

Микроэлектроника, 2019, T. 48, № 5, стр. 323-333
Исследование влияния технеция в составе катализаторов из соединений РЗЭ на процесс разложения моногидридов кремния и германия
А. А. Ковалевский 1, А. С. Строгова 1, *, Д. Ф. Кузнецов 1, Я. С. Воронец 1, С. В. Гранько 1
1 Белорусский государственный университет информатики и радиоэлектроники
220013 Минск, ул. П. Бровки, 6, Беларусь
* E-mail: strogova@bsuir.by
Поступила в редакцию 10.12.2018
После доработки 11.03.2019
Принята к публикации 20.03.2019
Аннотация
Установлено влияние технеция в составе катализаторов из соединений РЗЭ на процесс разложения моногидридов кремния и германия при выращивании эпитаксиальных пленок кремния и твердого раствора кремний-германий. Показано, что благодаря тому, что радиус элементарной ячейки технеция равен 1.358 Å и близок к радиусу элементарной ячейки платиновых элементов (1.37–1.38 Å), в составе катализаторов из соединений РЗЭ позволяет обеспечить постоянство удельной каталитической активности и высокую селективность разложения моносилана и моногермана до ${\text{SiH}}_{2}^{{{\text{**}}}},$ ${\text{GeH}}_{2}^{{{\text{**}}}}.$ Это, в свою очередь, позволяет проводить процесс выращивания эпитаксиальных пленок в две стадии и тем самым повысить их качество.
ВВЕДЕНИЕ
Общеизвестно, что при осаждении тонких пленок из газовой фазы нарушения в тонких пленках могут возникать из-за того, что реакция на поверхности подложки протекает в одну стадию. Исходные реагенты присутствуют на поверхности подложки наряду с созданными в результате реакции тонкими слоями кремния, германия- или оксида кремния; или нитрида кремния – в зависимости от того, какая реакция рассматривается. С другой стороны, дефекты в пленке также образуют свободные связи, имеющиеся на поверхности полупроводниковой или другой подложки. Для создания бездефектных пленок необходимо осуществить разделение обеих стадий реакции, а именно, разделение этапа химического взаимодействия реагентов от этапа образования пленки на подложке. Эта проблема решается путем внедрения процессов термического разложения моногидридов в проточных системах. Однако в таких системах потеря не прореагировавшего моногидрида составляет более 40% [6–9]. Использование малоэффективных и труднореализуемых физических методов интенсификации химических реакций [1] также не дает экономии исходных материалов. Поэтому следует развивать химические методы интенсификации технологических процессов микроэлектроники, которые отличаются простотой реализации и высокой эффективностью, т.е. с использованием катализаторов. В этом аспекте определенный интерес представляют твердотельные катализаторы, располагаемые на расстоянии от 1 мм до 1 метра от поверхности подложки, на которой создается пленка.
Цель работы – создание бездефектных пленок кремния и германия посредством полного разделения этапа химического взаимодействия и этапа образования пленки в результате использования катализаторов на основе соединений редкоземельных элементов, содержащих технеций.
Указанная цель достигается тем, что при формировании тонких пленок кремния и германия из газовой фазы при разложении гидридов кремния и германия, они проходят химическую активацию на твердотельных катализаторах на основе соединений редкоземельных элементов, в качестве которых, преимущественно, используются оксиды, фториды, нитриды, селениды, теллуриды, фосфиды, бориды и бораты. В этом случае газовую смесь пропускают через катализаторы с высокой активностью и селективностью за счет введения в их состав технеция, радиус элементарной ячейки которого близок к радиусу элементарной ячейки платиновых элементов.
МЕТОДИКА И ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Выбор элементов с радиусом элементарной ячейки платиновых элементов обусловлен тем, что элементы с таким радиусом (1.358–1.380 Å) относятся к элементам с большим атомным радиусом и приводят к прямому дегидрированию моносилана и моногермана на катализаторе до активных радикалов ${\text{SiH}}_{2}^{{{\text{**}}}},$ ${\text{GeH}}_{2}^{{{\text{**}}}}.$
Использовался катализатор на полиимидном носителе, который готовился совместно с носителем следующим образом. В полиамидную кислоту с ароматическими звеньями с молекулярным весом 5 × 104 и кинетической вязкостью 2.4 × 10–4 м2 ∙ с–1 вносили ультрадисперсный (10–50 нм) порошок содержащий 99.5–84 вес. % оксида европия (фторида европия, сульфида европия, оксида гадолиния, фторида гадолиния или сульфида гадолиния) и 0.5–6.0 вес. % технеция по массе 1 : 50 по отношению к массе твердого продукта полиамидной кислоты. Затем полиамидную кислоту тщательно перемешивали в магнитной мешалке со скоростью 60 с–1 и наносили на полированную кварцевую подложку, по конфигурации соответствующую сечению торца кварцевого реактора, многократным окунанием при скорости извлечения подложки из полиамидной кислоты 5 см · мин–1. После этого кварцевая подложка с нанесенной полиамидной кислотой, содержащей ультрадисперсный порошок из вышеуказанных соединений РЗЭ и технеция, помещались в вакуумную термическую камеру. Степень вакуума поддерживалась равной 1.33 × 10–1 Па, а температура 598 К. В этих условиях в течение 25 мин проходил процесс имидизации полиамидной кислоты в полиимид. Причем формировались микропористые (с размером пор от 0.5 до 1.0 мкм) полиимидные пленки с удельной площадью поверхности 97 м2 · г–1. Полученная таким образом полиимидная пленка представляла собой микропористую мембрану, которая устанавливалась на входе моногидридов с аргоном в реактор. Полиимидная мембрана с катализатором располагалась на расстоянии 100 мм от подложкодержателя. Области чистого полиимида в катализаторе на основе соединений редкоземельных элементов с технецием на полиимидном носителе не вовлекаются в связывание соединений РЗЭ и технеция, поэтому в рабочем состоянии они, как правило, в качестве пористого материала служат для транспорта реагентов и продуктов реакции.
Процессы разложения моносилана проводились в горизонтальной установке наращивания эпитаксиальных слоев “Унипак-1010” с индукционным нагревом. Полиимидная мембрана с ПМК располагалась на расстоянии 100 мм от подложкодержателя, что позволяло поддерживать на катализаторе температуру 473 К.
Для осаждения эпитаксиальных пленок кремния использовались пластины кремния ЭКДБ-10 с ориентацией (111), диаметром 150 мм, отвечающие по качеству техническим условиям ЕТО.035.124ТУ.
Учитывая тот факт, что полиимидная мембрана, как носитель катализатора, для реагентов представляет определенное сопротивление – парциальное давление аргона как газоносителя было выбрано равным 3 × 105 Па, а парциальное давление моногидридов (моносилан, моногерман) 10–50 Па. Аргон подогревался до 473 К с тем расчетом, чтобы в области катализатора поддерживалась именно такая температура, что способствовало интенсивному разложению моногидридов кремния и германия на катализаторе до ${\text{SiH}}_{2}^{{{\text{**}}}},$ ${\text{GeH}}_{2}^{{{\text{**}}}}.$ Температура подложек за счет ВЧ-нагрева в процессе наращивания тонких эпитаксиальных пленок кремния, твердого раствора кремний-германий поддерживалась равной 973 К. Скорость роста пленок при этом составляла 0.5–0.6 мкм ∙ мин–1. Подготовка подложек перед наращиванием проводилась согласно широко используемым приемам в эпитаксиальной технологии. Наращивались эпитаксиальные пленки толщиной 1.5, 2.5, 3.0, 4.5, 6.0 мкм, предназначенные для производства интегральных микросхем (ИС, БИС, СИС).
Средняя плотность дислокаций исходных пластин, используемых при осаждении тонких эпитаксиальных пленок кремния, составляла 10 см–2.
В качестве газа носителя и восстановителя использовали водород, пропущенный через палладиевый фильтр. Температура подложек в процессе осаждения контролировали с помощью платинородиевой термопары с точностью ±0.5°, а также оптическим пирометром China Work Ltd. (Япония) с точностью ±2.5°.
Пластины равномерно размещали по изотермической зоне реактора на расстоянии 5 мм друг от друга. В качестве исходных реагентов использовались моносилан SiH4, моногерман GeH4, фосфин РН3, арсин AsН3, диборан В2Н6 и аргон Аr марки ОС. Ч.
Толщина пленок кремния определялась методом косого и сферического шлифов и методом воздушного зазора с точностью ±1 нм, используя сканирующий электронный микроскоп S-4800 (Hitachi, Япония).
Продукты реакции разложения (дегидрирования) SiH4 анализировали методами газовой хромотографии, масс-спектрометрии и инфракрасной спектроскопии. Морфологию, химический состав катализаторов и полученных пленок исследовали с помощью растровой электронной микроскопии (РЭМ – PhilipsXL 30 SFEG и HitachiS-4800), просвечивающей электронной микроскопии (ПЭМ – PhilipsCM – 30), ИК-спектроскопии (Perkin – Elmer Spectrum One), электронной Оже-спектроскопии (ЭОС –спектрометр ЗРHI 660), спектроскопии комбинационного рассеяния (КР – твердотельный лазер Nicolet Spectrometer“NECSUS 720” с длиной волны λ = = 1064 нм и мощностью 0.1–3 Вт) и рентгеновской фотоэлектронной спектроскопии (РФЭС – спектрометр ЭС 2401 с полусферическим анализатором электронов). Для получения информации о химическом состоянии элементов в объеме пленки при использовании РФЭС для возбуждения фотоэмиссии применяли монохроматизированное MgKα излучение мощностью 300 Вт, направленное под углом 45° к анализируемой поверхности. Регистрацию спектров проводили при давлении остаточных газов в камере спектрометра не хуже 8 × 10–8 Па. Спектры калибровали по пику 1s-уровня углерода с энергией связи 285.0 эВ, обусловленного эмиссией фотоэлектронов из атомов углерода в С–Н группах. Химическое состояние элементов поверхности образца оценивали по сдвигам энергетических пиков в спектрах высокого разрешения С1s-линий. Для изменения глубины анализа в объеме образца использовали распыление поверхности маломощным пучком ионов аргона.
Поэлементный состав полученных эпитаксиальных пленок определяли с помощью микрозондового рентгеноспектрального анализа на установке “Cameca – MBX”.
Полноту разложения моногидридов α определяли как отношение объема прореагировавшего моногидрида Vп к общему его объему в газовой фазе Vоб: α = (Vп/Vоб) × 100%.
Для сравнительной оценки активности катализаторов, содержащих добавки технеция, реакция разложения моносилана была проведена с использованием также катализаторов на основе оксидов, фторидов и сульфидов европия и гадолиния без добавок технеция.
Оценка качества тонких эпитаксиальных пленок производилась в соответствии с требованиями технических условий ЕТО.035.097 ТУ.
Проводилась оценка: плотности дислокаций, дефектов упаковки, бугорков, шипов, следов кристаллитов, дефектов роста типа пирамид в эпитаксиальном слое – для 80% просмотренных полей.
Для оценки качества тонких эпитаксиальных пленок использовалось химическое травление в селективном травителе (50 г CrO3 + 100 мл H2O) 100 мл HF = 1 : 1, микроскопы ММУ-1, ММУ-3, МИМ-7, МИИ-4, МБС-1, МБС-2, МИИ-10, МИМ-8М.
При этом дефекты упаковки и дислокации в эпитаксиальных слоях кремния с кристаллографической ориентацией (111) выявлялись химическим травлением в селективном травителе указанного состава. В местах выхода дефектов упаковки на поверхность эпитаксиального слоя вытравливались фигуры в форме треугольников, V-образных и одиночных линий. В местах выхода дислокаций на поверхность эпитаксиального слоя вытравливались фигуры в виде треугольников, имеющих вершину. Плотность дефектов определялась путем подсчета соответствующих фигур травления на поверхности образца под микроскопами ММУ-3, МИМ-7 при увеличении 200–500×. Площадь одной площадки контроля не превышала 3 × 10–3 см–2 (поле зрения микроскопа).
Nизм = n/NсхK, где n – учтенное количество дефектов упаковки или дислокаций, K – пересчетный коэффициент, равный 1/S, где S – площадь области подлежащей обсчету; Nсх – число просмотренных участков.
В случае, если после дополнительного травления в течение 25 с размеры фигур травления на дефектах упаковки и дислокациях становились примерно равными и трудноразличимыми по размерам, подсчитывалась общая плотность треугольных фигур травления. Для определения плотности дислокаций из общей плотности фигур травления вычиталась плотность дефектов упаковки после травления в течение 5 с. Для определения плотности таких дефектов роста, как бугорки, применялись микроскопы МБС-1 и МБС-2, а для определения их высоты калька бумажная натуральная марки А. Контроль наличия и определение количества дефектов роста (бугорков) проводился путем визуального просмотра поверхности эпитаксиального слоя под люминесцентным источником света. Высота бугорков определялась путем сканирования кальки по поверхности эпитаксиального слоя в области видимых неровностей. Размеры бугорков считались критическими, если полоска кальки при сканировании задерживалась на них. Количество выявленных бугорков подсчитывалось визуально. Плотность бугорков вычислялась по формуле N = n/S, где n – количество выявленных бугорков, S – площадь эпитаксиальной структуры в см2 (без учета краевой кромки шириной 3 мм).
Количество выявленных шипов определялось аналогичным образом, а их высота – в микроинтерферометре Линника МИИ-4 и МИИ-10. При работе с белым светом H = 0.27K, где K – количество интерференционных колец, а при работе с монохроматическим светом H = 1/2λK.
Трипирамиды – дефекты роста в эпитаксиальных пленках, видимые без травления и наблюдаемые с помощью металлографического микроскопа МБС-1 с увеличением 50–88× при освещении через второй бинокуляр. В поле микроскопа трипирамиды наблюдаются в виде звездообразных холмиков. Высота трипирамид определялась на микроскопе МИМ-8М с помощью микрометра окулярного винтового МОВ-1.
Плотность следов кристаллитов на кремниевых эпитаксиальных структурах оценивалась путем визуального наблюдения под микроскопом мелких локальных углублений, образующихся в местах кристаллизации оксида на подложке. Эти углубления имеют характерную огранку, которая сохраняется и после эпитаксии. Для определения плотности кристаллитов на кремниевой структуре использовались микроскопы ММУ-1, ММУ-3 и микроинтерферометр Линника МИИ-4, объект – микрометр СМП и окуляр-микрометр МОВ-1-16х. Средняя плотность следов кристаллитов для пленки не менее 80% просмотренных полей, имеющих минимальную плотность, определялась по формуле
где N – количество учтенных полей (не менее 5), 2.2 × 10–3 – поле зрения микроскопа, в см–2. Размер кристаллита измерялся с помощью окулярного микрометра МОВ-1-16х, цена деления которого определялась с помощью объект микрометра.РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Механизм активации процесса разложения моногидридов катализаторами представляет собой не весьма простое взаимодействие компонентов системы между собой. При такой постановке задачи осуществляется химическая активация реакционных газов, причем химические связи этих газов поддерживаются в неустойчивом состоянии, тем самым ускоряется процесс осаждения и образования пленки из продуктов реакции разложения. Под воздействием катализаторов реакции протекают по новому реакционному пути в результате промежуточного химического взаимодействия реагирующих веществ с катализатором. При этом ускорение реакции в направлении образования определенного продукта достигается за счет роста вероятности образования активных комплексов на стадиях нового реакционного пути при равновесном распределении энергии в системе. Как хорошо известно [1–4], в случае газовых, как гомогенных, так и гетерогенных реакций, скорости превращения достигаются за счет резкого увеличения концентрации активных образований, по сравнению с равновесными, благодаря использованию свободной энергии реакции. Для предвидения каталитического действия очень существенна правильная оценка роли химического состава и структурных нарушений кристаллов катализатора. Долгое время очень большое значение придавалось особым каталитическим свойствам атомов, расположенных в углах или в ребрах кристаллов катализаторов и вблизи всякого рода структурных дефектов. В работах [3–5] весьма четко акцентируется внимание на постоянстве удельной каталитической активности платины, палладия и никеля при вариации размера кристаллитов от 1 до 20 нм на различных носителях. В целом можно заключить, что решающее значение для каталитических свойств имеет химический состав, а не структурные нарушения или положение атомов в кристаллах катализатора. При этом, конечно, имеется ввиду не усредненный химический состав, а состав соединений или твердых растворов, образующих катализатор. При наличии нескольких фаз может играть существенную роль развитие поверхности контактов между фазами, особенно в случае сложных, многостадийных реакций [1–5]. Вывод о постоянстве удельной каталитической активности катализаторов одинакового химического состава является, тем не менее, лишь первым приближением при выборе катализатора. Проблему предвидения каталитического действия можно рассматривать в основном как проблему связи каталитических свойств с химическим составом твердых катализаторов [6–9]. Корреляция между каталитической активностью и энергией связи водорода справедлива не только для простых гидридов, но и для более сложных соединений, например, ди- и трисилана. Это открывает возможность регулирования каталитической активности гидридов путем вариации состава. Энергия связи водорода определяется в основном более электроотрицательным металлом, но может существенно меняться при вариации электроотрицательного второго катиона. Так, в платино-никелевых катализаторах с ростом электроотрицательного катиона при переходе от кальция через алюминий и хром к железу энергия связи водорода уменьшается, а каталитическая активность возрастает. Основной принцип катализа заключается в том, что в каталитической реакции катализатор изменяет механизм реакции на энергетически более выгодный, то есть снижает энергию активации. Катализатор образует с молекулой одного из реагентов промежуточное соединение, в котором ослаблены химические связи. Это облегчает его реакцию со вторым реагентом. Важно отметить, что катализаторы ускоряют обратимые реакции, как в прямом, так и в обратном направлениях. Энергия активации (Е) – это разность энергий активного комплекса и расходных реагирующих молекул. Если предположить, что реакция не разрушает равновесного распределения энергий между молекулами, то по закону Аррениуса вероятность образования активного комплекса, а следовательно и скорость реакции в первом приближении пропорциональна exp(–E/RT), где R – газовая постоянная, равная 1.987 кал/град · моль; T – абсолютная температура. Энергия активации каталитических реакций значительно меньше, чем для тех же реакций в отсутствие катализатора. Благодаря этому обеспечивается их ускорение по сравнению с не каталитическим [6–9]. Снижение энергии активации обусловлено тем, что при катализе реакция протекает по новому механизму, складывающемуся из элементарных реакций с меньшими энергиями активации, чем некаталитическая. В нашем случае катализатор вступает в химическое взаимодействие с гидридами кремния и германия. При этом образуются более реакционно способные промежуточные частицы (свободные радикалы), чем исходные вещества. Активные промежуточные частицы реагируют в дальнейшем таким образом, что их превращения приводят в итоге к образованию конечных продуктов и регенерации катализатора. Ускорение реакции в присутствии катализатора достигается за счет того, что максимальное значение свободной энергии, которое реагирующая система достигает при движении от начального состояния к конечному, для каталитического маршрута ниже, чем для не каталитического. Поверхность катализатора неоднородна (рис. 1). На ней имеются так называемые активные центры, на которых главным образом и протекают каталитические реакции. Гидриды кремния и германия как реагирующие вещества адсорбируются на этих центрах, в результате чего увеличивается концентрация их на поверхности катализатора, а это отчасти приводит к ускорению реакции. Но главная причина возрастания скорости реакции – сильное повышение химической активности адсорбированных молекул.
Рис. 1.
Структура катализатора (а–в) и рентгенограмма (г) продуктов разложения моносилана на его поверхности. Содержание технеция в составе катализатора 4 вес. %: а – Gd2O3; б – Gd2S3; в – EuF3.

Под действием катализатора у адсорбированных молекул ослабляются связи между атомами, и они становятся более реакционноспособными. И в этом случае реакция ускоряется благодаря снижению энергии активации (рис. 2) (в том числе за счет образования поверхностных промежуточных соединений).
Рис. 2.
Зависимость скорости осаждения эпитаксиальных пленок кремния от температуры подложки в Аррениусовых координатах с использованием катализаторов: 1 – Gd2O3(Tс); 2 – EuF3(Tc); 3 – Gd2S3; 4 – без катализатора.
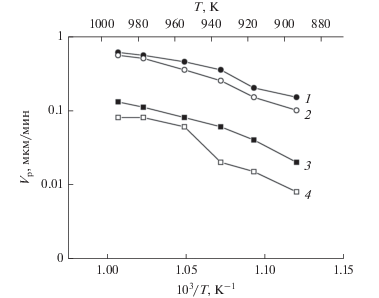
Оценка энергии активации проводилась по уравнению:
(1)
$E_{{{\text{а к }}}}^{*} = - 8.3 \times {{10}^{{ - 3}}} \times \frac{{2.3\lg \frac{{{{V}_{2}}}}{{{{V}_{1}}}}}}{{\left( {\frac{1}{{{{T}_{2}}}} - \frac{1}{{{{T}_{1}}}}} \right)}},\,\,\,\,{{к Д ж } \mathord{\left/ {\vphantom {{к Д ж } {м о л ь }}} \right. \kern-0em} {м о л ь }},$По сути, энергия активации – это то избыточное количество энергии (по сравнению со средней величиной), которой должна обладать молекула в момент столкновения, чтобы быть способной к данному химическому взаимодействию. В большинстве своем увеличение ТП способствует усилению такого взаимодействия. При повышении ТП скорость химических реакций увеличивается весьма значительно.
Фундаментальные исследования процесса разложения SiH4 и GeH4 показали, что реакции их термического разложения являются многомаршрутными и многостадийными, зависящими от условий разложения. Процесс разложения SiH4 и GeH4, может протекать по двум маршрутам – гетерогенному и гомогенному, имеющим разные кинетические временные характеристики [8]. Эти различия позволяют разделить составляющие процесса. Величины энергии активации гетерогенного ($E_{{{\text{a}}\left( {{\text{г е т }}} \right)}}^{*}$) и гомогенного ($E_{{{\text{a}}\left( {{\text{г о м }}} \right)}}^{*}$) процессов различны и зависят от температуры подложки при разложении гидридов. Эти особенности разложения моногидридов характерны и в случае использования катализаторов [6–8].
Скорость реакции сильнее изменяется с температурой на тех катализаторах в реакциях, которых энергия активации больше. И наоборот, если в данной реакции энергия активации незначительна, то скорость этой реакции слабо изменяется с температурой (рис. 2). Многочисленные исследования, посвященные процессу разложения SiH4 и GeH4, в реакторах атмосферного давления при температуре 550–900°С без использования катализаторов свидетельствуют, что $E_{{{\text{a}}\left( {{\text{г е т }}} \right)}}^{*}$ ≈ 200–224 кДж/моль, а $E_{{{\text{a}}\left( {{\text{г о м }}} \right)}}^{*}$ ≈ 230–235 кДж/моль [8, 10].
Использование технеций содержащих катализаторов типа – Gd2O3, Gd2S3, EuF3 позволяет добиться значительного снижения энергии активации по сравнению с теми же реакциями в отсутствие катализатора (рис. 2). Энергия активации каталитических реакций значительно меньше и составляет для Gd2O3 (4 вес. % Tс) 53.95 кДж/моль (1), EuF3 (4 вес. % Tc) 78.85 кДж/моль (2), Gd2S3 83.05 кДж/моль (3), и для сравнения без катализатора 224 кДж/моль (4) рис. 2. Благодаря этому обеспечивается их ускорение в области равных температур и меньшая зависимость скорости роста эпитаксиальных пленок с температурой. Снижение энергии активации обусловлено тем, что при катализе реакция протекает по новому механизму, складывающемуся из элементарных реакций с меньшими энергиями активации, чем некаталитическая. В нашем случае катализатор вступает в химическое взаимодействие с гидридами кремния и германия. При этом образуются более реакционноспособные промежуточные частицы (свободные радикалы), чем исходные вещества. Активные промежуточные частицы реагируют в дальнейшем таким образом, что их превращения приводят в итоге к образованию конечных продуктов и регенерации катализатора. Ускорение реакции в присутствии катализатора достигается за счет того, что максимальное значение свободной энергии, которое реагирующая система достигает при движении от начального состояния к конечному, для каталитического маршрута ниже, чем для некаталитического.
Эффективная энергия активации, рассчитанная нами по уравнению (1), с использованием экспериментальных данных для случая, когда в качестве катализатора использовался Gd2O3 (4 вес. % Tс) в процессе разложения SiH4 составляет величину 53.95 кДж/моль (1), при использовании EuF3 (4 вес. % Tc) 78.85 кДж/моль (2), а при использовании Gd2S3 в отсутствие в составе катализатора Tc эффективная энергия активации процесса разложения моносилана составляет 83.05 кДж/моль (3), и для сравнения без катализатора 224 кДж/моль (4) рис. 2. Снижение содержания Tс в составе катализатора ниже 0.5 вес. % и выше 6 вес. % при прочих равных условиях приводит к увеличению энергии активации до 83.05–224 кДж/моль. Нижняя граница количественного содержания (0.5 вес. %) технеция в составе катализатора на основе соединений РЗЭ, через который пропускают газовую смесь, определяет минимальную селективность (95–98%) разложения моногидридов до ${\text{SiH}}_{2}^{{{\text{**}}}},$ ${\text{GeH}}_{2}^{{{\text{**}}}}$ без потери активности катализатора. Верхняя граница количественного содержания технеция (6.0 вес. %) в составе катализатора из соединений РЗЭ, через который пропускают газовую смесь, определяет то количество вводимого элемента, при котором селективность и активность катализатора остаются неизменными. Выше этого значения возможно уменьшение селективности и отравление катализатора, т.е. уменьшение его активности особенно в области повышенных температур на катализаторе >473 K. В этом случае происходит быстрое насыщение активных центров металлом, и катализатор теряет как активность, так и селективность.
Таким образом, содержание технеция в составе катализатора на основе соединений РЗЭ при разложении SiH4 и GeH4 в количестве менее 0.5 вес. % и более 6.0 вес. % имеет отрицательный эффект, поскольку в обоих случаях происходит снижение селективности и, как результат, исключается возможность реализации полного разделения этапов химического взаимодействия и образования пленки, что подтверждается получением дефектных эпитаксиальных пленок кремния (табл. 1).
Таблица 1.
Степень дефектности тонких эпитаксиальных пленок, полученных без использования и с использованием катализаторов
| Гидридный способ получения | Соеди-нение РЗЭ | Tс, вес. % | Дислокации, см–2 | Дефекты упаковки, см–2 | Бугор-ки см–2 |
Шипы, см–2 | Трипира-миды, см–2 | Кристал-литы, см–2 |
|---|---|---|---|---|---|---|---|---|
| Толщина эпитаксиальных пленок 1.5 мкм | ||||||||
| Базовый | Hет | Hет | 1.2 × 103 | 80 | 10 | 8 | 5 | 40 (10 мкм) |
| с РЗЭ | Eu2O3 | Hет | 1.3 × 102 | 200 | 100 | 60 | 30 | 200 |
| Gd2O3 | Hет | 1.6 × 102 | 80 | 100 | 50 | 40 | 200 | |
| EuF3 | Hет | 1.3 × 102 | 260 | 180 | 60 | 25 | 160 | |
| GdF3 | Hет | 1.4 × 102 | 180 | 100 | 100 | 30 | 150 | |
| Gd2S3 | Hет | 1.6 × 102 | 180 | 100 | 100 | 40 | 200 | |
| Eu2S3 | Hет | 1.4 × 102 | 200 | 140 | 100 | 25 | 160 | |
| с РЗЭ и Tc | Eu2O3 | 0.4 | 60 | 1 | 2 | Hет | 6 | 100 |
| 0.5 | 15 | Hет | Hет | Hет | Hет | 10 | ||
| 3.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.5 | 25 | 4 | Hет | 8 | 4 | 60 | ||
| Gd2O3 | 0.4 | 55 | 22 | 4 | 2 | 4 | 15 | |
| 0.5 | 20 | 4 | Hет | Hет | Hет | 5 | ||
| 3.0 | 18 | Hет | Hет | Hет | Hет | Hет | ||
| 6.0 | 15 | Hет | Hет | Hет | Hет | 5 | ||
| 6.5 | 62 | 31 | 4 | 6 | 2 | 5 | ||
| EuF3 | 0.4 | 80 | 25 | 60 | 2 | 4 | 60 | |
| 0.5 | 20 | 4 | 10 | Hет | Hет | 35 | ||
| 3.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.5 | 25 | 15 | 4 | 8 | 4 | Hет | ||
| Базовый | Hет | Hет | 1.2 × 103 | 80 | 10 | 8 | 5 | 40 (10 мкм) |
| с РЗЭ и Tc | GdF3 | 0.4 | 40 | 25 | 6 | 1 | 6 | 25 |
| 0.5 | 15 | 6 | Hет | Hет | Hет | 5 | ||
| 3.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.5 | 100 | 20 | 1 | 4 | 4 | Hет | ||
| Gd2S3 | 0.4 | 56 | 30 | 2 | 1 | 6 | 15 | |
| 0.5 | 21 | Hет | Hет | Hет | Hет | 15 | ||
| 3.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.5 | 112 | 16 | 2 | 2 | 4 | 40 | ||
| Eu2S3 | 0.4 | 90 | 55 | 2 | 6 | 9 | 40 | |
| 0.5 | 20 | 6 | Hет | Hет | Hет | 20 | ||
| 3.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.0 | 20 | Hет | Hет | Hет | Hет | Hет | ||
| 6.5 | 35 | 28 | 1 | 8 | 4 | 10 | ||
| Толщина эпитаксиальных пленок 3.0 мкм | ||||||||
| с РЗЭ и Tc | Eu2O3 | 0.4 | 60 | 10 | 2 | 1 | 6 | 95 |
| 0.5 | 15 | 2 | Hет | Hет | Hет | 30 | ||
| 3.0 | 15 | Hет | Hет | Hет. | Hет | Hет | ||
| 6.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.5 | 31 | 4 | Hет | 6 | 4 | 40 | ||
| Базовый | Hет | Hет | 1.2 × 103 | 80 | 10 | 8 | 5 | 40 (10 мкм) |
| с РЗЭ и Tc | Gd2O3 | 0.4 | 55 | 25 | 4 | 2 | 4 | 15 |
| 0.5 | 20 | 2 | Hет | Hет | Hет | 5 | ||
| 3.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.5 | 60 | 31 | Hет | 6 | 2 | 5 | ||
| Толщина пленок 6.0 мкм | ||||||||
| Eu2O3 | 0.4 | 100 | 16 | 3 | 4 | 6 | 60 | |
| 0.5 | 22 | 6 | 10 | Hет | Hет | 10 | ||
| 3.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.5 | 120 | 24 | 3 | 4 | 8 | 10 | ||
| GdF3 | 0.4 | 40 | 25 | 6 | 1 | 6 | 25 | |
| 0.5 | 15 | 6 | Hет | Hет | Hет | 5 | ||
| 3.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.5 | 100 | 20 | 1 | 4 | 4 | Hет | ||
| Gd2O3 | 0.4 | 60 | 25 | 4 | 4 | 6 | 25 | |
| 0.5 | 20 | Hет | Hет | Hет | Hет | 15 | ||
| 3.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.5 | 84 | 82 | 6 | 4 | 6 | 15 | ||
| Eu2S3 | 0.4 | 90 | 55 | 2 | 6 | 9 | 40 | |
| 0.5 | 20 | 6 | Hет | Hет | Hет | 20 | ||
| 3.0 | 15 | Hет | Hет | Hет | Hет | Hет | ||
| 6.0 | 20 | Hет | Hет | Hет | Hет | Hет | ||
| 6.5 | 35 | 28 | 1 | 8 | 4 | 10 | ||
Таким образом, как малое ≤0.5 вес. %, так и высокое ≥6 вес. % содержание технеция в составе катализаторов на основе соединений редкоземельных элементов способствуют усилению температурной зависимости скорости роста (Vp) эпитаксиальных слоев кремния.
Энергия активации процесса разложения SiH4 для условий наших экспериментов показала, что в отсутствии Tc в составе катализатора ее величина составляет 83.95–224 кДж/моль.
Положительный эффект действия катализатора с высокой активностью и селективностью на процесс разложения гидридов заключается в следующем: Радиус элементарной ячейки технеция равен 1.358 Å и близок к радиусу элементарной ячейки платиновых элементов (1.37–1.38 Å). Благодаря этому технеций в составе катализаторов на основе соединений РЗЭ позволяет обеспечить постоянство удельной каталитической активности и высокую селективность разложения моносилана и моногермана до ${\text{SiH}}_{2}^{{{\text{**}}}},$ ${\text{GeH}}_{2}^{{{\text{**}}}}.$
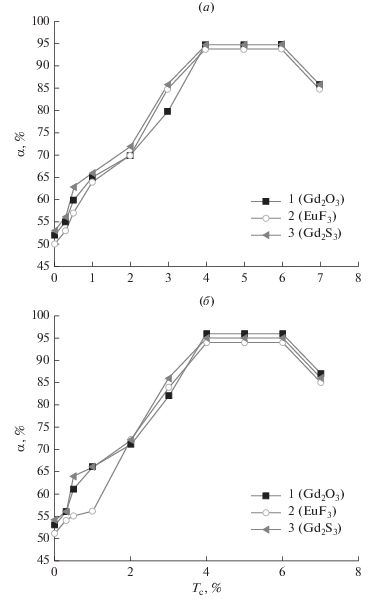
На рис. 3 приведены зависимости полноты разложения моносилана и моногермана от содержания технеция в соединениях РЗЭ, которые подтверждают эффективность влияния технеция на процесс разложения моногидридов.
Рис. 3.
Зависимость полноты разложения (α) моносилана (а) и моногермана (б) от количественного содержания (N) технеция в составе катализатора. Температура катализатора 473 К, подложки 993 K.
Взаимодействие моносилана и моногермана непосредственно с катализатором начинается на стадии адсорбции молекул гидридов на активном центре катализатора. Таким центром может быть оборванная химическая связь (валентность) на поверхности, как соединения, так и на поверхности зерна (кристаллита) металла (технеция). Таких оборванных валентностей может быть две и более на атом технеция. В связи с этим в процессе адсорбционного взаимодействия происходит отрыв не менее двух атомов водорода в молекуле гидрида с последующей перегруппировкой в адсорбционном слое и образованием связей водорода с катализатором и активных радикалов ${\text{SiH}}_{2}^{{{\text{**}}}},$ ${\text{GeH}}_{2}^{{{\text{**}}}}$ в газовой фазе. Последние десорбируют с поверхности катализатора и уносятся газовым потоком, в нашем случае аргоном, в зону подложек. Следовательно, на поверхности катализатора имеют место следующие стадии:
(2)
${\text{Si}}{{{\text{H}}}_{{4\left( {{\text{г а з }}} \right)}}},\,\,\,\,{\text{Ge}}{{{\text{H}}}_{{4\left( {{\text{г а з }}} \right)}}} \to {\text{Si}}{{{\text{H}}}_{{4\left( {{\text{а д с }}} \right)}}},\,\,\,\,{\text{Ge}}{{{\text{H}}}_{{4\left( {{\text{а д с }}} \right)}}};$(3)
${\text{Si}}{{{\text{H}}}_{{{\text{4}}\left( {{\text{а д с }}} \right)}}}{\text{,}}\,\,\,\,{\text{Ge}}{{{\text{H}}}_{{4\left( {{\text{а д с }}} \right)}}} \to {\text{SiH}}_{{2\left( {{\text{а д с }}} \right)}}^{{{\text{**}}}},\,\,\,\,{\text{GeH}}_{{2\left( {{\text{а д с }}} \right)}}^{{{\text{**}}}};$(4)
${\text{SiH}}_{{2\left( {{\text{а д с }}} \right)}}^{{{\text{**}}}},\,\,\,\,{\text{GeH}}_{{2\left( {{\text{а д с }}} \right)}}^{{{\text{**}}}} \to {\text{SiH}}_{{2\left( {{\text{г а з }}} \right)}}^{{{\text{**}}}},\,\,\,\,{\text{GeH}}_{{2\left( {{\text{г а з }}} \right)}}^{{{\text{**}}}}.$Такой механизм реакции в области катализатора позволяет полностью разделить этапы химического взаимодействия и образования пленки непосредственно на подложке, и как результат, создать бездефектные пленки кремния, германия и твердого раствора SiGe [8].
Исследуемые катализаторы, являясь инициаторами реакций разложения моногидридов кремния и германия, приводят к полному превращению их в целевые продукты – кремний, германий и водород при температуре подложки на 200–300° ниже, чем при традиционных технологических процессах.
В таблице приведены значения плотности дислокаций, дефектов упаковки, дефектов роста (плотности бугорков, шипов, трипирамид и следов кристаллитов в эпитаксиальных пленках кремния толщиной 1.5, 3.0, 6.0 мкм, полученных c использованием катализаторов без технеция и с технецием. Формировались сплошные эпитаксиальные слои n-типа проводимости. Всего была запущена 171 партия, по три партии на каждый процесс, по пять пластин в партии. В таблице приведены усредненные результаты по трем партиям каждого из 57 процессов.
Как следует из анализа результатов таблицы, получение тонких пленок кремния из газовой фазы при разложении гидридов кремния, включающий химическую активацию реакционной смеси с помощью твердотельных катализаторов на основе соединений редкоземельных элементов, содержащих от 0.5 до 6.0 вес. % элемента, радиус элементарной ячейки которого близок к радиусу элементарной ячейки платиновых элементов, т.е. технеция, позволяет получить практически бездефектные слои кремния в результате полного разделения этапа химического взаимодействия и этапа образования пленки по сравнению с базовым процессом и процессом с использованием катализатора без технеция. Такие слои кремния представляют несомненный интерес при создании эпитаксиальных структур со “скрытыми” слоями, так как позволяют повысить процент выхода эпитаксиальных структур непосредственно на операции эпитаксии с 86 до 96–98%, тем самым уменьшив коэффициент запуска на эпитаксии с 1.16 до 1.04. При программе 1.0 млн пластин это позволяет сократить запуск на 120 тысяч пластин. Из табл. 1 также очевидна правильность выбора нижнего и верхнего пределов количественного содержания элемента, радиус элементарной ячейки которого близок к радиусу элементарной ячейки платиновых элементов. Исследуемый процесс получения тонких эпитаксиальных пленок кремния может быть широко использован для создания кремниевых эпитаксиальных структур со “скрытыми” диффузионными слоями и ионно-имплантированными слоями, применяемых при производстве биполярных микросхем с инжекционным питанием, а также различного рода БИС и СИС. Благодаря практически полному отсутствию дефектов слоев создается реальная возможность получения элементов ИМС субмикронных размеров. Данный технологический процесс может также быть широко использован для создания тонких поликристаллических пленок кремния, используемых в качестве затвора К/МДП – микросхем, в качестве плавких перемычек в электрически программируемых постоянных ЗУ, а также при создании аморфных пленок кремния, используемых в качестве межкомпонентной изоляции. Преимущества исследованного технологического процесса перед известными базовыми процессами очевидны и бесспорны как в техническом, так и в экономическом аспектах.
ЗАКЛЮЧЕНИЕ
Катализаторы из соединений РЗЭ с технецием, позволяют обеспечить постоянство удельной каталитической активности и высокую селективность разложения моносилана и моногермана до ${\text{SiH}}_{2}^{{{\text{**}}}},$ ${\text{GeH}}_{2}^{{{\text{**}}}},$ они являясь инициаторами реакций разложения моногидридов кремния и германия, приводят к полному превращению их в целевые продукты – кремний, германий и водород при температуре подложки на 200–300° ниже, чем при традиционных технологических процессах.
Получение тонких пленок кремния из газовой фазы при разложении гидридов кремния, включающий химическую активацию реакционной смеси с помощью твердотельных катализаторов на основе соединений редкоземельных элементов, содержащих от 0.5 до 6.0 вес. % элемента, радиус элементарной ячейки которого близок к радиусу элементарной ячейки платиновых элементов, т.е. технеция, позволяет получить практически бездефектные слои кремния в результате полного разделения этапа химического взаимодействия и этапа образования пленки по сравнению с базовым процессом и процессом с использованием катализатора без технеция.
Исследуемый процесс получения тонких эпитаксиальных пленок кремния может быть широко использован для создания кремниевых эпитаксиальных структур со “скрытыми” диффузионными слоями и ионно-имплантированными слоями, применяемых при производстве биполярных микросхем с инжекционным питанием, а также различного рода БИС и СИС.
Список литературы
Боресков Г.К. Катализ. Новосибирск: Наука, 1971. 267 с.
Ганкин В.Ю., Ганкин Ю.В. Новая общая теория катализа. Л.: 1991.
Гейтс Б.К., Кетцир Дж., Шуйт Г. Химия каталитических процессов. М.: Мир, 1981.
Мелихов И.В. Тенденции развития нанохимии // Российский химический журн. (Журнал Российского химического общества им. Д.И. Менделеева). 2002. Т. XLVI. № 5. С. 7–14.
Швец В.Ф. Введение в химию каталитических реакций // Соросовский образовательный журнал. 1996.
Ковалевский А.А. Способ получения тонких пленок из газовой фазы. А. с. СССР № 782604, М. Кл2 H01L 21/20. 1980.
Ковалевский А.А. Способ получения тонких пленок из газовой фазы. А. с. СССР. № 1102413. М. Кл4H01L 21/20. 1984.
Комар О.М., Ковалевский А.А., Строгова А.С. Кремний германиевые наноструктурированные пленки и нанокластеры. LAPLAMBERT Academic Publishing. 2016. 345 с.
Комар О.М., Ковалевский А.А., Строгова А.С. Полупроводниковый дисилицид титана. LAPLAMBERT Academic Publishing. 2016. 225 с.
Колешко В.М., Ковалевский А.А. Поликристаллические пленки полупроводников в микроэлектронике. Мн. Наука и техника, 1978. 345 с.
Дополнительные материалы отсутствуют.
Инструменты
Микроэлектроника