

Журнал физической химии, 2020, T. 94, № 9, стр. 1337-1342
Особенности деструкции лигнина в древесине под действием озона
Н. А. Мамлеева a, *, А. Н. Харланов a, В. В. Лунин a
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
* E-mail: mamleevana@bk.ru
Поступила в редакцию 02.11.2019
После доработки 02.11.2019
Принята к публикации 12.11.2019
Аннотация
Методом УФ-спектроскопии диффузного отражения исследованы образцы древесины сосны, озонированной в присутствии различного содержания воды. Показано, что при озонировании древесины сосны разрушаются полиароматические и стильбеновые структуры, гваяцильные производные, карбонил- и карбоксил- содержащие ароматические структуры лигнина. Деструкция ароматических структур протекает эффективно в древесине с содержанием воды более 60–65% в области расходов озона 2–3 ммоль/г. При содержании воды ниже 40% обработка озоном древесины сосны при низких расходах озона позволяет получить разнообразные модификации кислородсодержащих ароматических и полиароматических структур на поверхности ЛЦМ. ИК-спектры поглощения диоксанлигнина, полученного из исходного и озонированных образцов древесины, подтверждают разное направление превращений лигнина в зависимости от содержания воды в древесине.
Озон является эффективным окислителем ароматических соединений [1, 2], и в этой связи используется в процессах удаления остаточного лигнина в целлюлозно-бумажных производствах на стадии отбелки бумажной массы [3, 4]. Делигнификация растительной биомассы озоном представляет интерес как метод предварительной обработки, позволяющий увеличить выход моносахаридов при последующем ферментативном гидролизе лигноцеллюлозного материала (ЛЦМ) [5–8]. Этот подход представляет интерес и для получения целлюлозы, так как деструкция целлюлозы при озонировании ЛЦМ относительно невелика [9, 10].
Использование древесины в качестве исходного материала для обработки озоном вызывает интерес в связи со значительным количеством производственных отходов, которые могут служить сырьем для дальнейшей химической переработки [3, 4, 10, 11]. Перспективность этих исследований обусловлена также высоким содержанием целлюлозы в древесине, характеризующейся высокой степенью полимеризации [9].
Установлено [5–10], что необходимым условием делигнификации биомассы при обработке озоном является присутствие воды в структуре образца; от количества воды зависит количество поглощенного озона и глубина делигнификации биомассы, а оптимальное количество воды различно для разных типов растительных субстратов. Изучение процесса поглощения озона и определение содержания остаточного лигнина в озонированных образцах древесины показало, что для процесса обработки древесины сосны оптимальным является содержание воды 60–63%, когда достигается степень делигнификации древесины 40% [9, 12]. При озонировании древесины сосны образуются продукты озонолиза лигнина – глиоксалевая, муравьиная и щавелевая кислоты, которые также участвуют в реакциях окисления озоном [9, 12].
Результаты перечисленных работ указывают на уменьшение общего содержания ароматической компоненты, не давая никакой информации относительно типа структур, участвующих в реакциях с озоном. Учитывая многообразие ароматических структур, присутствующих в различных видах растительной биомассы, для получения более конкретной информации по этому вопросу может быть полезен метод УФ-спектроскопии, с помощью которого ранее в работах [13–15] охарактеризованы основные структуры, входящие в состав лигнинов.
В данной работе исследована деструкция лигнина в древесине сосны при озонировании. Рассмотрена роль содержания воды в древесине как фактора, регулирующего глубину и направление превращений лигнина биомассы под действием озона. С этой целью использован недеструктивный метод изучения превращений лигнина растительной биомассы – УФ-спектроскопия диффузного отражения (УФДО). Анализ превращений лигнина при озонировании древесины проведен также с помощью ИК-спектров диоксанлигнина, выделенного из образцов древесины.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исследуемого материала использовали опилки древесины сосны (Pinus silvestris) с размером частиц 0.315–0.63 мм и содержанием воды (moisture content – МС) от 7 до 80 ± 1% относительно массы абсолютно сухой древесины (а.с.д). МС = (${{m}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$/mа.с.д) × 100%. Приготовление образцов проводили согласно [10]. Масса образцов составляла 0.42–0.45 г с объемом 0.75 ± 0.02 мл.
Озонирование проводили в проточной установке в термостатированном (25°С) реакторе с неподвижным слоем по методике [10]. Количество поглощенного озона (Q(t)) в момент времени t рассчитывали из кинетических кривых зависимости концентрации озона, согласно [10]. Сразу после обработки озоном образцы промывали водой и сушили на воздухе.
Для получения диоксанлигнина (ДЛ) использовали методику [11]. Навеску древесины кипятили (1 ч) в диоксане в присутствии каталитических количеств НCl. Раствор ДЛ в диоксане отфильтровывали и выпаривали на воздухе, затем высушенный образец использовали для приготовления таблеток ДЛ с КBr (0.1% образца ДЛ). ИК-спектры образцов ДЛ регистрировали на приборе Equinox 55/S (Bruker) в интервале 400–4000 см–1 в режиме пропускания.
Спектры УФДО образцов древесины (300 мг) регистрировали на приборе Specord M-40 с интегрирующей сферой в диапазоне 220–820 нм. При регистрации спектров в качестве эталона использовали образец BaSO4 (его отражение принимается за 100%).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
На рис. 1 приведены кривые зависимости удельного поглощения озона от продолжительности озонирования в опытах с различным содержанием воды в образце. Все кривые выходят постепенно на насыщение, что указывает на замедление реакций озона с ЛЦМ и их постепенное прекращение, которое объясняли торможением процесса поглощения озона реакциями с продуктами окисления биомассы [10]. Из рис. 1 видно, что увеличение содержания воды в исходном образце позволяет достигнуть более высоких значений количества поглощенного озона в ходе обработки озоном.
Рис. 1.
Кинетические кривые зависимости удельного поглощения для образцов древесины сосны с различным содержанием воды; МС, %: 7 (1), 25 (2), 35 (3), 45 (4), 65 (5), 80 (6).
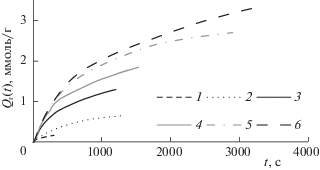
В табл. 1 представлены основные характеристики исследованных образцов – содержание воды в образце (МС, %) и количество поглощенного озона (Qr). Образец № 1 – исходная древесина сосны.
Таблица 1.
Характеристики образцов озонированной древесины: содержание воды в образце (МС, %) и удельное поглощение озона (Qr, ммоль/г)
| № | МС, % | Qr, ммоль/г | № | МС, % | Qr, ммоль/г |
|---|---|---|---|---|---|
| 1 | 7 | 0 | 7 | 35 | 1.1 |
| 2 | 7 | 0.2 | 8 | 40 | 1.3 |
| 3 | 15 | 0.3 | 9 | 45 | 1.6 |
| 4 | 20 | 0.5 | 10 | 60 | 1.9 |
| 5 | 25 | 0.6 | 11 | 65 | 2.5 |
| 6 | 30 | 0.7 | 12 | 80 | 3.2 |
На рис. 2 представлены спектры УФДО для исходного образца и некоторых озонированных образцов рис. 1.
Рис. 2.
УФДО спектры (а) и дифференциальные спектры УФДО (б) образцов древесины сосны. На рисунках указаны номера образцов из табл. 1.
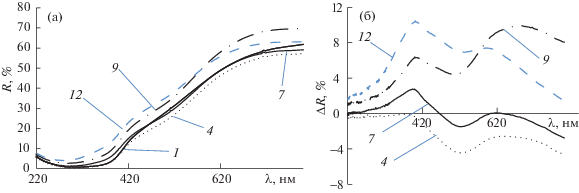
В исследуемом образце древесины сосны присутствует 28% лигнина [9]. Лигнины хвойных пород древесины характеризуются присутствием гваяцильных, сиренгильных и фенильных структур, в лигнинах также присутствуют конденсированные ароматические системы. Кислородсодержащие структуры лигнина, в основном, представлены простыми эфирными связями, спиртовыми и фенольными гидроксильными группами [11].
Спектры УФДО образцов древесины приведены на рис. 2а. Хромофорные группы лигнина характеризуются поглощением в широкой области спектра с максимумами от 205 до 650 нм [13–15]. Из рис. 2а видно, что для ряда озонированных образцов возрастает отражение во всей области спектра, происходит “обесцвечивание” образца. Для анализа спектров диффузного отражения, поглощения и пропускания часто используют разностные спектры, позволяющие получить более подробную картину изменений, вызванных тем или иным воздействием на исследуемый образец. На рис. 2б приведены разностные спектры УФДО озонированных образцов относительно спектра исходного образца древесины сосны. Разностные спектры показывают, что при низких расходах озона структуры, поглощающие в области 220–400 нм и в области ∼500 нм, не разрушаются. Значение ΔR уходит для этих образцов в отрицательную область (спектр образца 4 рис. 2б). Разрушение этих структур характерно для более высоких значений удельного поглощения озона.
При Qr 1.5–2.5 ммоль/г под действием озона происходит разрушение хромофоров, поглощающих во всей области спектра. В области 550–650 нм поглощают придающие окраску древесине конденсированные полиароматические и хиноидные структуры [13–15]. Видно, что эти структуры, а также поглощающие при 390–400 нм стильбены и стильбенхиноны, разрушаются озоном.
Для количественного и полуколичественного анализа по спектрам диффузного отражения наиболее корректно использовать функцию Кубелки–Мунка F(R), представляющую собой отношение коэффициента поглощения к коэффициенту рассеяния среды и определяемую по формуле:
Значения F(R) для длин волн 278, 300, 250 и 400 нм, где наблюдаются максимумы поглощения структурных моделей лигнина, ранее охарактеризованные в работах [13–15], приведены на рис. 3.
Рис. 3.
Зависимость параметра F(R) при длине волны 278, 300 нм (а) и 250 и 400 нм (б) спектров ДО древесины сосны от удельного поглощения озона.
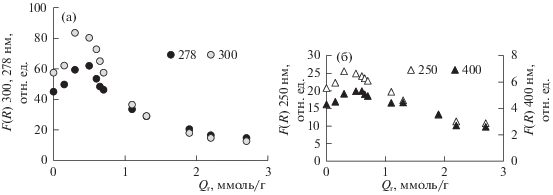
Из рис. 3 видно, что в области значений удельного поглощения озона 1.5–2.5 ммоль/г значение F(R) при λ = 250, 278, 300 и 400 нм заметно снижается. Это свидетельствует о разрушении структур гваякола, гваяцилпропана, гваяцилэтанола, гваяцилпропанола (поглощение при 278 нм), полиароматических структур с поглощением при 390–400 нм, а также структур с поглощением при 300 нм [13–15].
При низких значениях расходов озона (<1 ммоль/г) возрастает F(R) при 300 нм, где поглощают карбонил-содержащие ароматические структуры (ацетогвакон, гваяцилпропанон, ванилин) (рис. 3а). Отметим, что перечисленные соединения характеризуются также высокими значениями экстинкции в области 270–280 нм. Значение F(R) при длине волны 250 нм также проходит через максимум при Qr < 1 ммоль/г (рис. 3б), что объясняется модифицированием ароматики древесины с образованием карбоксилсодержащих ароматических соединений (например, ванилиновой кислоты). При увеличении Qr эти структуры также разрушаются.
Зависимости рис. 3 показывают симбатный характер изменения оптической плотности в разных областях спектра, что не позволяет сделать вывод относительно преимущественного разрушения озоном тех или иных структур. Отмеченные на рис. 3 максимумы оптической плотности относятся к образцам, полученным озонированием древесины с содержанием воды менее 40% (образцы № 1–8 табл. 1), что указывает на связь наблюдаемого эффекта с условиями озонирования.
ИК-спектры диоксанлигнина из исходной и озонированной древесины (в табл. 1 им соответствуют образцы № 1, 4 и 10) приведены на рис. 4.
Рис. 4.
ИК-спектры диоксанлигнина, выделенного из древесины, обработанной озоном: Qr, ммоль/г: 0 (1), 1.9 (2), 0.5 (3); МС, %: 60 (2), 20 (3).
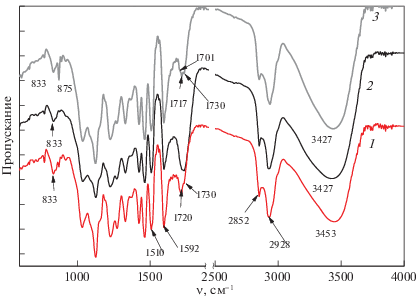
В ИК-спектре ДЛ из исходного образца наблюдаются полосы скелетных колебаний ароматических колец (1510, 1592 см–1); видна интенсивная полоса при 1720–1730 см–1 валентных С=О-колебаний с алифатическими заместителями. В области 850–880 см–1 и 800–855 см–1 проявляются внеплоскостные деформационные колебания С–Н-связей гваяцильных колец различной степени и характера замещения [16]. Интенсивные полосы при 2852 и 2928 см–1 характерны для асимметричных и симметричных валентных С–Н колебаний в метильных группах лигнина. Широкая полоса с максимумом поглощения 3453 см–1 относится к валентным колебаниям О–Н-групп, связанным водородной связью [16].
В спектре образца ДЛ из древесины, озонированной при содержании воды 60% (образец № 10 в табл. 1), интенсивность полос скелетных колебаний С–С-связей в ароматических кольцах (1510, 1592 см–1) уменьшается, а интенсивность полосы при 1730 см–1 заметно увеличивается (спектры 1 и 2 на рис. 4). Отмеченное изменение характерно для превращений лигнина при озонировании [17], когда разрушение ароматики озоном вызывает возрастание содержания карбоксильных групп с алифатическим заместителем.
Спектр 3 относится к диоксанлигнину, полученному из образца № 4 (МС 20%, Qr 0.5 ммоль/г). В отличие от спектра 2 в спектре 3 полоса поглощения валентных С=О-колебаний карбоксильных групп (1730 см–1) не возрастает. Видно плечо при 1701 см–1 (конъюгированные альдегиды и карбоновые кислоты [16]), и полоса 1717 см–1 валентных колебаний С=О в неконъюгированных кетонах и сложных эфирах [16]. Интенсивность скелетных колебаний ароматического кольца уменьшается мало. Заметно изменение интенсивностей деформационных С–Н-колебаний (833, 875 см–1). Из спектра 3 можно заключить, что в данном случае происходит конденсация ароматических колец лигнина, при этом уменьшается экстинкция полосы ароматических С–С-колебаний, происходит изменение состава заместителей ароматических систем. Аналогичные процессы наблюдали при озонировании воздушно-сухого порошка лигнина [17]. Таким образом, данные ИК-спектров ДЛ согласуются с выводами об образовании полиароматических структур и ароматических структур с кислородсодержащими группами.
Спектры 1–3 рис. 4 показывают возрастание интенсивности полосы О–Н-колебаний и смещение ее в область более низких значений волновых чисел у озонированных образцов. Обе характеристики связаны с высоким содержанием в ДЛ кислородсодержащих групп, вступающих в образование водородных связей. Изменений интенсивности и положения полос валентных колебаний алифатических С–Н-групп у озонированных образцов не наблюдается, что согласуется с относительной инертностью озона по отношению к алифатическим связям [1].
Данные УФДО и ИК-спектров свидетельствуют о различном направлении превращений лигнина в древесине под действием озона, в зависимости от содержания воды. Это объясняется ролью воды в процессе озонирования биомассы, которая в основном, сводится к увеличению размеров доступной реагенту поверхности, вследствие набухания биомассы. Кроме того, присутствие воды в пористой структуре субстрата обеспечивает доступность функциональных групп биомассы реагенту, растворенному в воде [5, 6, 10, 12].
Показано [5–10, 18], что для эффективного поглощения озона древесиной и ее делигнификации необходим значительный избыток воды относительно точки насыщения волокна (ТНВ), соответствующей содержанию воды 30%. В условиях избытка содержания воды относительно ТНВ система характеризуется наличием двух межфазных границ. Первая граница: газовая фаза (с озоном в озон-кислородной смеси) – водная фаза, вторая граница раздела: водная фаза с растворенным озоном – поверхность ЛЦМ.
В условиях избытка содержания воды относительно ТНВ реакции озона с лигнином в биомассе идут по схеме озонолиза с разрушением ароматических систем и образованием низкомолекулярных кислот [12, 19].
По данным спектров УФДО, разрушение всех типов ароматических структур в древесине наблюдается при МС 60–80%, мало изменяясь при МС выше 60–65%. Это согласуется с представлением, что оптимум содержания воды для делигнификации древесины сосны находится в области 60–63% [9, 12].
При озонировании ненабухшего образца (МС ≤ 30%) реакции с озоном идут на внешней поверхности, на границе с газовой фазой. Происходит гетерогенное разложение озона, которое сопровождается генерацией атомарного кислорода [20]. Радикальные процессы вызывают образование на поверхности полимера древесины алкильных, алкоксильных, алкилпероксидных и ароматических радикалов [18]. В ходе реакций с участием этих частиц происходит образование новых структур, в частности конденсация и полимеризация ароматических структур ЛЦМ, отмеченная нами в [21] при изучении озонированных образцов методом термического анализа. Радикальные процессы приводят к окислению боковых заместителей ароматических колец с образованием ароматических кетонов, альдегидов и кислот. Это нашло свое отражение на рис. 2–4.
Таким образом, являясь фактором, определяющим размер доступной озону поверхности, содержание воды регулирует вклад реакций, протекающих на внутренней поверхности ЛЦМ с участием озона, растворенного в воде, и свободно-радикальных реакций, которые осуществляются на внешней поверхности ЛЦМ. Первые идут по механизму озонолиза [19], характерным для реакций озона с ароматическими соединениями в растворе, и приводят к деструкции лигнина до низкомолекулярных продуктов; результатом вторых является модифицирование ароматики с образованием кислородсодержащих ароматических и полиароматических структур.
Показано [18], что на процесс делигнификации древесины озоном влияет сушка образца, сопровождающая опыты по озонированию, которые осуществляются в проточной установке. В ходе озонирования из-за сушки древесины содержание воды постепенно становится ниже ТНВ. Это приводит к отмеченному выше сокращению внутренней поверхности древесины и постепенному возрастанию вклада процессов на границе раздела: газ–поверхность ЛЦМ. Наличие градиента содержания воды в реакторе вдоль слоя образца создает условия для возникновения в ходе озонирования участков с содержанием воды ниже ТНВ. В результате, для образцов, исследованных в работе, процесс делигнификации по схеме озонолиза комбинируется со свободно-радикальными реакциями, которые сопровождают обработку образцов озоном и негативно влияют на проведение делигнификации биоматериала.
Данные спектров УФДО подтвердили сделанный ранее вывод, что делигнификация древесины наиболее эффективно идет при озонировании образцов с содержанием воды более 60–65% в области расходов озона 2–3 ммоль/г. Показано, что содержание воды позволяет регулировать не только глубину делигнификации, но и направление превращений ароматических структур. Содержание воды ниже 40% неперспективно для проведения делигнификации. В этих условиях обработка озоном древесины сосны при низких расходах озона позволяет получить разнообразные модификации ароматических, кислородсодержащих ароматических и полиароматических структур на поверхности ЛЦМ. Результаты по образованию окисленных ароматических соединений могут быть интересны для получения новых полимерных материалов на основе озонированных ЛЦМ.
Список литературы
Разумовский С.Д., Заиков Г.Е. Озон и его реакции с органическими соединениями (кинетика и механизм). М.: Наука, 1974. 322 с.
Audran G., Marque S.R.A., Santelli M. // Tetrahedron. 2018. V. 74. P. 6221.
Дёмин В.А., Шерешовец В.В., Монаков Ю.Б. // Успехи химии. 1999. Т. 68. № 11. С. 1029.
Roncero M.B., Colom J.F., Vidal T. // Carbohydr. Polym. 2003. V. 51. P. 243.
Li C., Wang L., Chen Z., Li Y. et al. // Biores. Technol. 2015. V. 183. P. 240.
Travaini R., Otero M.D.M., Coca M. et al. // Biores. Technol. 2013. V. 133. P. 332.
Ben'ko E.M., Chukhchin D.G., Lunin V.V. // Russ. J. Phys. Chem. A. 2017. V. 91. P. 2092.
Бенько Е.М., Мамлеева Н.А., Чухчин Д.Г. и др. // Журн. физ. химии. 2020. Т. 94. № 8. C. 1153.
Мамлеева Н.А., Харланов А.Н., Чухчин Д.Г. и др. // Химия растительного сырья. 2019. № 1. С. 85. https://doi.org/10.14258/jcprm.2019015143
Mamleeva N.A., Autlov S.A., Bazarnova N.G., Lunin V.V. // Russ. J. Bioorg. Chem. 2016. V. 42. P. 694.
Holladay J.E., Bozell J.J., White J.F., Johnson D. Top Value-Added Chemicals from Biomass. V. II. 2007. USA. http://www.ntis.gov/ordering.htm
Мамлеева Н.А., Бабаева Н.А., Харланов А.Н., Лунин В.В. // Журн. физ. химии. 2019. Т. 93. № 1. С. 28.
Боголицын К.Г., Линдберг И. // Химия древесины. 1986. № 4. С. 56.
Попова Н.Р., Боголицын К.Г. Физикохимия лигнина / Ред. Боголицын К.Г., Лунин В.В. Архангельск, Россия: Изд-во Архангельский гос. техн. ун-т, 2009.
Боголицын К.Г. // Журн. прикл. спектр. 1987. Т. 46. С. 1024.
Schwanninger M., Rodrigues J.C., Pereira H., Hinterstoisser B. // Vibr. Spectr. 2004. V. 36. C. 23.
Мамлеева Н.А., Ковалева В.В., Бенько Е.М. и др. // Журн. физ. химии. 2001. Т. 75. № 1. С. 57.
Мамлеева Н.А., Кустов А.Л., Лунин В.В. // Там же. 2018. Т. 92. № 9. С. 1402.
Bailey P.S. Ozonation of Aromatic Compounds. in Ozonation in Organic Chemistry. V. 2. Nonolefinic compounds. N.Y.: Acad. Press, 1982. P. 31.
Лунин В.В., Попович М.П., Ткаченко С.Н. Физическая химия озона. М.: Изд-во МГУ, 1998.
Мамлеева Н.А., Шумянцев А.В., Лунин В.В. // Журн. физ. химии. 2020. Т. 94. № 3. C. 404.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии