

Журнал высшей нервной деятельности им. И.П. Павлова, 2020, T. 70, № 4, стр. 488-499
β-АМИЛОИД И ЛИТИЙ ВЛИЯЮТ НА ВЕЛИЧИНУ ФАЗИЧЕСКИХ ВЫБРОСОВ ДОФАМИНА В ОБОЛОЧКЕ ПРИЛЕЖАЩЕГО ЯДРА
В. Н. Мухин 1, *, И. Р. Боровец 1, В. В. Сизов 1, К. И. Павлов 1, В. М. Клименко 1
1 Федеральное государственное бюджетное научное учреждение “Институт экспериментальной медицины”
Санкт-Петербург, Россия
* E-mail: Valery.Mukhin@gmail.com
Поступила в редакцию 13.06.2019
После доработки 21.11.2019
Принята к публикации 16.12.2019
Аннотация
По данным литературы, в патогенезе болезни Альцгеймера имеет значение нарушение дофаминергической секреции в оболочке прилежащего ядра. Вероятно, его причина – повышение уровня токсических форм β-амилоида в мозге. Как и другие патогенетические проявления БА, данное нарушение может быть опосредовано повышением активности киназы гликоген-синтазы 3 (GSK3). Цель исследования – изучить влияние β-амилоида на высвобождение дофамина в оболочке прилежащего ядра. Изменение уровня дофамина в оболочке прилежащего ядра in vivo регистрировали у 38 наркотизированных крыс-самцов линии Вистар методом быстро-сканирующей циклической вольтамперометрии до экспериментального воздействия и в течение 1.5 ч после него. Способность дофаминергических нейронов секретировать дофамин оценивали по амплитуде увеличения его уровня в ответ на электрическую стимуляцию вентральной области покрышки (VTA). Для моделирования нейротоксических процессов, характерных для БА, в желудочковую систему мозга крыс вводили раствор β-амилоида (фрагмент 25–35). Роль GSK3 в реализации эффектов β-амилоида оценивали путем блокирования активности этого фермента с помощью ингибитора – лития. Раствор хлорида лития в “терапевтической” дозе 10.4 мг/кг вводили внутрибрюшинно сразу после инъекции β-амилоида. Увеличение стимулированных выбросов дофамина свидетельствовало о том, что в течение полутора часов после введения как β-амилоида, так и лития способность нейронов вентральной области покрышки секретировать дофамин в оболочку прилежащего ядра повышена. Более того, сравнение с контрольной группой показало, что данные вещества нивелируют ослабление секреторной функции, вызванное условиями эксперимента. Литий, введенный одномоментно с β-амилоидом, ослабляет его действие, что свидетельствует об участии GSK3 в этом процессе.
Болезнь Альцгеймера (БА) – хроническое нейродегенеративное заболевание, проявляющееся нарушением памяти, мышления, пространственной ориентации, речи и расстройствами эмоциональной сферы. Исследования последних десятилетий выявили основные компоненты патогенеза БА: дисфункцию базальной холинергической системы, нарушение синаптической пластичности и уменьшение численности нейронов в различных отделах мозга. В патогенезе БА может иметь значение дисфункция мезолимбического нервного пути, секретирующего дофамин в различные структуры мозга, включая оболочку прилежащего ядра [D’Amelio et al., 2018; Šimić et al., 2017].
Нарушение дофаминергической передачи в прилежащем ядре при БА подтверждается клиническими наблюдениями, исследованиями post mortem и экспериментами на животных.
Согласно клиническим наблюдениям, дофаминергические лекарственные средства, предназначенные в первую очередь для лечения болезни Паркинсона, дают положительный эффект и при БА (леводопа, ротиготин, селегилин) [Koch et al., 2014; Martorana et al., 2009; Martorana et al., 2013; Miklya, 2016]. Кроме того, в вентральном стриатуме больных понижена плотность D2-подобных дофаминовых рецепторов [Joyce et al., 1998].
Исследования post mortem показали низкую плотность D2-подобных дофаминовых рецепторов в прилежащем ядре, коррелирующую со снижением плотности M-холинорецепторов в этой структуре [Rinne et al., 1986], а также низкое содержание дофаминового транспортера [Murray et al., 1995].
При моделировании БА на животных в прилежащем ядре выявлено снижение базального уровня дофамина и амплитуды его повышений [Nobili et al., 2017; Preda et al., 2007]. Снижение выявляется на раннем этапе действия β-амилоида и поэтому, вероятнее всего, является следствием функциональных изменений, а не утраты нейронов в вентральной области покрышки [Krashia et al., 2019]. Положительный эффект галантамина (холинергического лекарственного средства, применяющегося при БА) опосредован активацией мезолимбического пути [Wang et al., 2006]. При инкубации органотипических срезов мозга крыс с β-амилоидом 1-42 снижается численность дофаминергических нейронов вентрального отдела среднего мозга [Hochstrasser et al., 2013].
Различают два типа изменений уровня дофамина в мозге: тонические (долговременные изменения продолжительностью до нескольких десятков минут) и фазические (быстрые колебания продолжительностью до нескольких секунд). В норме фазические колебания (увеличения или уменьшения) уровня дофамина в оболочке прилежащего ядра инициируют закрепление или утрату модели поведения (т.е. научение или забывание), а также ее мобилизацию в соответствующих условиях [Козунова и др., 2018; Майоров, 2018; Doll, Daw, 2016; Keiflin et al., 2019]. Теоретически изменения характеристик фазических колебаний могут приводить к ослаблению памяти – ведущему симптому БА, что делает их изучение весьма актуальным. Однако исследований в этой области практически нет. Лишь в одной работе на трансгенной модели БА показано, что амплитуда фазических повышений уровня дофамина в оболочке прилежащего ядра понижена [Nobili et al., 2017]. Но эта работа не может полноценно характеризовать функцию мезолимбического пути, так как выполнена ex vivo на срезах мозга крыс.
С проблемой наличия и модуса нарушений фазических колебаний уровня дофамина в оболочке прилежащего ядра тесно связан вопрос о том, каков механизм их возникновения. Данные литературы позволяют предполагать, что их непосредственной причиной является повышенный уровень в мозге токсических форм β-амилоида – особого пептида, секретируемого в мозге нейронами [Wei et al., 2010], астроцитами [Busciglio et al., 1993]. В настоящее время установлено, что этот фактор является центральным звеном патогенеза БА на клеточно-молекулярном уровне [Мухин, 2013; Мухин, Клименко, 2014; Karran, De Strooper, 2016]. О его влиянии на дофаминергическую передачу можно предположить на основании того, что в стриатуме больных выявлено избыточное накопление Aβ [Duyckaerts et al., 2009]. Кроме того, введение Aβ в желудочки мозга крыс уменьшает амплитуду тонических изменений секреторной активности дофаминергических путей [Itoh et al., 1996; Preda et al., 2007].
Цель исследования – изучить влияние β-амилоида в токсичной форме на величину фазических повышений уровня внеклеточного дофамина в оболочке прилежащего ядра.
Среди молекулярно-клеточных нарушений, возникающих вследствие избыточного накопления Aβ, – повышение активности фермента киназы гликоген-синтазы 3 (GSK3). Предполагают существование порочного круга молекулярных сигналов в нейронах. Избыток β-амилоида повышает активность GSK3, что в свою очередь приводит к увеличению продукции нейротоксичных форм Aβ и к последующим нарушениям структуры и функции мозга [Maqbool et al., 2016]. GSK3 – перспективная молекулярная мишень терапии болезни Альцгеймера [Llorens-Marítin et al., 2014]. Участвует ли GSK3 в механизме дофаминергических нарушений в оболочке прилежащего ядра при болезни Альцгеймера, неизвестно. Изучить роль GSK3 как посредника при воздействии Aβ на секреторную активность дофаминергических нейронов в оболочке прилежащего ядра – дополнительная задача исследования.
МЕТОДИКА
Эксперимент проведен на 38 крысах-самцах линии Вистар средней массой 364 г (SD = 63 г) в соответствии с этическими принципами работы с лабораторными животными, изложенными в Директиве Европейского парламента и Совета Европейского Союза 2010/63/EC от 22 сентября 2010 г., одобренной комиссией по биоэтике ФГБНУ ИЭМ.
Крыс наркотизировали уретаном, 1.5 г которого разводили в 1 мл изотонического раствора хлорида натрия и вводили внутрибрюшинно в дозе 1.5 г/кг.
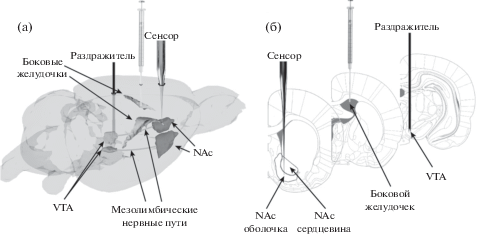
Секреторную активность дофаминергических нейронов оценивали по изменению уровня дофамина в межклеточном пространстве оболочки прилежащего ядра in vivo методом быстро-сканирующей циклической вольтамперометрии [Мухин и др., 2017]. Преимущество данного метода – высокая степень временного и пространственного разрешения [González-Mora et al., 2017]. Временное разрешение определяется частотой сканирования – 10 раз в секунду, пространственное разрешение определяется размерами сенсора – углеволокно длиной 100 мкм и толщиной 7 мкм. Сенсорный электрод вводили в левое полушарие мозга стереотаксически по координатам AP = 1.3 мм; L = 1.6–1.7 мм (от брегмы); H = 7.6 мм (от поверхности мозга) (рис. 1). Референтный электрод – хлорированную серебряную проволоку – располагали в контралатеральном полушарии.
Рис. 1.
Схема эксперимента – реконструкция расположения электродов в мозге крысы. Раздражитель (стимулирующий электрод) находится в вентральной области покрышки, сенсорный электрод – в прилежащем ядре, игла шприца – в левом боковом желудочке; VTA – вентральная область покрышки, NAc –прилежащее ядро. (а) – Реконструкция на объемной модели мозга крысы выполнена на основе трехмерного электронного атласа мозга крысы “3d Brain Atlas Reconstructor”, доступного в интернете по адресу http://www.3dbar.org [Majka et al., 2012]. (б) – Реконструкция на срезах мозга крысы выполнена на основе стереотаксического атласа [Paxinos, Watson, 2005].
Fig. 1. Schematic diagram of the experimental setup. Reconstruction of the electrodes locations in the rat brain. Stimulating electrode is located in the ventral tegmental area (VTA). Working electrode is in the n. accumbens (NAc). (a) – Reconstruction made using the 3D electronic atlas of the rat brain available online at http://www.3dbar.org [Majka et al., 2012]. (б) – Reconstruction made using the stereotaxic atlas [Paxinos, Watson, 2005].
Способность дофаминергических нейронов секретировать дофамин в оболочку прилежащего ядра оценивали по амплитуде увеличения его уровня в ответ на стимуляцию вентральной области покрышки (VTA). Стимуляция производилась электрическими импульсами, подаваемыми в VTA через биполярный металлический электрод, введенный в мозг с помощью стереотаксического аппарата по координатам от брегмы AP = –4.9 мм; L = 1.0 мм; H = 8.3 мм (рис. 1). На электрод подавалась пачка из 60 прямоугольных импульсов тока силой 200–300 мкА и продолжительностью 2 мс, следующих с частотой 60 Гц [Jones et al., 2006, с модификациями].
Перед началом эксперимента производили пробные электростимуляции, добиваясь путем коррекции положения сенсорного и стимулирующего электродов выраженных и стабильных вольтамперометрических признаков выброса дофамина (рис. 2). Наличие таких признаков являлось функциональным подтверждением правильности расположения электродов.
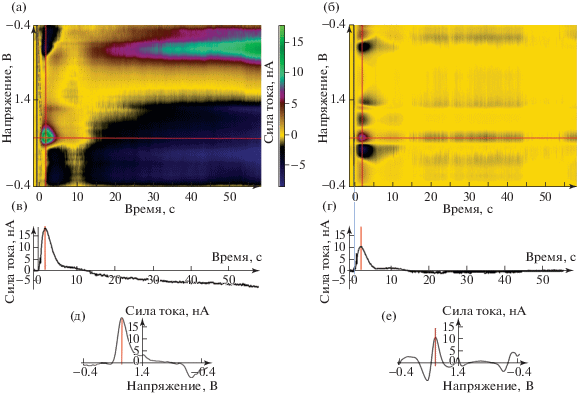
Рис. 2.
Фрагмент вольтамперометрической записи (60 с) (крыса 37). (а), (в), (д) – исходная запись; (б), (г), (е) – тот же фрагмент, восстановленный на основе фактора дофамина, выделенного методом главных компонент. (а) и (б) – динамика разностных вольтамперограмм во времени; вычитаемая вольтамперограмма (обозначена вертикальной черной линией) и начало координат по оси абсцисс расположены непосредственно перед началом стимулирующих импульсов; в течение первой секунды записи видны артефакты импульсов электрического раздражения; видна волна прибавки тока, обусловленная окислением дофамина на поверхности углеволокна; пик волны окисления дофамина обозначен перекрестьем горизонтальной и вертикальной красных линий. (в) и (г) – динамика тока при напряжении сканирующего импульса, соответствующем окислению дофамина; после анализа главных компонент динамика тока (на диаграмме (г)) приблизительно соответствует динамике дофамина около конца сенсорного электрода (диаграммы (в) и (г) соответствуют горизонтальной линии на вольтамперограммах (а) и (б)). (д) и (е) – вольтамперограммы, зарегистрированные в момент максимума волны прибавки тока на напряжении дофамина после электрического раздражения (соответствуют вертикальным красным линиям на диаграммах (а) и (б)).
Fig. 2. Fragment of a voltammetric recording (60 s). (а), (в), (д) – original data; (б), (г), (е) – the same data reconstructed from the principal component that relates to dopamine. (а) and (б) – dynamics in time of the difference voltammetric recordings; subtrahend voltammogram (indicated by a vertical black line) and the origin along the abscissa are located immediately before the beginning of the stimulating pulses; during the initial second, there are artifacts due to the pulses of electrical stimulation; a wave of current increase due to the oxidation of dopamine on the surface of the carbon fiber is showed; the peak of the dopamine oxidation wave is indicated by the crosshair of the horizontal and vertical red lines. (в) and (г) – dynamics of current at the voltage of dopamine oxidation; dynamics of the current after the principal component analysis (diagram (г)) approximately corresponds to the dynamics of dopamine near the end of the working electrode (diagrams (в) and (г) which correspond to the horizontal lines on the voltammetric recording diagrams (а) and (б)). (д) and (е) – voltammograms recorded at maximal increase in current at the dopamine voltage (correspond to the vertical red lines in the voltammetric recording diagrams (а) and (б)).
Морфологический контроль расположения электродов не проводился, так как при калибровке электрода после эксперимента (что дает более точные результаты) его выполнить технически невозможно. Трек от конца сенсорного электрода, по данным литературы, при морфологическом исследовании не виден, так как электрод слишком тонкий. Создание электрической метки через сенсорный электрод могло привести к его разрушению, что сделало бы невозможным калибровку электрода после измерений. Введение металлического электрода вместо сенсорного после измерений для создания электрической метки не дало бы в наших условиях гарантии попадания в то же место, где был сенсорный электрод. Кроме того, стала бы невозможной очень желательная калибровка сенсорного электрода сразу после измерения, так как она в пространственном плане проводилась в той же небольшой экранированной камере, где и стереотаксический аппарат с крысой. Оболочка прилежащего ядра является достаточно крупной структурой мозга (миллиметры), поэтому вероятность неправильного введения электрода очень мала. В то же время мы не ставили перед собой задачу измерения в какой-либо конкретной малой точке данных структур. Случайные небольшие отклонения электродов неизбежно дали случайное изменения величины уровня дофамина, что “учтено” при статистическом анализе. Морфологический контроль в подобного рода исследованиях не всегда делают и другие авторы [Takmakov et al., 2010; Phillips et al., 2003; Robinson et al., 2001; Walker et al., 1999].
Электростимуляция производилась каждые три минуты. У каждой крысы предварительно регистрировали фоновый уровень вызванных выбросов дофамина – три стимула. Затем вводили вещества и продолжали регистрацию в течение 90 мин, произведя за это время 30 стимулов.
Для моделирования патологических процессов, характерных для БА, в желудочковую систему мозга крыс вводили раствор β-амилоида (фрагмент 25–35). Выбор этого фрагмента обусловлен его значительной ролью в патогенезе заболевания [Kaminsky et al., 2010; Kubo et al., 2003; Xu et al., 2015], а его водные растворы имеют нейротоксические свойства, аналогичные целой молекуле [Hensley et al., 1994; Millucci et al., 2010]. Способность Aβ проникать в структуры мозга сразу после интрацеребровентрикулярного введения и оставаться там в течение нескольких часов показана на примере фрагмента 1–40 и 1–42 [Preda et al., 2007].
β-амилоид (фрагмент 25–35, производитель Sigma) растворяли в воде для инъекций (1 мг/мл) и выдерживали в термостате 4 сут при температуре 37°C для агрегации (в таком состоянии он проявляет большую нейротоксичность) [Delobette et al., 1997; Pike et al., 1993]. Аликвоты раствора хранили при температуре –20°C. 10 мкл размороженного раствора вводили в левый боковой желудочек мозга с помощью микрошприца по координатам от брегмы AP = –0.8 мм, L = 1.5 мм; H = 3.8 мм (рис. 1). Сразу после введения проводилась регистрация изменений секреторной активности мезолимбической системы, так как на более поздних стадиях возможно возникновение структурных изменений, препятствующих изолированной оценке функциональных нарушений [Krashia et al., 2019].
Роль GSK3 в реализации эффектов Aβ оценивали путем блокирования активности этого фермента с помощью ингибитора – лития. Литий не является селективным ингибитором GSK3. Однако показано, что эффекты лития, наблюдаемые in vivo на человеке и млекопитающих, обусловлены его влиянием на GSK3 [Freland, Beaulieu, 2012; O’Brien, Klein, 2009]. По данным литературы, дозы лития в экспериментах на животных делятся на 2 группы: малые и большие. Большие дозы (больше 100 мг/кг хлорида лития) токсичны и используются в качестве аверсивного стимула при выработке условной рефлекторной аверзии. Малые дозы соответствуют используемым в терапии и составляют от 3 до 60 мг/кг хлорида лития. Литий в малых дозах имеет терапевтический потенциал при болезни Альцгеймера [Hampel et al., 2019]. 0.15 M раствор хлорида лития в “терапевтической” дозе 1.63 мл/кг (10.4 мг/кг) вводили внутрибрюшинно сразу после инъекции β-амилоида. В качестве контроля вместо хлорида лития вводили изотонический раствор хлорида натрия.
С учетом наличия или отсутствия экспериментальных факторов (Aβ и литий) сформированы четыре группы крыс (полный факторный эксперимент). Группа 1 (n = 10) – введение только Aβ (вместо лития вводился физиологический раствор), группа 2 (n = 9) – введение только лития (вместо Aβ вводился физиологический раствор), группа 3 (n = 9) –введение Aβ и лития, группа 4 (n = 10) – введение физиологического раствора вместо Aβ и вместо лития.
Известно, что во время вольтамперометрического сканирования, помимо дофамина, на силу тока влияют и другие факторы, такие как изменение pH и импеданса регистрирующей системы. В связи с этим прямая оценка изменений уровня дофамина на участках записи продолжительностью больше нескольких секунд невозможна. Для этого необходим хемометрический анализ записи, позволяющий выделить влияние дофамина из общей композиции влияний. Такой анализ был осуществлен методом главных компонент на основе разложения по собственным значениям. Анализ проведен с помощью разработанного авторами приложения Voltammograph. Из всех компонент выбраны только те, которые связаны с влиянием дофамина. Критерий связи – наличие изменений тока после стимула на тех же участках вольтамперограммы, что и на исходной записи. На основе выбранных компонент выполнено восстановление матрицы вольтамперограммы в исходной размерности (амперы) (рис. 2). Такая методика, пусть и с оговорками, касающимися точности измерений, все же позволяет использовать содержащуюся в вольтамперометрических записях информацию об изменении базального уровня дофамина [DeWaele et al., 2017].
Восстановленные матрицы отражали только влияние дофамина (лишены влияния прочих электрохимических факторов). На их основе определяли динамику уровня дофамина во времени по изменению силы тока при напряжении, соответствующем пику окисления дофамина на исходной записи. Значения силы тока были переведены в значения концентрации дофамина на основе калибровочных записей, выполненных in vitro после каждого эксперимента на растворах дофамина (Sigma) концентрацией 0.5, 1 и 2 мкМ.
Для оценки динамики базального уровня дофамина из всей записи выбраны 33 значения его концентрации непосредственно перед каждой стимуляцией VTA (т.е. с шагом 3 мин). Для оценки величины стимулированного выброса дофамина определен его прирост в момент максимума волны увеличения после каждой из 33 стимуляций VTA.
Таким образом, для статистической обработки получены две матрицы данных: матрица значений базального уровня дофамина и матрица амплитуд стимулированных выбросов. В матрицах представлены данные по каждой крысе и каждому из 33 моментов времени.
Для оценки влияния факторов эксперимента проводился анализ на основе смешанных линейных моделей. Этот вид анализа более чем дисперсионный анализ на основе общих линейных моделей подходит для данных об изменении во времени уровня дофамина, поскольку они, являясь по своей природе лонгитудинальными, сильно зависят не только от факторов эксперимента, но и от объекта наблюдения (крысы) [Четвериков, 2015; Garcia, Marder, 2017]. Анализ выполнен в компьютерной среде R с использованием пакетов программ lme4, lmerTest и car [Bates et al., 2015; Kuznetsova et al., 2017; R Core Team, 2019]. В качестве зависимой переменной использованы значения уровня дофамина в каждый из 33 моментов времени. Помимо фиксированных факторов (Aβ и литий), в модели включен случайный фактор объекта наблюдения (крысы) как имеющий случайную константу.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЙ
Электростимуляция вызывала кратковременные усиления тока, свидетельствующие о том, что во внеклеточной среде оболочки прилежащего ядра происходят фазические повышения уровня дофамина (рис. 2).
У крыс контрольной группы (группа 4) амплитуда вызванных повышений дофамина постепенно уменьшается от стимула к стимулу и к концу эксперимента составляет в среднем треть от первоначальной (для фактора времени F(1, 32) = 4.83; p < 2 × 10–16). Этот эффект обусловлен, по-видимому, влиянием факторов эксперимента: наркоз, введение вещества в мозг, повторяющиеся электростимуляции. Достоверным он становится, начиная с 9–15-й минуты (рис. 3, 4). Соответствующих данному эффекту изменений базального уровня дофамина не наблюдалось.
Рис. 3.
Выброс дофамина в оболочке прилежащего ядра в зависимости от периода времени после введения β-амилоида и лития по группам крыс. Данные представлены как оценки, полученные на основе смешанной линейной модели ± стандартные ошибки. Статистическая значимость контрастов для контрольной группы (группа 4) указана по сравнению с исходным уровнем, для группы 1 (Aβ) и группы 2 (литий) – по сравнению с контрольной группой, для группы 3 (оба фактора вместе) – по сравнению с уровнем групп 1 и 2. Обозначения: + – p ≤ 0.05, * – p ≤ 0.01.
Fig. 3. Release of dopamine in the n. accumbens shell depending on the period of time after administration of amyloid β and lithium in the rat groups. Data is expressed as the estimates ± standard errors obtained using mixed linear model analysis. Statistical significance of contrasts for the control group (group 4) is determined in comparison with the baseline; for the group 1(Aβ) and the group 2 (lithium) is determined in comparison with the control group; for the group 3 (both factors together) – in comparison with the group 1 and the group 2. + denote p ≤ 0.05, * denote p ≤ 0.01.
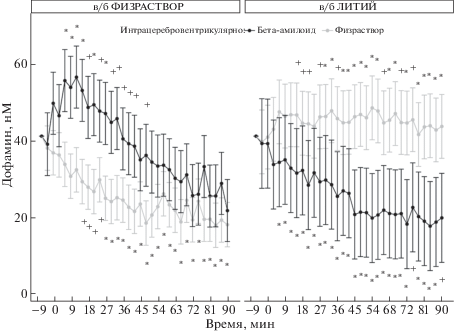
Рис. 4.
Изменение волны выброса дофамина в ответ на экспериментальные воздействия у крыс разных групп (примеры вольтамперометрических записей, анализ главных компонент). Примечания. На каждой диаграмме представлены две кривые: до воздействия и через 12 мин после него (момент наибольших отличий от контрольной группы). Начало координат по оси абсцисс расположено непосредственно перед началом стимулирующих импульсов. В течение первой секунды записи видны артефакты импульсов электрического раздражения.
Fig. 4. Change in the dopamine release wave in response to the experimental impacts in the rats of different groups (examples of voltammetric recordings, principal component analysis). Two curves are presented in each figure: before exposure and 12 minutes after that (the moment of the largest difference from the control group). The origin along the abscissa are located immediately before the beginning of the stimulating pulses. There are artifacts of pulses of electrical stimulation during the initial second.
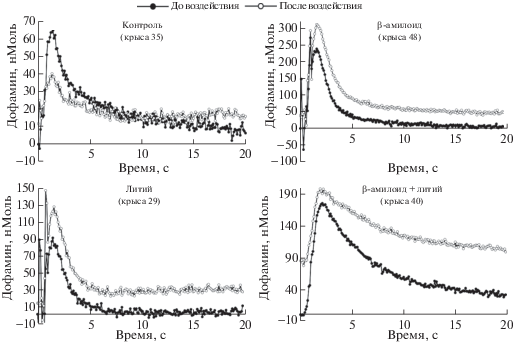
Введение крысам раздельно Aβ или лития (группы 1 и 2) увеличивало амплитуду вызванных повышений дофамина по сравнению с исходным уровнем и контрольной группой на 20 нМ (рис. 3, 4). Сила влияния зависела от времени, прошедшего после введения (взаимодействие между тремя факторами статистически значимо, (F(1, 32) = 2.28; p = 2 × 10–13)). Анализ контрастов позволил определить, что влияние Aβ и лития статистически достоверно с 6-й минуты после введения каждого из веществ. Максимум эффекта Aβ и лития наступает с 12-й минуты после введения. Влияние Aβ заметно до 48-й минуты, а лития – до конца наблюдения (90 мин) (p ≤ 0.05).
С 6-й по 90-ю минуту (весь период действия веществ) наблюдается взаимодействие между Aβ и литием (p ≤ 0.05), выражающееся в резком снижении стимулированного выброса дофамина до уровня контрольной группы при действии обоих веществ вместе (группа 3) (рис. 3, 4).
Влияние Aβ и лития на базальный уровень дофамина в оболочке прилежащего ядра выявить не удалось. По-видимому, влияние Aβ на величину стимулированного выброса дофамина не связано с изменением его базального уровня, а наблюдающиеся изменения базального уровня случайны.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Исследование показало, что в течение полутора часов после введения Aβ способность нейронов вентральной области покрышки секретировать дофамин в оболочку прилежащего ядра повышена по сравнению с контрольной группой, а с 12-й по 50-ю минуту – и по сравнению с исходным уровнем.
Тот факт, что в отличие от нашего предыдущего исследования [Мухин и др., 2017], в этой работе удалось выявить влияние Aβ, обусловлен рядом методических усовершенствований. В частности, измерения проводились в одной и той же точке мозга, без изменения позиции электрода, что исключало возможность погрешности при его возврате на исходную позицию; измерения проводились чаще (каждые 3 мин, не через 15, 30 и 60 мин после введения Aβ); применен метод статистического анализа (смешанные линейные модели), исключающий влияние на данные эффективности электростимуляции, которая значительно различалась у разных крыс.
По данным литературы, Aβ не увеличивает, а уменьшает величину вызванных выбросов дофамина в оболочке прилежащего ядра. Расхождение результатов объясняется, по-видимому, различиями в условиях экспериментов. Подавление выявлено не при остром, как в нашем исследовании, а при хроническом воздействии Aβ (модель на трансгенных мышах Tg2576) и не в условиях in vivo, как в настоящем исследовании, а на срезах мозга [Nobili et al., 2017]. Острое воздействие Aβ (фрагмент 1-40), введенного в желудочковую систему мозга крыс, тоже уменьшает выбросы дофамина в оболочку прилежащего ядра [Preda et al., 2007]. Но в этой работе идет речь не о фазических, а о тонических, долговременных изменениях уровня дофамина.
Увеличение стимулированных выбросов дофамина, вызванное острым воздействием Aβ, не сопровождалось изменением базального уровня. Хроническое воздействие избытка β-амилоида приводит, по данным литературы, к иному результату. В упоминавшемся уже исследовании показано, что у мышей Tg2576 в оболочке прилежащего ядра снижен базальный уровень дофамина на срезах мозга и in vivo [Nobili et al., 2017].
Увеличение способности к высвобождению дофамина развивается в течение первых десятков минут после введения Aβ. В работах других авторов тоже показана возможность быстрого развития различных изменений под влиянием Aβ. Так, в течение первого часа после интрацеребровентрикулярного введения Aβ 25–35 у животных развивается нарушение долговременной потенциации в поле CA1 гиппокампа [Freir et al., 2001; Gengler et al., 2007; Uzakov et al., 2015]. На культуре гиппокампальных нейронов крысы Aβ 25–35 в течение первого часа после введения повышает вероятность возникновения возбуждающих постсинаптических потенциалов и потенциалов действия [Brorson et al., 1995].
Быстро развивающиеся эффекты связывают с тем, что в течение первого часа воздействия Aβ в нервных клетках увеличивается уровень кальция. Усиление кальциевого тока, входящего в нейроны коры мозга крысы, опосредовано инсулиновой сигнальной цепью с участием молекул PI3K и p85 [Wang et al., 2016]. В гиппокампе крыс этот процесс связан с NMDA-рецепторами [Brorson et al., 1995] и кальциевыми каналами L-типа [Rovira et al., 2002]. Усиление кальциевого тока может быть обусловлено и тем, что молекулы Aβ 25–35 взаимодействуют с липидами клеточных мембран, образуя каналы, которые могут быть проницаемы для ионов кальция [Kandel et al., 2019]. Данные механизмы могут лежать в основе эффекта Aβ, обнаруженного в нашем исследовании.
Aβ нивелирует ослабление секреторной функции, вызванное условиями эксперимента, что может быть проявлением его нейропротективного действия. Нейропротективное действие Aβ, наряду с его нейротоксическим влиянием, широко обсуждается в литературе [например, Brothers et al., 2018; Giuffrida et al., 2009; Parihar, Brewer, 2010].
Литий, введенный совместно с Aβ, подавляет его действие. Несмотря на то что литий действует на активность многих ферментов, считается, что влияние лития на поведение животных опосредовано в первую очередь GSK3 [Freland, Beaulieu, 2012; O’Brien, Klein, 2009]. Этот результат соответствует данным литературы о том, что действие Aβ на уровне клетки опосредовано киназой гликоген-синтазы 3 (GSK3) [Maqbool et al., 2016].
Литий в “терапевтической” дозе увеличивает вызванные фазические выбросы дофамина в оболочке прилежащего ядра. В литературе аналогичных данных нет. Однако известно, что литий в токсических и “терапевтических” дозах влияет на величину фазических выбросов и базальный уровень дофамина в других структурах мозга. Так, в сердцевине прилежащего ядра острое воздействие токсических доз лития уменьшает величину фазических выбросов дофамина [Fortin et al., 2016] или не влияет на нее, а уменьшает хроническое воздействие [Can et al., 2016].
Влияние лития на базальный уровень дофамина не выявлено. Этот результат соответствует данным литературы: острое воздействии лития (дозы 0.5–2 meq/L жидкости для диализа) не влияет на базальный уровень дофамина в прилежащем ядре и префронтальной коре в течение нескольких часов после начала введения [Baptista et al., 1993].
ЗАКЛЮЧЕНИЕ
Через несколько минут после повышения уровня β-амилоида в мозге крыс увеличиваются вызванные фазические выбросы дофамина в оболочке прилежащего ядра. Вероятно, клеточно-молекулярный механизм включает в себя фермент киназу гликоген-синтазы 3. Результаты исследования могут способствовать более глубокому пониманию патогенеза болезни Альцгеймера и разработке способов ее лечения и профилактики.
Список литературы
Козунова Г.Л., Воронин Н.А., Венедиктов В.В., Строганова Т.А. Обучение с подкреплением: роль непосредственной обратной связи и внутренней модели ситуации. Журнал высшей нервной деятельности им. И.П. Павлова. 2018. 68 (5): 602–613.
Майоров В.И. Функции дофамина в инструментальном условном рефлексе. Журнал высшей нервной деятельности им. И.П. Павлова. 2018. 68 (4): 404–414.
Мухин В.Н., Сизов В.В., Павлов К.И., Клименко В.М. β-амилоид 25-35 подавляет секреторную активность дофаминергических систем мозга крыс. Российский физиологический журнал им. И.М. Сеченова. 2017. 103 (12): 1350–1360.
Мухин В.Н. Патогенетические механизмы дисфункции базальной холинергической системы при болезни Альцгеймера. Российский физиологический журнал им. И.М. Сеченова. 2013. 99 (7): 793–804.
Мухин В.Н., Клименко В.М. Механизмы нарушения долговременной потенциации при болезни Альцгеймера. Медицинский академический журнал. 2014. 14 (1): 42–51.
Четвериков А.А. Линейные модели со смешанными эффектами в когнитивных исследованиях. Российский журнал когнитивной науки. 2015. 2 (1): 41–51.
Baptista T., Teneúd L., Contreras Q., Burguera J.L., Burguera M., Hernández L. Effects of acute and chronic lithium treatment on amphetamine-induced dopamine increase in the nucleus accumbens and prefrontal cortex in rats as studied by microdialysis. J. Neural Transm. Gen. Sect. 1993. 94 (2): 75–89.
Bates D., Mächler M., Bolker B., Walker S. Fitting Linear Mixed-Effects Models Using lme4. Journal of Statistical Software. 2015. 67 (1): 1–48.
Brorson J.R., Bindokas V.P., Iwama T., Marcuccilli C.J., Chisholm J.C., Miller R.J. The Ca2+ influx induced by β-amyloid peptide 25–35 in cultured hippocampal neurons results from network excitation. Journal of Neurobiology. 1995. 26 (3): 325–338.
Brothers H.M., Gosztyla M.L., Robinson S.R. The Physiological Roles of Amyloid-β Peptide Hint at New Ways to Treat Alzheimer’s Disease. Front. Aging Neurosci. 2018. 10. Article 118.
Busciglio J., Gabuzda D.H., Matsudaira P., Yankner B.A. Generation of beta-amyloid in the secretory pathway in neuronal and nonneuronal cells. PNAS. 1993. 90 (5): 2092–2096.
Can A., Frost D.O., Cachope R., Cheer J.F., Gould T.D. Chronic lithium treatment rectifies maladaptive dopamine release in the nucleus accumbens. J. Neurochem. 2016. 139 (4): 576–585.
D’Amelio M., Puglisi-Allegra S., Mercuri N. The role of dopaminergic midbrain in Alzheimer’s disease: Translating basic science into clinical practice. Pharmacol. Res. 2018. 130: 414–419.
Delobette S., Privat A., Maurice T. In vitro aggregation facilities beta-amyloid peptide-(25-35)-induced amnesia in the rat. Eur. J. Pharmacol. 1997. 319 (1): 1–4.
DeWaele M., Oh Y., Park C., Kang Y.M., Shin H., Blaha C.D., Bennet K.E., Kim I.Y., Lee K.H., Jang D.P. A baseline drift detrending technique for fast scan cyclic voltammetry. Analyst. 2017. 142 (22): 4317–4321.
Doll B.B., Daw N.D. The expanding role of dopamine. eLife. 2016. 5): e15963.
Duyckaerts C., Delatour B., Potier M.-C. Classification and basic pathology of Alzheimer disease. Acta Neuropathologica. 2009. 118 (1): 5–36.
Fortin S.M., Chartoff E.H., Roitman M.F. The Aversive Agent Lithium Chloride Suppresses Phasic Dopamine Release Through Central GLP-1 Receptors. Neuropsychopharmacology. 2016. 41 (3): 906–915.
Freir D.B., Holscher C., Herron C.E. Blockade of long-term potentiation by beta-amyloid peptides in the CA1 region of the rat hippocampus in vivo. J. Neurophysiol. 2001. 85 (2): 708–713.
Freland L., Beaulieu J.-M. Inhibition of GSK3 by lithium, from single molecules to signaling networks. Front Mol Neurosci. 2012. 5: 14.
Garcia T.P., Marder K. Statistical Approaches to Longitudinal Data Analysis in Neurodegenerative Diseases: Huntington’s Disease as a Model. Curr Neurol Neurosci Rep. 2017. 17 (2). Article 14.
Gengler S., Gault V.A., Harriott P., Hölscher C. Impairments of hippocampal synaptic plasticity induced by aggregated beta-amyloid (25-35) are dependent on stimulation-protocol and genetic background. Exp Brain Res. 2007. 179 (4): 621–630.
Giuffrida M.L., Caraci F., Pignataro B., Cataldo S., De Bona P., Bruno V., Molinaro G., Pappalardo G., Messina A., Palmigiano A., Garozzo D., Nicoletti F., Rizzarelli E., Copani A. Beta-amyloid monomers are neuroprotective. J. Neurosci. 2009. 29 (34): 10582–10587.
González-Mora J.L., Salazar P., Martín M., Mas M. Monitoring Extracellular Molecules in Neuroscience by In Vivo Electrochemistry: Methodological Considerations and Biological Applications. In Vivo Neuropharmacology and Neurophysiology Neuromethods. / Ed. A. Philippu. New York, NY: Springer New York, 2017. 181–206 pp.
Hampel H., Lista S., Mango D., Nisticò R., Perry G., Avila J., Hernandez F., Geerts H., Vergallo A. Lithium as a Treatment for Alzheimer’s Disease: The Systems Pharmacology Perspective. Journal of Alzheimer’s Disease. 2019. 69 (3): 615–629.
Hensley K., Carney J.M., Mattson M.P., Aksenova M., Harris M., Wu J.F., Floyd R.A., Butterfield D.A. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. PNAS. 1994. 91 (8): 3270–3274.
Hochstrasser T., Hohsfield L.A., Sperner-Unterweger B., Humpel C. β-Amyloid induced effects on cholinergic, serotonergic, and dopaminergic neurons is differentially counteracted by anti-inflammatory drugs. Journal of Neuroscience Research. 2013. 91 (1): 83–94.
Itoh A., Nitta A., Nadai M., Nishimura K., Hirose M., Hasegawa T., Nabeshima T. Dysfunction of cholinergic and dopaminergic neuronal systems in beta-amyloid protein–infused rats. J. Neurochem. 1996. 66 (3): 1113–1117.
Jones S.R., Mathews T.A., Budygin E.A. Effect of moderate ethanol dose on dopamine uptake in rat nucleus accumbens in vivo. Synapse. 2006. 60 (3): 251–255.
Joyce J.N., Murray A.M., Hurtig H.I., Gottlieb G.L., Trojanowski J.Q. Loss of Dopamine D2 Receptors in Alzheimer’s Disease with Parkinsonism But Not Parkinson’s or Alzheimer’s Disease. Neuropsychopharmacology. 1998. 19 (6): 472–480.
Kaminsky Y.G., Marlatt M.W., Smith M.A., Kosenko E.A. Subcellular and metabolic examination of amyloid-β peptides in Alzheimer disease pathogenesis: Evidence for Aβ25–35. Experimental Neurology. 2010. 221 (1): 26–37.
Kandel N., Matos J.O., Tatulian S.A. Structure of amyloid β 25–35 in lipid environment and cholesterol-dependent membrane pore formation. Sci Rep. 2019. 9 (1): 1–12. Article 2689.
Karran E., De Strooper B. The amyloid cascade hypothesis: are we poised for success or failure? J. Neurochem. 2016. (139): 237–252.
Keiflin R., Pribut H.J., Shah N.B., Janak P.H. Ventral Tegmental Dopamine Neurons Participate in Reward Identity Predictions. Current Biology. 2019. 29 (1): 93–103. e3.
Koch G., Di Lorenzo F., Bonnì S., Giacobbe V., Bozzali M., Caltagirone C., Martorana A. Dopaminergic Modulation of Cortical Plasticity in Alzheimer’s Disease Patients. Neuropsychopharmacology. 2014. 39 (11): 2654–2661.
Krashia P., Nobili A., D’Amelio M. Unifying Hypothesis of Dopamine Neuron Loss in Neurodegenerative Diseases: Focusing on Alzheimer’s Disease. Front Mol Neurosci. 2019. 12. Article 123.
Kubo T., Kumagae Y., Miller C.A., Kaneko I. β-Amyloid Racemized at the Ser26 Residue in the Brains of Patients with Alzheimer Disease: Implications in the Pathogenesis of Alzheimer Disease. J Neuropathol Exp Neurol. 2003. 62 (3): 248–259.
Kuznetsova A., Brockhoff P.B., Christensen R.H.B. lmerTest Package: Tests in Linear Mixed Effects Models. Journal of Statistical Software. 2017. 82 (1): 1–26.
Llorens-Marítin M., Jurado J., Hernández F., Ávila J. GSK-3β, a pivotal kinase in Alzheimer disease. Front Mol Neurosci. 2014. 7: 1–11.
Majka P., Kublik E., Furga G., Wójcik D.K. Common Atlas Format and 3D Brain Atlas Reconstructor: Infrastructure for Constructing 3D Brain Atlases. Neuroinform. 2012. 10 (2): 181–197.
Maqbool M., Mobashir M., Hoda N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. European Journal of Medicinal Chemistry. 2016. 107: 63–81.
Martorana A., Di Lorenzo F., Esposito Z., Lo Giudice T., Bernardi G., Caltagirone C., Koch G. Dopamine D2-agonist Rotigotine effects on cortical excitability and central cholinergic transmission in Alzheimer’s disease patients. Neuropharmacology. 2013. 64: 108–113.
Martorana A., Mori F., Esposito Z., Kusayanagi H., Monteleone F., Codecà C., Sancesario G., Bernardi G., Koch G. Dopamine Modulates Cholinergic Cortical Excitability in Alzheimer’s Disease Patients. Neuropsychopharmacology. 2009. 34 (10): 2323–2328.
Miklya I. The significance of selegiline/(−)-deprenyl after 50 years in research and therapy (1965–2015). Molecular Psychiatry. 2016. 21 (11): 1499–1503.
Millucci L., Ghezzi L., Bernardini G., Santucci A. Conformations and Biological Activities of Amyloid Beta Peptide 25–35. Current Protein and Peptide Science. 2010. 11 (1): 54–67.
Murray A.M., Weihmueller F.B., Marshall J.F., Hurtig H.I., Gottleib G.L., Joyce J.N. Damage to dopamine systems differs between Parkinson’s disease and Alzheimer’s disease with parkinsonism. Annals of Neurology. 1995. 37 (3): 300–312.
Nobili A., Latagliata E.C., Viscomi M.T., Cavallucci V., Cutuli D., Giacovazzo G., Krashia P., Rizzo F.R., Marino R., Federici M., De Bartolo P., Aversa D., Dell’Acqua M.C., Cordella A., Sancandi M., Keller F., Petrosini L., Puglisi-Allegra S., Mercuri N.B., Coccurello R., Berretta N., D’Amelio M. Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer’s disease. Nat Commun. 2017. 8: 1–14. Article 14727.
O’Brien W.T., Klein P.S. Validating GSK3 as an in vivo target of lithium action. Biochem Soc Trans. 2009. 37 (Pt5): 1133–1138.
Parihar M.S., Brewer G.J. Amyloid Beta as a Modulator of Synaptic Plasticity. J Alzheimers Dis. 2010. 22 (3): 741–763.
Paxinos G., Watson C. The Rat Brainin Stereotaxic Coordinates. San Diego: Elsevier Academic Press, 2005. 207 c.
Phillips P.E.M., Stuber G.D., Heien M.L.A.V., Wightman R.M., Carelli R.M. Subsecond dopamine release promotes cocaine seeking. Nature. 2003. 422 (6932): 614–618.
Pike C.J., Burdick D., Walencewicz A.J., Glabe C.G., Cotman C.W. Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly state. J. Neurosci. 1993. 13 (4): 1676–1687.
Preda S., Govoni S., Lanni C., Racchi M., Mura E., Grilli M., Marchi M. Acute β-Amyloid Administration Disrupts the Cholinergic Control of Dopamine Release in the Nucleus Accumbens. Neuropsychopharmacology. 2007. 33 (5): 1062–1070.
R Core Team. R: The R Project for Statistical Computing. 2019.
Rinne J.O., Säkö E., Paljärvi L., Mölsä P.K., Rinne U.K. Brain dopamine D-2 receptors in senile dementia. J. Neural Transmission. 1986. 65 (1): 51–62.
Robinson D.L., Phillips P.E., Budygin E.A., Trafton B.J., Garris P.A., Wightman R.M. Sub-second changes in accumbal dopamine during sexual behavior in male rats. Neuroreport. 2001. 12 (11): 2549–2552.
Rovira C., Arbez N., Mariani J. Abeta(25–35) and Abeta(1–40) act on different calcium channels in CA1 hippocampal neurons. Biochem. Biophys. Res. Commun. 2002. 296 (5): 1317–1321.
Šimić G., Babić Leko M., Wray S., Harrington C.R., Delalle I., Jovanov-Milošević N., Bažadona D., Buée L., de Silva R., Di Giovanni G., Wischik C.M., Hof P.R. Monoaminergic neuropathology in Alzheimer’s disease. Progress in Neurobiology. 2017 151: 101–138.
Takmakov P., Zachek M.K., Keithley R.B., Bucher E., McCarty G.S., Wightman R.M. Characterization of Local pH Changes in Brain Using Fast-Scan Cyclic Voltammetry with Carbon Microelectrodes. Anal Chem. 2010. 82 (23): 9892–9900.
Uzakov S.S., Ivanov A.D., Salozhin S.V., Markevich V.A., Gulyaeva N.V. Lentiviral-mediated overexpression of nerve growth factor (NGF) prevents beta-amyloid [25–35]-induced long term potentiation (LTP) decline in the rat hippocampus. Brain Research. 2015. (1624): 398–404.
Walker Q.D., Rooney M.B., Wightman R.M., Kuhn C.M. Dopamine release and uptake are greater in female than male rat striatum as measured by fast cyclic voltammetry. Neuroscience. 1999. 95 (4): 1061–1070.
Wang D., Noda Y., Zhou Y., Mouri A., Mizoguchi H., Nitta A., Chen W., Nabeshima T. The Allosteric Potentiation of Nicotinic Acetylcholine Receptors by Galantamine Ameliorates the Cognitive Dysfunction in Beta Amyloid 25–35 I.c.v.-Injected Mice: Involvement of Dopaminergic Systems. Neuropsychopharmacology. 2006. 32 (6): 1261–1271.
Wang Y., Liu L., Hu W., Li G. Mechanism of soluble beta-amyloid 25–35 neurotoxicity in primary cultured rat cortical neurons. Neuroscience Letters. 2016. (618): 72–76.
Wei W., Nguyen L.N., Kessels H.W., Hagiwara H., Sisodia S., Malinow R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nature Neuroscience. 2010. 13 (2): 190–196.
Xu P., Li Z., Wang H., Zhang X., Yang Z. Triptolide Inhibited Cytotoxicity of Differentiated PC12 Cells Induced by Amyloid-Beta25–35 via the Autophagy Pathway. PLOS ONE. 2015. 10 (11): e0142719.
Дополнительные материалы отсутствуют.
Инструменты
Журнал высшей нервной деятельности им. И.П. Павлова