотрены с позиций моделирования свойств и теории образования кристаллизации новых фаз.
Цель настоящей работы – установление кинетических закономерностей процессов нуклеации и формирования термодинамических фаз на примере двумерных кристаллов GaSxSe1 –x (0 ≤ х ≤ 1) с учетом флуктуаций.
2. ТЕОРИЯ, МОДЕЛЬ И МЕТОДИКА РАСЧЕТА
Условие перенасыщения или переохлаждения само по себе не является достаточной причиной для начала кристаллизации системы. Прежде чем кристаллы смогут развиваться, в растворе должно существовать несколько мельчайших твердых тел (ядер), которые действуют как центры кристаллизации. Нуклеация может происходить самопроизвольно или может быть вызвана искусственно.
В настоящее время нет общего согласия по номенклатуре нуклеации. Поэтому далее будем использовать термин “нуклеация” (первичный) для всех случаев зародышеобразования в системах, не содержащих кристаллическое вещество. С другой стороны, ядра часто генерируются вблизи кристаллов, присутствующих в перенасыщенной системе. Такое поведение можно принять как “вторичное” зарождение.
Таким образом, можем рассмотреть простую схему нуклеации: однородный (спонтанный) и гетерогенный (вызванный посторонними частицами), а также вторичный (индуцируется кристаллами).
Кинетика зарождения и роста термодинамической фазы. Для изложения методики обоснованного моделирования процессов, в частности, в материаловедении необходимо проводить контекстно ориентированный расчет соответствующих уравнений. Их последующее компьютерное интегрирование способствовало бы выявлению закономерностей нелинейных эффектов, например, эволюции термодинамических фаз при нуклеации [19–24] и роста [25–32]. Теория нуклеации позволяет определить скорости образования новой фазы в единице объема материнской фазы в изучаемом метастабильном состоянии. В развитие этой теории вначале заметный вклад внесли, в частности, авторы работ [19–21].
Ниже рассмотрим важные условия процессов зарождения и роста зародышей в закрытой термодинамической системе и разработанную нами нелинойную физико-химическую модель для описания эволюционной функции распределения частиц. Получены данные с применением численного подхода для процессов образования и роста новой фазы с учетом влияния температурно-зависимых факторов.
В теории нуклеации процессы зарождения и роста зародышей в системе часто рассматриваются с использованием квазимолекулярных реакций. Эти реакции предполагают присоединение одиночных частиц (“мономеров”) к зародышам, что приводит к постепенному росту “кластеров”. Кинетика образования “кластеров” частиц рассмотрена также в классической теории. В процессе кристаллизации метастабильные кластеры формируются флуктуациями, и только кластеры с размерами, случайно превышающими критический размер, непрерывно растут. После некоторого времени релаксации рассматриваемая система приближается к устойчивому состоянию. Идея рассмотрения фазового перехода в указанной модели сводится к анализу квазимолекулярных реакций. Такие реакции предполагают присоединение одиночных “мономеров” к зародышам, что приводит к их постепенному росту. Количество зародышей критического размера, образующихся в единицу времени в единице объема, определяет скорость зародышеобразования (нуклеации).
Гомогенное зародышеобразование. Как именно образуется стабильное кристаллическое ядро в однородной жидкости, неизвестно с достоверностью. Образование кристаллических ядер представляет собой сложный процесс, который можно предусмотреть. Составляющие систему молекулы могут коагулировать, сопротивляясь тенденции к повторному растворению, но они также могут ориентироваться в фиксированную решетку. Число молекул в стабильном кристаллическом ядре может варьироваться от десяти до нескольких тысяч. Тем не менее, стабильное ядро вряд ли может быть результатом одновременного столкновения необходимого количества молекул, так как это будет стохастическим явлением. Скорее всего, это могло произойти из последовательности соeдинеия “мономеров” в виде бимолекулярных добавок $A + A\,\,~ \leftrightarrow {{A}_{2}}$ с образованием “кластеров” ${{A}_{{n - 1}}} + A~\,\, \leftrightarrow {{A}_{n}}$ (критического размера).
Дальнейшие молекулярные добавления к критическому “кластеру” могут привести к зародышеобразованию и последующему росту ядра. Аналогично, ионы или молекулы в растворе могут взаимодействовать с образованием короткоживущих “кластеров”. Изначально могут образовываться короткие цепи или плоские монослои, и в конечном итоге образуется кристаллическая решетчатая структура. Процесс конструирования, который происходит очень быстро, может продолжаться только в локальных областях с очень высокой перенасыщенностью, и многие из “субъядер” не достигают зрелости; они просто перерастворяются, потому что нестабильны. Если, однако, ядро вырастает за пределы определенного критического размера, оно становится стабильным.
Согласно теории гомогенной нуклеации число зародышей, возникающих в единицу времени на границе “кристалл–среда”, равно
(1)
$J = K~{\text{exp}}\left( {{{ - \Delta {{G}_{a}}} \mathord{\left/ {\vphantom {{ - \Delta {{G}_{a}}} {{{k}_{{\text{B}}}}T}}} \right. \kern-0em} {{{k}_{{\text{B}}}}T}}} \right){\text{exp}}\left( {{{ - 16\pi {{{{\Omega }}}^{2}}{{g}^{3}}} \mathord{\left/ {\vphantom {{ - 16\pi {{{{\Omega }}}^{2}}{{g}^{3}}} {3{{k}_{{\text{B}}}}T\Delta {{\mu }^{2}}}}} \right. \kern-0em} {3{{k}_{{\text{B}}}}T\Delta {{\mu }^{2}}}}} \right),$Скорость зарождения частицы $J\left( t \right),$ согласно [22] приближается к устойчивому состоянию по уравнению
(2)
$\begin{gathered} J\left( t \right) = ~{{J}_{s}}\left[ {1 - {\text{exp}}\left( {{{ - t} \mathord{\left/ {\vphantom {{ - t} {{{t}_{{{\text{lag}}}}}}}} \right. \kern-0em} {{{t}_{{{\text{lag}}}}}}}} \right)} \right], \\ {1 \mathord{\left/ {\vphantom {1 {(6.3a\left( {{{l}_{c}}} \right){{Z}^{2}})}}} \right. \kern-0em} {(6.3a\left( {{{l}_{c}}} \right){{Z}^{2}})}} \geqslant {{t}_{{{\text{lag}}}}} \geqslant {1 \mathord{\left/ {\vphantom {1 {(12a\left( {{{l}_{c}}} \right){{Z}^{2}})}}} \right. \kern-0em} {(12a\left( {{{l}_{c}}} \right){{Z}^{2}})}}, \\ \end{gathered} $Так как градиент скорости стационарного состояния в области |$\left| {l - l{\text{*}}} \right| < Z$ мал, то формированный “кристалл–кластер” будет перемещаться в этой области случайным блужданием с частотой скачка $a(l*)$. Время, необходимое кластеру для прохождения расстояния ${1 \mathord{\left/ {\vphantom {1 Z}} \right. \kern-0em} Z}$ путем случайного блуждания, будет определяться задержкой во времени, которое оценивается как ${{t}_{{{\text{lag}}}}} = {1 \mathord{\left/ {\vphantom {1 {2a}}} \right. \kern-0em} {2a}}(l*){{Z}^{2}}.$
Эффективный поиск условий управления процессами в многофазных системах требует теоретического и численного анализа процесса. Недостатком известных работ по моделированию многокомпонентных систем является то, что для описания зарождения фаз часто применяется классическая теория образования новой фазы. Такая модель не позволяет описать функции распределения зародышей, например, по окончании стадии нуклеации. Таким образом, в этой теории взаимное влияние зарождения и роста не учтено в полной мере.
В современной теории образования новой фазы можно рассматривать эволюцию всей функции распределения как на стадии зарождения, так и на стадии роста. Ограничения этой теории связаны с тем, что она привязана к общей реализации метода формирования фазы, и в ней не рассмотрены с конкретных позиций различные режимы зарождения и возникающие при этом стадии роста и структуры многокомпонентных фаз.
До сих пор мало работ, посвященных результатам проведения аналитического и численного моделирования процессов, возникающих при формирование многокомпонентных наночастиц в полупроводниковых растворах при различных режимах подачи компонентов. В применении к фазовым переходам 1-го рода методами физической кинетики с использованием, в частности, стохастических уравнений можно изучить распределение зародышей новой фазы в процессе их роста. Например, нуклеацию можно характеризовать функцией распределения частиц по радиусам, эволюцию которой можно описать дифференциальным уравнением типа Фоккера–Планка (Ф–П) [33–36].
Такой подход позволяет адекватно описать, например, скорости зарождения в стационарном режиме. Однако, в теории нуклеации стадия гетерогенного роста не рассматривается, и эту стадию невозможно корректно описать в рамках эволюционного уравнения Ф–П.
В ценной работе [37], посвященной в основном анализу фазовых переходов (коалесценция) в моногокомпонентных системах, не рассматриваются подходы и задачи, связанные с численным моделированием закономерностей процессов нуклеации и формирования термодинамических фаз, в частности, двумерных полупроводниковых кристаллов с учетом флуктуаций.
Нуклеация описывается с помощью функции распределения частиц по их радиусам $\varphi \left( {r,t} \right)$. Если принять, что функция нормирована на количество частиц в единице объема, то для такой функции эволюционное уравнение Ф–П имеет вид:
(3)
${{\varphi }_{0}} = {{K}_{0}}{\text{exp}}\left( { - \frac{{\Delta G}}{{{{k}_{{\text{B}}}}T}}} \right),$Нелинейные процессы нуклеации и рост кристаллов в термодинамической системе обычно проходят при флуктуациях. Такие процессы в данной работе нами исследованы с применением уравнения Ф–П. Изучали термодинамические и кинетические закономерности нуклеации и роста с учетом блуждания зародышей (частиц) в пространстве размеров с использованием уравнения Ф–П.
Динамику изменения свойств указанных процессов описывали разработанной нами моделью, где свойства характеризуются через случайное блуждание частиц. Эволюцию функции распределения по размерам частиц аппроксимировали уравнением Ф–П с учетом заданных условий кристаллизации на примере 2D термодинамической фазы.
Процесс образования 2D фазы исследовали в термодинамической системе, где принимали, что во всем объеме температура и концентрация компонентов постоянны. Допускали, что кристаллизация в системе описывается уравнением Ф–П, основанным на эволюции функции $\varphi $ распределения кристаллов по размеру (r) во времени (t). Тогда кинетическое уравнение Ф–П для плотности распределения запишется в следующем виде
(4)
$\frac{{\partial \varphi }}{{\partial t}} = - \frac{{\partial \left( {\varphi L} \right)}}{{\partial r}} + \frac{\partial }{{\partial r}}\left( {p\frac{{\partial \left( {\varphi L} \right)}}{{\partial r}}} \right),$С учетом того, что формированные кристаллы имеют ограниченный размер в рамках рассмотренной модели граничные условия выбирали с учетом плотности распределения процесса кристаллизации. Кинетическое уравнение (4), характеризующее процесс, после этого дополнили начальным условием и уравнением баланса. Модель, содержащую систему нелинейных дифференциальных уравнений, на примере процессов 2D-нуклеации и роста GaSxSe1 –x решали разностным методом [38].
В системе GaS–GaSe твердые растворы GaSxSe1 –x плавятся конгруэнтно, т.е. без разложения и не имеют фазовых переходов [12]. Фазовые превращения всегда сопровождаются изменением энтальпии, и этот тепловой эффект легко наблюдается в эксперименте, например, из кривых термического анализа (ТА) сплавов изучаемой системы. Рассмотрим кривые ТА сплавов системы GaS–GaSe. Гладкая кривая охлаждения следует до тех пор, пока не произойдет фазовая реакция, когда сопровождающий тепловой эффект вызывает остановку или изменение наклона. На рис. 1а показана типичная кривая ТА для чистого GaS и/или GaSe. АВ – кривая охлаждения для однородной жидкой фазы. В точке ${\text{B}}$ вещество начинает кристаллизоваться и система остается при постоянной температуре, до полного затвердевания в точке ${\text{C}}$. Затем твердое вещество охлаждается со скоростью, обозначенной кривой ${\text{CD}}$. Жидкая фаза может охлаждаться ниже точки кристаллизации, и некоторые системы могут выдерживать значительные степени переохлаждения (пунктирная кривая).
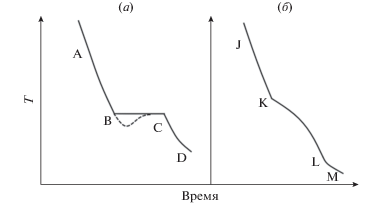
На рис. 1б показана типичная кривая ТА охлаждения для системы GaS–GaSe, которая образует непрерывный ряд твердых растворов. Первый участок ${\text{JK}}$ на кривой соответствует началу кристаллизации, и это представляет точку на ликвидусе $~T--x$ фазовой диаграммы GaS–GaSe. Второй участок ${\text{KL}}$ соответствует завершению кристаллизации и представляет точку на солидусе.
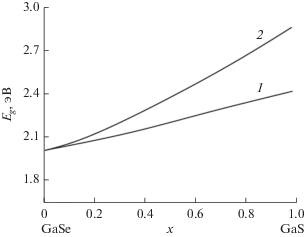
Однако, из экспериментальных данных следует, что в зависимости от состава, структуры (GaХ, где Х=S, Se имеют различные политипы) и условий кристаллизации формированные структуры и физические свойства GaSxSe1 –x заметно отличаются (рис. 2).
Рис. 2.
Концентрационные зависимости ширины запрещенной зоны ${{E}_{g}}$ твердых растворов GaSxSe1 – x (x = 0–1) для двух политипов кристаллов. 1 – $\varepsilon $‑GaS(Se)политип, 2 – $\beta $-GaS(Se)политип.
Рельеф поверхности кристаллов GaSxSe1 –x указывает на образование разнообразных 2D наноструктур на поверхности. Выделяются два типа неоднородностей: протяженные структуры и локальные 2D-нанообъекты. Скорость роста GaSxSe1 –x из расплава больше, чем скорость роста из растворов.
3. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Гомогенное зародышеобразование и рост. Процесс гомогенной кристаллизации, как известно, состоит из двух процессов: зарождения (нуклеация) центров кристаллизации и роста кристаллов из этих центров. Необходимым условием для роста кристалла является перевод среды роста (расплав, раствор) в неравновесное состояние путем ее переохлаждения либо пересыщения растворенным веществом.
Рассмотрим изменение общей свободной энергии Гиббса $~\Delta {{G}_{{{\text{gen}}}}}$ системы при переходе расплава в кристаллическое состояние. Твердый раствор заданного состава примем как квази- однокомпонентный. Тогда согласно термодинамической теории кристаллизации твердых тел величина $~\Delta {{G}_{{{\text{gen}}}}}$ однокомпонентной системы меняется за счет двух вкладов. Первая часть $~\Delta {{G}_{{{\text{gen}}}}}$ обусловлена понижением энергии системы при кристаллизации. Энергия системы $\Delta {{G}_{{{\text{общ}}}}}$ уменьшается на величину ${{G}_{1}} = V\Delta {{G}_{V}}\sim {{n}^{3}}$ ($V$ – объем зародыша, $\Delta {{G}_{V}}$ – удельная разность свободных энергий при переходе расплава в кристаллическое состояние, ${{n}^{3}}$ – число частиц). Вторая часть $\Delta {{G}_{{{\text{gen}}}}}$ наоборот увеличивается на величину, равную работе по образованию поверхности раздела ${{G}_{2}} = s\sigma \sim {{n}^{2}}$ ($\sigma $ – удельное поверхностное натяжение на границе “кристалл–жидкость”, $s$ – поверхность зародыша). Если предположить, что каждое ядро имеет сферическую форму радиуса $r$, тогда $\Delta {{G}_{{{\text{gen}}}}} = {{r}^{3}}\Delta {{G}_{V}} + 6{{r}^{2}}\sigma $. “Кластеры” до критических размеров ($r < r{\text{*}}$) характеризуют зародыши, а “кластеры” с радиусом больше $r{\text{*}}$ – ядра. Отсюда можно сделать вывод о том, что зародыши с размером больше критического вызывают уменьшение $\Delta {{G}_{{{\text{gen}}}}}$, т.е. они будут устойчивыми и способными к росту. Таким образом, в первом приближении процесс возникновения новой, упорядоченной фазы согласуется с критериями термодинамической теории формирования новой фазы.
В процессе зарождения новой фазы наряду с термодинамическим вкладом важно учесть также кинетический вклад. Запишем скорость гомогенного зародышеобразования, которая характеризует общее число зародышей кристаллической фазы. Для числа зародышей, возникающих в единице объема расплава в одну секунду, в стационарном приближении скорость нуклеации $~~J~$ равна
Расположение частиц в кристалле примем как упорядоченное, тогда их энтропия ${{S}_{c}}$ будет меньше энтропии ${{S}_{m}}$ в неупорядоченной среде (расплаве, растворе, паре). Поэтому снижение температуры $T$ при постоянном давлении $p$ приведет к тому, что химический потенциал вещества в кристалле ${{\mu }_{c}}$ станет меньше его потенциала ${{\mu }_{m}}$ в исходной среде. Т.e. ${{\mu }_{c}} = \,\,~{{\varepsilon }_{c}} - T{{S}_{c}} + p{{\Omega }_{c}} < {{\mu }_{m}}$ = $~ = {{\varepsilon }_{m}} - T{{S}_{m}} + p{{{{\Omega }}}_{m}}$, где $~{{\varepsilon }_{c}},~{{\Omega }_{c}}$, ${{\varepsilon }_{m}},~{{\Omega }_{m}}$ – энергии взаимодействия частиц и удельный объем вещества в кристаллическом и неупорядоченном состояниях (фазах). Другими словами кристаллизация (фазовый переход первого рода) сопровождается выделением теплоты кристаллизации: $\Delta {{H}_{c}} = T\left( {{{S}_{m}} - {{S}_{c}}} \right)$, а также скачком удельного объема $\Delta {{\Omega }} = {{{{\Omega }}}_{m}} - {{{{\Omega }}}_{c}}$. При невысоких давлениях $p\Delta \varphi $ мал, и при ${{\mu }_{c}} = {{\mu }_{m}}$ изменение энтальпии системы будет равно $\Delta H = {{\varepsilon }_{m}} - {{\varepsilon }_{c}}$. Т.е. величина $\Delta H$ является мерой изменения энергии связи между частицами при кристаллизации. При кристаллизации, например, простых полупроводниковых фаз из расплава $\Delta \Omega \approx \left( {0.05 - 0.15} \right){{\Omega }_{m}}$ и может иметь различные знаки.
Гетерогенное зародышеобразование и рост. В гетерогенной системе образование термодинамически стабильной фазы в закрытой системе также происходит путем кристаллизации из материнской фазы. Формирование такой фазы проходит три стадии: нуклеация, диффузионный рост зародышей и коалесценция. Описание этих стадий можно осуществить с учетом теории фазовых переходов.
В многокомпонентных системах условия ${{\mu }_{c}}\left( {p,T,{{C}_{c}}} \right)$ = ${{\mu }_{m}}\left( {p,T,{{C}_{m}}} \right)$ для каждого из компонентов кристалла и среды определяют связь $p,T,C$ (концентрации компонентов), при которых кристалл находится в равновесии со средой. Такая связь геометрически представляется, в частности, в виде $~p{\kern 1pt} - {\kern 1pt} T{\kern 1pt} - {\kern 1pt} x~$ равновесной фазовой диаграммы состояния системы. Разность $\Delta \mu = {{\mu }_{m}} - {{\mu }_{c}}$ является мерой отклонения от равновесия и характеризует термодинамическую часть движущей силы кристаллизации.
В реальных условиях в многокомпонентных системах при наличии центров кристаллизации может происходить не самопроизвольная (гетерогенная) кристаллизация. Эволюция дисперсной фазы при нуклеации в закрытой системе происходит по сложному механизму. При этом возможно образование метастабильных фаз и эволюция дисперсных частиц с кристаллизацией твердой фазы.
Механизмы превращений указанных фаз и кинетика кристаллизации подчиняются нелинейным законам. При этом параметры процесса кристаллизации определяются граничными условиями и коэффициентами кинетических уравнений. Кинетические коэффициенты характеризуют вероятность присоединения и/или выделения одной частицы в единицу времени соответственно. Кинетические коэффициенты процессов роста фазы определяются также факторами диффузионного роста и растворения выделений второй фазы. При этом свойства формируемых фаз также зависят от предыстории процесса кристаллизации.
Рассмотрим гомогенизированный твердый раствор GaSxSe1 –x, например, на основе соединения GaSe со слоистой структурой. Такой раствор, закаленный в метастабильной области Т–х фазовой диаграммы системы GaS–GaSe [12], некоторое время может остаться в метастабильном состоянии. В конечном итоге система достигает термодинамического равновесия. При одной из равновесных концентраций компонентов могут образовываться некоторые нанокластеры в матрице GaSe, т.е. возможна гетерогенная 2D-нуклеация.
Однако, самопроизвольная 2D-нуклеация обычно происходит в высокочистом жидком расплаве при больших степенях переохлаждения [25–27]. Источником нуклеации могут являться твердые частицы, например, неметаллические включения, которые всегда присутствуют в расплаве. Чем больше примесей, тем больше центров нуклеации гетерогенной кристаллизации, тем мельче будет кристаллическое зерно. Для идентификации активных центров нуклеации в экспериментах важно контролировать степень переохлаждения. Существует ряд возможных центров и механизмов нуклеации, которые могут действовать как катализаторы на рост кристаллов.
К таким катализаторам в рассматриваемом нами процессе роста 2D кристаллов можно отнести, например, первичные фазы, возникающие при охлаждении расплавов и изменяемые границы раздела частиц с жидкой добавкой. Такие фазы создают центры нуклеации, возникающие в результате образования остаточной твердой фазы (кристалла) во включениях или поверхностных слоях кристаллов.
В гетерогенной кристаллизации полупроводниковых систем в качестве центров кристаллизации часто выступают находящиеся в расплаве, например, оксиды и интерметаллические соединения, образуемые примесями, шероховатости стенок сосуда-кристаллизатора, в котором находится расплав, вводимые в расплав специальные “затравки–кристаллы”.
Из вышеуказанного и анализа экспериментальных данных следует, что важным является знание закономерностей протекания процессов нуклеации и роста фаз многокомпонентных систем на различных поверхностях. Для гетерогенной 2D-нуклеации на подложке можно выделить следующие закономерности.
– Возникновение кристаллических зародышей наблюдается большей частью на дефектах, таких как царапины и границы зерен.
– Скорость зарождения кристаллов в несколько раз ниже на монокристаллических подложках в сравнении с поликристаллическими подложками того же вещества.
– С увеличением размеров первоначально образовавшихся зародышей частота появления новых зародышей на участках поверхностей, не покрытых кристаллом, снижается.
– Скорость роста удаленных друг от друга микрокристаллов выше у зародышей, растущих при близком расположении друг к другу.
Рассмотрим процесс образования твердой частицы из жидкой фазы на плоской поверхности или границе раздела. Если обе фазы “смачивают” эту плоскую поверхность, то должны возникать три межфазные энергии (силы поверхностного натяжения) (рис. 3). Угол $\theta $ – это угол контакта между кристаллическим осадком и инородной твердой поверхностью, которая соответствует углу смачивания в жидко-твердых системах.
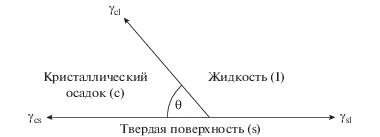
Указанные фазы будут существовать на двухфазных границах ${{\gamma }_{{si}}},{{\gamma }_{{sl}}},{{\gamma }_{{li}}},$ и $\theta $ – угол смачивания (угол между ${{\gamma }_{{si}}}$ и $~{{\gamma }_{{sl~~}}}$). В равновесном состоянии на поверхности должен выполняться баланс сил поверхностного натяжения: ${{\gamma }_{{li}}} = {{\gamma }_{{si~}}} + {{\gamma }_{{sl~~}}}{\text{cos}}\theta $. Тогда критический радиус и изменения свободной энергии Гиббса рассмотренного процесса однородного образования фаз можем определить из теории образования новой фазы
(6)
$r* = - \frac{{2~{{\gamma }_{{sl~~}}}}}{{\Delta {{G}_{v}}}}\Delta G_{{{\text{hom}}}}^{*} = \left( {\frac{{16\pi \gamma _{{sl}}^{3}}}{{3\Delta G_{v}^{2}}}} \right)f\left( \theta \right).$Поскольку $~{{\gamma }_{{sl~~}}}$ такая же поверхностная энергия, как и $\gamma $ в уравнении для гомогенного образования, то $r{\text{*}}$ для гетерогенного зарождения такой же, как и для гомогенного. А энергия активации барьера для гетерогенного зародышеобразования меньше, чем однородного барьера на величину функции $f\left( \theta \right)$. Т.е. в процессе гетерогенного образования для преодоления энергетического барьера нужно затратить меньше энергии, чем для гомогенного, и, таким образом, гетерогенное образование частиц должно происходить легче.
Другими словами, наличие подходящего инородного тела или “соответствующей” поверхности может вызвать зародышеобразование при степенях переохлаждения, меньших, чем те, которые требуются для самопроизвольного зародышеобразования. Тогда общее изменение свободной энергии, связанное с образованием критического зародыша в гетерогенных условиях Гиббса ${{\Delta }}G_{{{\text{het}}}}^{{\text{*}}}$, должно быть меньше соответствующего изменения свободной энергии, ${{\Delta }}G_{{{\text{hom}}}}^{{\text{*}}}$, связаной с гомогенной нуклеацией. Эти наблюдаемые закономерности 2D-нуклеации на подложке свидетельствуют о том, что зарождение происходит не в объеме жидкой или газовой фазы, а непосредственно на поверхности используемых подложек. При этом изменение критической свободной энергии Гиббса ${{\Delta }}G_{{{\text{het}}}}^{{\text{*}}}$ образования ядра на подложке соответствует уравнению
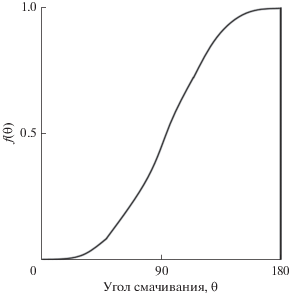
где $\Delta G_{{{\text{hom}}}}^{{\text{*}}}$ – изменение критической свободной энергии зародышеобразования при спонтанной кристаллизации, $\theta $ – угол контакта (смачивание) подложки с ядром. Здесь фактор $f\left( \theta \right)$ меньше единицы (рис. 4).Рис. 4.
Отношение свободных энергий гомогенного и гетерогенного зародышеобразования в зависимости от угла контакта с подложкой.
Представим, что зародышеобразование происходит в стационарном потоке (Jс), т.е. в потоке, скорость которого в любом месте рассматриваемой системы, например, жидкости никогда не изменяется. В таком потоке в пространстве размера через барьер 2D-нуклеации скорость зародышеобразования определяется из кинетической формулы:
(8)
$\begin{gathered} ~{{J}_{c}}\sim 4\pi {{r}^{2}}Z\exp \left[ { - \frac{{{{\Delta }}G_{n}^{*}}}{{{{k}_{{\text{B}}}}T}}} \right]{\text{exp}}\left( { - {{t}_{i}} - t} \right), \\ Z = {{\left( { - \frac{1}{{2\pi {{k}_{{\text{B}}}}T}}{{{\left. {\frac{{{{d}^{2}}G}}{{d{{r}^{2}}}}} \right|}}_{r}}} \right)}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}, \\ \end{gathered} $Вероятностные процессы при нуклеации и росте. С применением эвалюционного уравнения ФП рассмотрим особенности составленной нами физико-химической модели, которая учитывает вероятностные процессы при нуклеации и росте. Примем, что распределение дисперсной фазы происходит по времени и размерам $~\varphi \left( {r,t} \right)$. Для гетерогенных систем такие флуктуационные процессы мало изучены. Дисперсная фаза, например, твердые растворы, может образоваться путем фазового перехода из неупорядоченного состояния (из расплава, пара и раствора) системы. Примем, что кристаллизация осуществляется в два этапа. Сначала возникает микроскопический зародыш кристаллической фазы, которая в дальнейшем растет. При значительной степени пересыщения или переохлаждения системы вероятность зарождения новой фазы увеличивается.
На примере системы GaS–GaSe [12], с учетом экспериментальных данных рассмотрим случай, когда кристаллизация твердых растворов GaSxSe1 –x происходит из однородно пересыщенного раствора в термодинамически закрытой системе. При этом раствор имеет ограниченный объем $V$ в момент времени $t$ = 0. Если концентрация дисперсной фазы $Q$ превышает растворимость компонентов системы, то происходит 2D-нуклеация и рост кристаллов (твердая фаза). При такой кристаллизации возможно образование различных модификаций твердой фазы.
Примем, что в системе при кристаллизации не происходит химическая реакция и поддерживается постоянная температура раствора. Перемешивание раствора не приводит к раскалыванию и агрегированию. Массовая кристаллизация происходит спонтанной нуклеацией, т.е. зарождением центров кристаллизации, ростом кристаллов и растворением частиц дисперсной фазы.
Нуклеация включает образование и рост кластеров из молекул исходного раствора. Формирование кластеров происходит вплоть до размера, при котором возможно выделить грань кристалла как структурный элемент, ответственный за рост. В таком случае интенсивность нуклеации, скорости роста кристаллов и растворения частиц дисперсной фазы определяются степенью пересыщения раствора и размером грани кристалла.
С учетом вышеуказанной физико-химической модели, содержащей суть гетерогенного формирования фазы и кинетическое уравнение Ф–П, определим закономерности процессов 2D-нуклеации и роста GaSxSe1 –x. Полученную систему уравнения вида (4) рассматриваемой нами модели решали численным методом, где использовали равномерную сетку с шагом $h$ по размеру и $t$ по времени. Перенос решения j-го слоя на ($j + 1$)-й осуществляли чисто неявной разностной схемой, после чего пересчитывали функцию $G$. Разностная схема для уравнения типа (4) имеет вид:
(9)
$\begin{gathered} \frac{{\varphi _{i}^{{j + 1}} - \varphi _{i}^{j}}}{\tau } = - \frac{{{{G}^{j}}}}{{2h}}\left( {\varphi _{{i + 1}}^{{j + 1}} - \varphi _{{i - 1}}^{{j + 1}}} \right) + \\ + \,\,\frac{{p{{G}^{j}}}}{{{{h}^{2}}}}~\left( {\varphi _{{i + 1}}^{{j + 1}} - 2\varphi _{i}^{{j + 1}} + \varphi _{{i - 1}}^{{j + 1}}} \right). \\ \end{gathered} $Уравнение (9) имеет порядок аппроксимации $O\left( {\tau + {{h}^{2}}} \right)$. Разностное уравнение для левого краевого условия имеет вид
(10)
$\varphi _{0}^{{j + 1}}{{G}^{j}} - p{{G}^{j}}\frac{{\varphi _{1}^{{j + 1}} - \varphi _{0}^{{j + 1}}}}{h} + p{{G}^{j}}\frac{{\varphi _{0}^{j}2\varphi _{1}^{j} + \varphi _{2}^{j}}}{{{{h}^{2}}}} = {{\eta }^{j}}.$Полученное уравнение имеет порядок аппроксимации $O\left( {\tau + \tau h + {{h}^{2}}} \right)$. Разностные уравнения решали методом прогонки, который применим в силу диагонального преобладания. Разностные схемы (9) и (10) являются устойчивыми по правой части. После переноса решения на (j + 1)-й слой вычисляли новое значение концентрации ${{C}_{k}}$, а также параметры $G$ и $\eta $. Вычисление интеграла производили методом трапеций – его точность имеет порядок $O({{h}^{2}})$, что соответствует аппроксимации использованной разностной схемы. Полученное значение концентрации ${{C}_{k}}$ повторно использовали для нахождения решения на (j + 1)-м слое. Данную процедуру повторяли заданное (фиксированное) число раз. Таким образом, кристаллизацию системы рассмотрели в виде совокупности физико-химической и математической моделей. Такая модель позволяет осуществить численную реализацию нелинейных кинетических уравнений разностной схемой. Численные эксперименты по изучению процесса формирования фазы GaSxSe1 –x проводили в среде программы Delphi.
Модельные расчеты и сравнение с экспериментом. Рассмотрим процессы 2D-нуклеации и роста GaSxSe1 –x на примере системы GaS–GaSe. Представим, что кристаллические грани 2D-фазы растут за счет формирования 2D ядер критического размера в отсутствии винтовых дислокаций, заканчивающихся на поверхности. Двумерные ядра образуются, когда отдельные единицы роста (например, атомы, молекулы, димеры) адсорбируются на поверхности кристалла, диффундируют и затем агломерируют. После того, как 2D ядро становится больше по размеру, чем его критический размер, оно становится термодинамически выгодным для присоединения единиц роста к этому ядру. В пересыщенном растворе 2D ядро большего размера, чем критическое, распространяется поперек грани, пока не достигает границы кристалла. Этими границами могут быть как край слоя 2D кристалла, так и фронт слоя под ним или фронт роста от другого ядра.
Обсуждение роста кристаллов начнем из затравочного кристалла, т.е. маленького кристалла с заданными поверхностями, который погружен в пересыщенную жидкость и находится в процессе роста. В рамках классической модели 2D-нуклеации, так называемой модели Косселя–Странского, предполжим, что кристаллы формируются слой за слоем, это требует двумерного зародышеобразования после завершения каждого слоя. Такое зародышеобразование сходно с образованием трехмерного ядра и может быть шагом, определяющим скорость для роста идеальных кристаллов.
Представим послойный механизм роста, где для того чтобы росла грань фазы, требуется, чтобы на ней образовался двумерный (плоский) зародыш роста. Тогда поверхность растущего кристалла, в частности, GaSxSe1 –x будет плоской и рост кристаллов произойдет при незначительном пересыщении или переохлаждении.
Проведенная нами оценка времени 2D-нуклеации твердых растворов GaSxSe1 –x разных размеров в вакуумированных кварцевых ампулах (при $p = {\text{const}}$) показывает, что быстро кристаллизуются лишь зародыши с радиусом более 1 мкм. Для среднедисперсных кристаллов время процессов нуклеации и кристаллизации измеряется минутами. Испарение же субмикронной жидкой капли происходит за доли секунды. Для грубодисперсных кристаллов процессы нуклеации и кристаллизации будут идти с одинаковой по порядку величины скоростью.
Для заданного физико-химического состава зародыша скорость нуклеации зависит от кривизны поверхности, параметра, характеризующего начальное распределение частиц по размерам; скорости укрупнения кристаллов. При специальных воздействиях скорость укрупнения может существенно меняться. В этом случае вид функции вероятности столкновений частиц изменится и будет включать в себя параметры воздействующего фактора.
Результаты проведенных расчетов с помощью используемой модели, содержащей уравнение (9) для случая отсутствия испарения ($p = {\text{const}}$) GaSxSe1 –x приведены ниже. Численное решение подбирали в виде гамма-распределения с допущением, что функция распределения соответствует состоянию равновесия в системе GaS–GaSe.
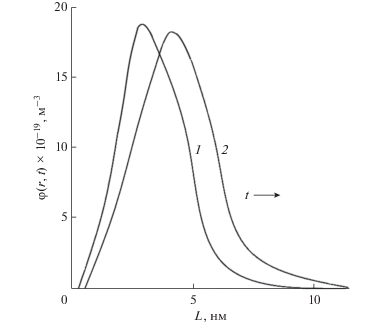
Параметры расчета и аппроксимации эволюции функции распределения частиц GaSxSe1 –x (х = 0.01) по размеру в момент времени нуклеации (t, с) были следующими 1 – 0.005 и 2 – 0.001. $\delta \left( t \right)$ = 5 и ${{K}_{L}}$ = 1 (рис. 5). Установлено, что, ускорение процессов укрупнения кристаллов приводит к различию вида функции распределения и количества формируемого кристалла. Принимали, что зависимость температуры от размера (радиуса) частиц описывается соотношением
где ${{r}_{0}}$ – начальный радиус частицы.Рис. 5.
Эволюция функции распределения кристаллов GaSxSe1– x (х = 0.3) по размеру в момент времени нуклеации (t, мин): 1 – 5 и 2 – 10.
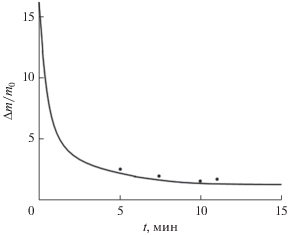
Для проверки адекватности предложенной модели проведено сравнение с экспериментом. Результаты полученного расчета (кривая) и эксперимента (точки) динамики отношения изменения массы ($\Delta m$) нанокристаллов GaSxSe1 –x к начальной среднеарифметической массе частиц ${{m}_{0}}$ приведены на рис. 6. Численные эксперименты указывают на то, что концентрация кристаллов заданного состава меняется от исходного пересыщения до равновесной концентрации за очень короткое время. Из зависимости концентрации формированных нанокристаллов, например, состава GaSxSe1 –x (х = 0.01) от времени следует, что основная масса нанокристаллов образуется в течение секунды от момента начала кристаллизации. Такой процесс изменения концентрации сопровождается снижением пересышения раствора. При постоянном числе образованных зародышей пересыщение заканчивается и затем происходит рост кристаллов с дальнейшем увеличением размера кристаллов. Таким образом, при использовании нелинейной модели необходимо учитывать концентрацию кристаллизующейся фазы, а также данные о пересыщении и размере кристаллов.
Рис. 6.
Зависимость концентрации нанокристаллов GaSxSe1 –x (х = 0.3) от времени; $\Delta m$ – масса частицырадиуса $r$; ${{m}_{0}}$ – среднеарифметическая масса частиц. Точки – эксперимент.
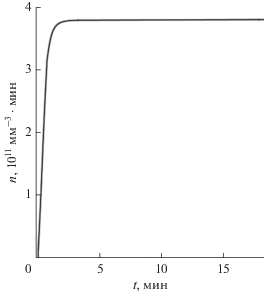
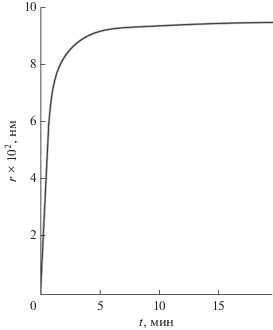
Аппроксимированы временные зависимости числа центров роста и среднего размера кристаллов GaSxSe1 –x. Из рис. 7, 8 следует, что ход распределения в эксперименте и в расчете совпадают, но расчетные кривые отличаются при больших радиусах частиц. Имеется качественное совпадение модельных расчетов с экспериментальными данными. Количественные отличия распределения частиц можно объяснить ограничениями физико-химической модели и метода расчета; в реальном процессе роста частиц вид эволюция функции распределения может искажаться. Поэтому для полноты и адекватности модели нужно учесть, в частности, влияние химического потенциала компонент, температуры, а также примесей на нуклеацию и рост кристаллов GaSxSe1 –x.
Сначала рассмотрим, как влияют активности компонентов на нуклеацию и кристаллизацию GaSxSe1 –x. Активности компонентов связаны с химическим потенциалом, поэтому разность химических потенциалов мономера в растворе и в массивном твердом теле равна $\Delta \mu = {{k}_{{\text{B}}}}T\ln \left( {{c \mathord{\left/ {\vphantom {c {{{c}_{0}}}}} \right. \kern-0em} {{{c}_{0}}}}} \right)$, где $c$ – концентрация “мономеров” в растворе, ${{c}_{0}}$ – концентрация насыщенного раствора. На зависимости $\Delta G = f\left( r \right)$ свободная энергия Гиббса как функция размера имеет максимум, поэтому на участке с радиусом $r > r{\text{*}}$ зародыши будут расти, а на участке $r < r{\text{*}}$ – они растворяются.
Концентрационно-температурные зависимости активности компонентов GaSxSe1 –x проанализируем в приближении регулярных растворов. В формированных кристаллах GaS и GaSe химическая связь между атомами преимущественно ковалентная. Поэтому растворимость в GaSxSe1 –x примесей с другим характером химической связи по механизму замещения ограничена. В GaS и GaSe атомная доля примесей не превышает ≤10–3. В системе GaS–GaSe определим коэффициент активности, например, моноселенида галлия $~\gamma _{{l,s}}^{{{\text{GaSe}}}}$
(12)
$RT{\text{ln}}\gamma _{{l,s}}^{{{\text{GaSe}}}} = \omega _{{l,s}}^{{{\text{GaSe}}}}{{(1 - x_{{l,s}}^{{{\text{GaSe}}}})}^{2}},$В системе GaS–GaSe значение $\gamma _{{l,s}}^{{{\text{GaSe}}}}$ зависит от выбранной модели раствора [12]. Если в системе жидкие и твердые растворы неидеальны, то значение $\gamma _{{l,s}}^{{{\text{GaSe}}}} = 1$. Равновесный коэффициент распределения $k_{0}^{{{\text{GaSe}}}}$ определим в области малых концентраций GaSe:
(13)
${\text{\;ln}}~k_{0}^{{{\text{GaSe}}}} = ~\frac{{\Delta H_{{\text{m}}}^{{{\text{GaSe}}}}}}{R}\left( {\frac{1}{{T_{{\text{m}}}^{{{\text{GaS}}}}}} - \frac{1}{{T_{{\text{m}}}^{{{\text{GaSe}}}}}}} \right) + ~\frac{{\omega _{l}^{{{\text{GaS}}}}}}{{RT_{{\text{m}}}^{{{\text{GaS}}}}}} - \frac{{\omega _{s}^{{{\text{GaS}}}}}}{{T_{{\text{m}}}^{{{\text{GaS}}}}}} = \frac{{\Delta H_{{\text{m}}}^{{{\text{GaSe}}}}}}{R}\left( {\frac{1}{{T_{{\text{m}}}^{{{\text{GaS}}}}}} - \frac{1}{{T_{{\text{m}}}^{{{\text{GaSe}}}}}}} \right) + ~\,\,\frac{{{{a}_{l}} - {{b}_{l}}T_{{\text{m}}}^{{{\text{GaS}}}} - \omega _{s}^{{{\text{GaS}} - {\text{GaSe}}}}}}{{RT_{{\text{m}}}^{{{\text{GaS}}}}}},$Формирование новых ядер зависит от температуры. Рассмотрим влияние градиента температуры на процесс 2D-нуклеации и роста с учетом концентрации примесей. При понижении температуры критический радиус $r{\text{*}}$ и соответствующая свободная энергия $\Delta G{\text{*}}$ системы уменьшаются. Т.е. при понижении температуры ниже равновесной температуры затвердевания (${{T}_{{\text{m}}}}$) 2D-нуклеация происходит легко. Число стабильных ядер $n{\text{*}}$ (имеющих радиусы больше, чем $r{\text{*}}$) является функцией температуры:
где ${{K}_{1}}$ – кинетическая константа, которая связана с общим числом ядер твердой фазы. Когда температура $T$ опускается ниже ${{T}_{{\text{m}}}}$, величина $n{\text{*}}$ возрастает.В полупроводниковых системах влияние градиента температуры на процесс 2D-нуклеации и роста с учетом концентрации примесей может быть значительным. Для учета влияния примесей на морфологическую нестабильность роста в качестве граничного случая возьмем минимальный градиент температуры ($dT$). Значение $dT$, который должен быть приложен к расплаву на границе 2D роста, чтобы предотвратить переохлаждение, можем определить из равновесной Т–х фазовой диаграммы
(15)
$\frac{{dT}}{{dx}} \geqslant m\left( {{{dC} \mathord{\left/ {\vphantom {{dC} {dx}}} \right. \kern-0em} {dx}}} \right)(1,20),$На 2D-нуклеацию и рост кристаллов из расплава влияют также и другие факторы, в частности, теплопроводности компонентов, анизотропия скорости роста, конвекционные потоки. Эти явления, в основном связаны с температурно-концентрационными переохлаждениями системы. Они заметно влияют на качество получения ориентированных кристаллов.
При росте кристаллов из растворов будет действовать другой механизм переохлаждения. Это происходит из-за разного коэффициента диффузии ($D$) компонентов из основной массы растворов. В этом случае из-за близко действующей диффузии температура оказывает влияние на образование ядер вторичных фаз. Влияние температуры на скорость диффузии, которая характеризуется величиной $D$, задается уравнением:
где ${{D}_{0}}$ – предэкспоненциальный множитель, который не зависит от Т, ${{Q}_{{\text{d}}}}$ – энергия активации диффузии. Кроме того, такой диффузионный эффект определяет частоту, с которой атомы из жидкой фазы присоединяются друг к другу(17)
${{\nu }_{{\text{d}}}} = {{K}_{2}}{\text{exp}}\left( { - \frac{{{{Q}_{{\text{d}}}}}}{{{{k}_{{\text{B}}}}T}}} \right),$С учетом влияния температуры на число стабильных ядер и указанного диффузионного эффекта скорость образования ядер в единице объема за 1 секунду ($\dot {N}$) определяется выражением
(18)
$\dot {N} = {{K}_{3}}n{\text{*}}{{\nu }_{{\text{d}}}} = {{K}_{1}}{{K}_{2}}{{K}_{3}}{\text{exp}}\left( { - \frac{{\Delta G{\text{*}}}}{{{{k}_{{\text{B}}}}T}}} \right){\text{exp}}\left( { - \frac{{{{Q}_{{\text{d}}}}}}{{RT}}} \right),$Учет диффузионного эффекта нарушает равновесную температуру ликвидуса на Т–х фазовой диаграмме GaS–GaSe, и в системе развивается переохлаждение.
Для гомогенного ядрообразования при охлаждении жидкости затвердевание начнется с уменьшением $T$ ниже равновесного затвердевания (или температуры плавления ${{T}_{{\text{m}}}}$) и произойдет переохлаждение. В этом случае степень переохлаждения $\Delta T$ для некоторых систем может быть значительной. Так, например, для полупроводникового германия при гомогенной нуклеации $\Delta T = $ 227°С.
Несмотря на то, что для гомогенного затвердевания требуется значительное переохлаждение, при гетерогенном зарождении фазобразование твердых частиц, например, GaSxSe1 –x (х = 0.3) начинается уже при охлаждении на несколько градусов. Т.е. если в системе существуют поверхности и границы раздела, то энергия активации процесса понижается и зарождение частиц второй фазы облегчается.
Рассмотрим рост фазы, который начинается в системе после того, как ядро превышает критический размер, $r{\text{*}}$, и превращается в стабильное ядро. Параллельно с ростом новой фазы зародышеобразование будет происходить в тех областях, где частицы новой фазы не встречаются. Рост частиц происходит за счет дальнодействующей атомной диффузии, которая включает несколько этапов: диффузия через “материнскую” фазу, через границу фаз, диффузия в зародыше. Поэтому скорость роста свободной энергии Гиббса будет определяться скоростью диффузии и ее зависимостью от температуры
(19)
$\dot {G} = {{K}_{g}}{\text{exp}}\left( { - \frac{{{{Q}_{{\text{d}}}}}}{{{{k}_{{\text{B}}}}T}}} \right).$Если принять, что диффузионная модель 2D роста обеспечивает поток частиц компонентов к поверхности, то с учетом общего числа ядер N уравнение (15) можно записать в виде
где $v$ – скорость роста, $\rho $ – плотность твердого вещества, М – молекулярная масса, D – коэффициент диффузии растворенного вещества в жидкой фазе.Для роста твердого раствора GaSxSe1 –x (х = 0.3) из жидкого раствора на основе GaSe с учетом опытных данных, если примем $v$ = 25 μм /мин и $D$ = 6 × 10–14 см2/с, то получим (${{dT} \mathord{\left/ {\vphantom {{dT} {dx}}} \right. \kern-0em} {dx}}~$)min = 15°C /см при 900°C. Здесь изменение значения (${{dT} \mathord{\left/ {\vphantom {{dT} {dx}}} \right. \kern-0em} {dx}}~$)min с температурой отражает минимальное изменение наклона линии ликвидуса на Т–х фазовой диаграамме системы GaS–GaSe. Оценки, представленные выше в системе роста кристаллов, сделаны с учетом неопределенного объема раствора. Если количество раствора ограничить, то растворенное вещество истощается с понижением его концентрации на внешней границе роста 2D кристалла. Для GaSxSe1–x(х = 0.3), например, толщиной 0.5 мм получим температурный градиент ~5°C/см (вместо вышеуказанного 15°C/см).
ЗАКЛЮЧЕНИЕ
На основе разработанной физико-химической модели рассмотрены стадии и закономерности нуклеации и кристализации двумерных термодинамических фаз. Моделирование процессов переноса частиц фаз проведено на примере формирования нанокристаллов твердых растворов GaSxSe1 –x (0 ≤ х ≤ 1) с учетом переохлажденного расплава в закрытых системах. С учетом уравнений процесса стационарной кристализации описаны скорости зарождения и роста новой фазы. В разработанной нелинейной модели нуклеации и кристаллизации двумерных 2D фаз использовано эволюционное уравнение типа Фоккера–Планка (Ф–П) в пространстве размеров. Выбраны термодинамические условия кристаллизации фаз системы GaS–GaSe, и на основе указанной модели аппроксимирована эволюция функции распределения частиц по размерам во времени. Численные данные получены решением модели, содержащей систему нелинейных дифференциальных уравнений Ф–П, методом конечных разностей. Использована неявная разностная схема, предполагающая разбиение протяженного объема раствора на независимые фрагменты. Численные аппроксимации проведены для нуклеации и роста 2D-нанокристаллов GaS0.3Se0.7. Показано, что в нелинейных моделях характер функции распределения существенно зависит от влияния химического потенциала компонентов, температуры, а также примесей на нуклеацию и рост кристаллов. Свойства формированных ядер частиц в системе зависит также от коэффициента активности, распределения примесей, температурно-зависящих факторов и диффузионного эффекта.
Список литературы
Лукичев В.Ф., Амиров И.И. Исследования и разработки в области микро- и наносистемной техники // История науки и техники. 2018. № 8. С. 92–99.
Wang T., Li J., Zhao Q., Yin Z., Zhang Y., Chen B., Xie Y., Jie W. High-Quality GaSe Single Crystal Grown by the Bridgman Method // Materials. 2018. V. 11. № 2. 186. P. 2–9. https://doi.org/10.3390/ma11020186
Ho C.H., Wang S.T., Huang Y.S., Tiong K.K. Structural and luminescent property of gallium chalcogenides GaSe1 −xSx layer compounds // J. Mater. Sci.: Mater. Electron. 2009. V. 20. P. S207–S210. https://doi.org/10.1007/s10854-007-9539-3
Bereznaya S., Korotchenko Z., Redkin R., Sarkisov S., Tolbanov O., Trukhin V., Gorlenko N., Sarkisov Y., Atuchin V. Broadband and narrowband terahertz generation and detection in GaSe1 −x Sx crystals // J. Opt. 2017. V. 19. № 11. P. 115503. https://doi.org/10.1088/2040-8986/aa8e5a
Kolesnikov N.N., Borisenko E.B., Borisenko D.N., Tereshchenko A.N., Timonina A.V. Synthesis and growth of GaSe1 –xSx (x = 0–1) crystals from melt. Phase Composition and properties // Inorganic Materials: Applied Research. 2018. V. 9. № 1. P. 66–69. https://doi.org/10.1134/S2075113318010173
Asadov S.M., Mustafaeva S.N., Lukichev V.F. Chargetransport in layer gallium monosulfide in direct and alternate electric fields // Russian Microelectronics.2019. V. 48. № 6. P. 422–427.https://doi.org/10.1134/S1063739719660016
Mustafaeva S.N., Asadov M.M. Currents of isothermal relaxation in GaS〈Yb〉 single crystals // Solid State Communications. 1983. V. 45. № 6. P. 491–494. https://doi.org/10.1016/0038-1098(83)90159-X
Asadov S.M., Mustafaeva S.N., Lukichev V.F., Guseinov D.T. Effect of the Composition on the Dielectric Properties and Charge Transfer in 2D GaS1–xSex Materials // Russian Microelectronics. 2019. V. 48. № 4. P. 203–207. https://doi.org/10.1134/S1063739719040024
Mustafaeva S.N., Asadov M.M., Ismailov A.A. Charge transfer over localized states in a TlS single crystal // Phys. Solid State. 2008. V. 50. № 11. P. 2040–2043. https://doi.org/10.1134/S1063783408110073
Mustafaeva S.N., Asadov M.M., Ismailov A.A. Dielectric and baric characteristics of TlS single crystal // Physica B: Physics of Condensed Matter. 2014. V. 453. P. 158–160. https://doi.org/10.1016/j.physb.2014.03.095
Mustafaeva S.N., Asadov M.M. High field kinetics of photocurrent in GaSe amorphous films // Materials Chemistry and Physics. 1986. V. 15. P. 185–189. https://doi.org/10.1016/0254-0584(86)90123-9
Asadov S.M., Mustafaeva S.N., Mammadov A.N. Thermodynamic assessment of phase diagram and concentration–temperature dependences of properties of solid solutions of the GaS-GaSe system // J. Thermal Analysis and Calorimetry. 2018. V. 133. № 2. P. 1135–1141. https://doi.org/10.1007/s10973-018-6967-7
Mustafaeva S.N., Asadov M.M., Ismailov A.A. Charge transfer along localized states in InSe and InSe〈Sn〉 single crystals // Low Temp. Phys. 2010. V. 36. № 4. P. 310–312. https://doi.org/10.1063/1.3388822
Mustafaeva S.N., Asadov M.M., Ismailov A.A. Effect of γ irradiation on the parameters of localized states in p‑InSe and n-InSe⟨Sn⟩ single crystals // Low Temp. Phys. 2010. V. 36. № 7. P. 642–644. https://doi.org/10.1063/1.3479690
Tan L., Liu Q., Ding Y., Lin X., Hu W., Cai M.-Q., Zhou H. Effective shape-controlled synthesis of gallium selenide nanosheets by vapor phase deposition // Nano Research. 2020. CN 11-5974/O4. https://doi.org/10.1007/s12274-020-2653-8
Jung C.S., Shojaei F., Park K., Oh J.Y., Im H.S., Jang D.M., Park J., Kang H.S. Red-to-ultraviolet emission tuning of two-dimensional gallium sulfide/selenide // ACS Nano. 2015. V. 9. № 10. P. 9585–9593.https://doi.org/10.1021/acsnano.5b04876
Li X.F., Lin M.W., Puretzky A.A., Idrobo J.C., Ma C., Chi M.F., Yoon M., Rouleau C.M., Kravchenko I.I., Geohegan D.B., Xiao K. Controlled vapor phase growth of single crystalline, two-dimensional GaSe crystals with high photoresponse // Sci. Rep. 2015. V. 4. 5497. P. 1–9. https://doi.org/10.1038/srep05497
Hu P.A., Wen Z.Z., Wang L.F., Tan P.H., Xiao K. Synthesis of few-layer GaSe nanosheets for high performance photodetectors // ACS Nano. 2012. V. 6. № 7. P. 5988–5994. https://doi.org/10.1021/nn300889c
Becker R., Döring W. Kinetischebehandlung der keimbildung in übersättigtendämpfen // Annalen Der Physik, 1935. V. 416. № 8. P. 719–752. https://doi.org/10.1002/andp.19354160806
Зельдович Я.Б. К теории образования новой фазы. Кавитация // Журн. экспериментальной и теоретической физики. 1942. Т. 12. № 11/12. С. 525–538.
Kashchiev D. Nucleation. Basic Theory with Applications. 1st Ed. Butterworth-Heinemann, Oxford. Amsterdam. Elsevier. 2003. 551 p. ISBN: 9780750646826
Saito Y., Honjo M., Konishi T., Kitada A. Time-Dependent nucleation rate // J. Physical Society of Japan. 2000. V. 69. № 10. P. 3304–3307. https://doi.org/10.1143/jpsj.69.3304
Anisimov M.P. Nucleation: theory and experiment // Russian Chemical Reviews. 2003. V. 72. № 7. P. 591–628. https://doi.org/110.1070/rc2003v072n07abeh000761
Nucleation in Condensed Matter: Applications in Materials and Biology. Kelton K.F. and Greer A.L. (Eds.). Pergamon Materials Series 15. Amsterdam. Elsevier. Pergamon. 2010. 726 p. ISBN:9780080421476
Flemings M.C. Solidification Processing. McGraw-Hill College. New York. 1974.364 p.ISBN-13: 978-0070212831
Chernov A.A. Modern crystallography III. Springer-verlag Berlin, Heidelberg, New York, Tokyo. 1984. 517 p. ISBN 3-540-11516-1. https://doi.org/10.1007/978-3-642-81835-6
Perepezko J.H., Hoffmeyer M.K., De Cicco M.P. Analysis of melt undercooling and crystallization kinetics // Metallurgical and Materials Transactions. A. 2015. V. 46. № 11. P. 4898–4907. https://doi.org/10.1007/s11661-015-2970-9
Ivanov A.O., Zubarev A.Yu. Non-linear evolution of a system of elongated droplike aggregates in a metastable magnetic fluid // Phys. A. 1998. V. 251. P. 348–367. https://doi.org/10.1016/S0378-4371(97)00561-X
Mullin J.W. Crystallization. 4th ed. Butterworth-Heinemann. 2001. 610 p. ISBN: 9780750648332
Herlach D.M. (Ed.). Phase Transformations in Multicomponent Melts. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. 2008. 450 p. ISBN: 9783527319947
Aaronson H.I., Enomoto M., Lee J.K. Mechanisms of Diffusional Phase Transformations in Metals and Alloys. BocaRaton. 1st Ed. CRC Press. Taylor & Francis Group. 2010. 685 p. ISBN-13: 978-1420062991
Rachah A., Noll D., Espitalier F., Baillon F. A mathematical model for continuous crystallization // Math. Methods Appl. Sci. 2016. V. 39. P. 1101–1120. https://doi.org/10.1002/mma.3553
Risken H. The Fokker–Planck Equation. Methods of Solutions and Applications. 2nd Edn. Springer-Verlag. Berlin. Heidelberg. 1996. 486 p. ISBN: 978-3-540-61530-9
Горбачевский А.Я. Численное исследование нелинейных моделей кpисталлизации // Математическое моделиование. 1999. Т. 11. № 8. С. 23–31.
Frank T.D. Nonlinear Fokker–Planck Equations. Fundamentals and Applications. Springer-Verlag. Berlin. Heidelberg. 2005. 413 p. ISBN: 978-3-540-21264-5
Makoveeva E.V., Alexandrov D.V. A complete analytical solution of the Fokker–Planck and balance equations for nucleation and growth of crystals // Phil. Trans. R. Soc. 2018. A 376: 20170327. https://doi.org/10.1098/rsta.2017.0327
Кукушкин С.А., Слезов В.В. Дисперсные системы на поверхности твердых тел (эволюционный подход): механизмы образования тонких пленок. СПб.: Наука, 1996. 304 с. ISBN: 5-02.-024856-8
Годунов С.К., Рябенький В.С. Разностные схемы. Введение в теорию. Изд. 2, перераб. и доп. М.: Наука, 1977. 440 с.
Дополнительные материалы отсутствуют.
Инструменты
Микроэлектроника