

Журнал физической химии, 2020, T. 94, № 9, стр. 1348-1354
Об особенностях протонирования ферроценилкарбоновых кислот кислотами Бренстеда
В. М. Фомин a, *, Н. Н. Шуклина a, М. Н. Климова a
a Национальный исследовательский Нижегородский государственный университет имени Н.И. Лобачевского
Нижний Новгород, Россия
* E-mail: niih325@bk.ru
Поступила в редакцию 02.12.2019
После доработки 02.12.2019
Принята к публикации 17.12.2019
Аннотация
Методом электронной спектроскопии показано, что протонирование ферроценилкарбоновой и ферроценилуксусной кислот сопровождается редокс-изомерией образующихся ферроценилкарбениевых ионов. Установлено влияние природы металлокомплексов, кислот Бренстеда и растворителя на скорость редокс-изомерии. Кинетическим методом показано, что стабилизация ацилиевого иона FcC+O (Fc – ферроценил) за счет координации с FcCOOH, обусловливает экстремальный характер зависимости скорости редокс-изомерии от концентрации FcCOOH.
Известно, что органические соединения, содержащие гидроксильную, винильную, карбонильную и карбоксильную функциональные группы, способны протонироваться сильными кислотами Бренстеда с образованием карбениевых и ацилиевых ионов [1]. Не являются исключением в этом смысле и производные ферроцена с теми же самыми функциональными группами [2]. На примере ферроценилметанола и ферроценилметилметанола FсCHROH (R – H, CH3), а также винилферроцена FсCH=CH2, было показано, что образующиеся при их протонировании α-ферроценилкарбениевые ионы FсC+HR проявляют способность к редокс-изомерии с образованием соответствующих катионов ферроцения, которые после их восстановления были выделены в виде димеров 1,2-диферроценилэтана и 1,2-диметилдиферроценилэтана [3, 4].
Карбокатион FсC+H2 можно зафиксировать на полосе поглощения с λmax = 600 нм, что позволило авторам [5, 6] изучить его свойства методом электронной спектроскопии и зафиксировать редокс-изомерию по накоплению катиона ферроцения.
Протонирование производных ферроцена оказывает существенное влияние на реакции с их участием, в частности процессы окисления молекулярным кислородом и пероксидом водорода в присутствии кислот, которое в зависимости от природы металлокомплекса может проявляться по-разному. Известно, что ферроценилуксусная кислота FcCH2COOH (соединение I) легко окисляется кислородом в присутствии трифторуксусной кислоты или вообще в отсутствие кислот по радикально-цепному механизму [7]. В присутствии хлорной кислоты поглощения кислорода реакционной смесью фактически не наблюдается, и радикально-цепной процесс не реализуется. Авторы [7] связывают такое влияние хлорной кислоты на окисление I с образованием ферроценилкарбениевых ионов при его протонировании, которые способны ингибировать цепной процесс за счет связывания пероксидных радикалов ${\text{RO}}_{2}^{ \bullet }$ (R – обобщенный фрагмент окисляемого соединения), ведущих цепной процесс, в комплекс донорно-акцепторного типа ${\text{Fc}}{{{\text{C}}}^{ + }}{{{\text{H}}}_{2}}{\kern 1pt} \cdots {\kern 1pt} {\text{OOR}}$. Аналогичная картина наблюдается при автоокислении ферроенилметанола FcCH2OH (II) в присутствии этой же кислоты [8]. При окислении формил- и ацетилферроцена кислородом [9] и пероксидом водорода [10] влияние протонирования металлокомплексов проявляется в виде экстремальной зависимости соответственно либо скорости поглощения кислорода, либо скорости накопления катиона ферроцения от концентрации кислоты.
С целью более детального исследования процесса протонирования производных ферроцена и явления редокс-изомериии образующихся при этом ферроценилкарбениевых ионов нами были изучены особенности превращения систем FсCH2СООН–НХ и FсCООН(III)–НХ, где НХ=HClO4(IV), СF3CООН(V), HCl(VI), в апротонных и протонсодержащих растворителях. Чтобы минимизировать возможное влияние кислорода воздуха на последующие процессы, все исследования проводили в атмосфере аргона.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Электронные спектры поглощения снимали на спектрометре ShimadzuUV-1800 в области 200–1100 нм в атмосфере аргона с использованием кварцевых кювет. Работу проводили в режиме сканирования по длине волны с последующей обработкой спектра. Использованные растворители имели квалификацию “х. ч.” и “ч. д. а.” и дополнительной очистке не подвергались11.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Прежде чем приступить к анализу результатов проведенных исследований, рассмотрим, каким образом происходит образование карбокатионов при протонировании III. По данным [11], образуются два типа карбокатионов за счет возможности взаимодействия протона с гидроксильной и карбонильной группами кислоты:
(1)
${\text{FcCOOH}} + {{{\text{H}}}^{ + }}\;\overset K \leftrightarrows \;{\text{Fc}}\mathop {\text{C}}\limits^{\text{ + }} {\text{O}} + {{{\text{H}}}_{2}}{\text{O}},$(2)
$\begin{gathered} {\text{FcCOOH}} + {{{\text{H}}}^{ + }}\;\overset {{{K}_{1}}} \leftrightarrows \;{\text{Fc}}{{{\text{C}}}^{ + }}{{({\text{OH}})}_{2}}\;\overset {{{K}_{2}}} \leftrightarrows \\ \overset {{{K}_{2}}} \leftrightarrows \;{\text{Fc}}\mathop {\text{C}}\limits^ + {\text{O}} + {{{\text{H}}}_{2}}{\text{O}}{\text{.}} \\ \end{gathered} $Все изложенное выше относится и к протонированию I, хотя есть принципиальное отличие, обусловленное возможностью фрагментации ацилиевого иона FcCH2C+O, ведущей к образованию более стабильного карбокатиона.
(3)
$\begin{gathered} {\text{FcC}}{{{\text{H}}}_{2}}{\text{COOH}} + {{{\text{H}}}^{ + }}\;\overset K \leftrightarrows \;{{{\text{H}}}_{2}}{\text{O}} + \\ + \;{\text{FcC}}{{{\text{H}}}_{2}}{{{\text{C}}}^{ + }}{\text{O}}\;\xrightarrow[{ - {\text{CO}}}]{}\;{\text{Fe}}\mathop {\text{C}}\limits^ + {{{\text{H}}}_{2}}. \\ \end{gathered} $Рис. 1.
Электронные спектры реакционных смесей, содержащих HClO4 и соединения I (1), II (2, 3) и III (4) в диоксане; $С_{{{\text{I}} - {\text{III}}}}^{0}$ = 0.001 М, $С_{{{\text{IV}}}}^{0}$ = 0.1 М (1, 2, 4); $~С_{{{\text{II}}}}^{0}$ = 0.05 М, $С_{{{\text{IV}}}}^{0}$ = 0.001 М (3); t = 15 с.
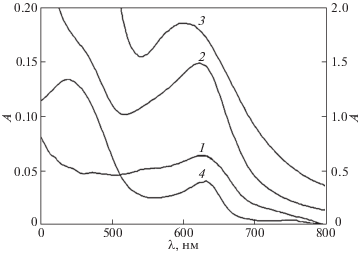
Обращает на себя внимание, что значение λmax полосы поглощения фиксируемого катиона ферроцения фактически совпадает с λmax полосы поглощения катиона ферроцения, образующегося при протонировании ферроценилметанола, из чего можно сделать вывод, что протонирование I действительно приводит к образованию карбокатиона FcC+H2, и далее катиона ферроцения Fc+C$^{ \bullet }$H2. В пользу этого вывода свидетельствуют и наблюдаемое мгновенное изменение окраски раствора I в диоксане с желтой до зеленой при приливании к нему кислоты IV и точно такое же изменение окраски раствора при смешении II и IV, связанное с образованием именно карбокатиона FcC+H2. Со временем растворы приобретают зеленовато-голубой оттенок за счет образования катиона ферроцения Fc+C$^{ \bullet }$H2 при редокс-изомерии карбокатиона.
Образование катиона ферроцения характерно и для взаимодействия III с IV в диоксане. На это указывает появление в электронном спектре реакционной смеси полосы поглощения с λmax = = 632 нм сразу после смешения реагентов (рис. 1). В этом случае следует сделать вывод о редокс-изомерии ацилиевого иона FcC+O. Полученные результаты достаточно убедительно свидетельствуют о том, что основным каналом образования катионов ферроцения при протонировании I, III является редокс-изомерия образующихся карбокатионов.
Выход катиона ферроцения при протонировании I–III существенно зависит от природы кислоты Бренстеда. На примере III показано, что при стократном избытке трифторуксусной кислоты по сравнению с металлокомплексом ($С_{{{\text{Fс}}}}^{^\circ }$ = 0.001 М, $С_{{{\text{НХ}}}}^{^\circ }$ = 0.1 М) образования катиона ферроцения практически не наблюдается, что указывает на ее низкую протонирущую способность. В присутствии соляной кислоты, которая по силе (рKα = ‒7) лишь незначительно уступает хлорной (рKα = –10), катион ферроцения также не образуется. Причина этого, скорее всего, заключается в том, что образующийся при протонировании III катион ацилия с высокой скоростью нейтрализуется анионом Cl–. С этим выводом согласуется наблюдаемое торможение скорости реакции, вплоть до ее полной остановки, при введении небольших добавок KI (0.005 М) в реакционную смесь.
Как следует из уравнений (1)–(3), введение воды в реакционную смесь должно приводить к снижению скорости редокс-изомерии карбокатиона, что в действительности и наблюдается на опыте.
Скорость накопления катиона ферроцения при протонировании металлокомплексов зависит как от их природы (рис. 2), так и от природы используемых растворителей (рис. 3). Из рис. 2 видно, что начальная скорость реакции ${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}}$ падает в ряду: II > I > III. Совершенно очевидно, что более низкая реакционная способность III по сравнению с II обусловлена более высокой стабильностью карбокатиона FсС+О по сравнению со стабильностью FсС+Н2 за счет возможности сопряжения вакантной р-орбитали атома углерода, принимающей участие в редокс-изомерии, в первом из карбокатионов не только с π-системой Ср-лиганда, но и с орбиталью неподеленной пары электронов кислорода. Образование FсС+Н2 при протонировании I протекает в результате двухстадийного процесса, описываемого уравнением (3), что и обусловливает положение I в указанном ряду реакционной способности металлокомплексов.
Рис. 2.
Кинетические кривые накопления катиона ферроцения при протонировании соединений I–III кислотой IV в диоксане; $С_{{{\text{I}} - {\text{III}}}}^{0}$ = 0.001 М, $С_{{{\text{IV}}}}^{0}$ = 0.1 М.
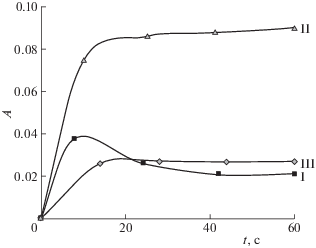
Рис. 3.
Влияние природы растворителя на выход катиона ферроцения при протонировании соединения III кислотой IV; $С_{{{\text{III}}}}^{0}$ = 0.001 М, $С_{{{\text{IV}}}}^{0}$ = 0.1 М; 1 – диоксан, 2 – этанол, 3 – диоксан–вода (1 : 1), 4 – ацетонитрил, 5 – ДМФА, 6 – ДМСО.
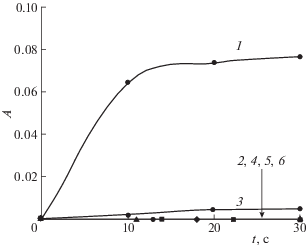
Из рис. 3 следует, что зависимость скорости накопления катиона ферроцения при протонировании соединений I–III от природы растворителей соответствует ряду: Diox $ \gg $ Diox-H2O ≥ EtOH ≥ ≥ MeCN ≥ ДМФА ≥ ДМСО, который практически повторяет ряд зависимости выхода карбокатиона FсС+Н2 от природы тех же самых растворителей при протонировании II [7], за исключением одного отличия, которое заключается в том, что образованию карбокатиона FсС+Н2 способствует не только диоксан, но и ацетонитрил, а редокс-изомерии – только диоксан. Это соответствие рядов растворителей служит веским доказательством зависимости скорости накопления катиона ферроцения от концентрации карбокатиона при редокс-изомерии последнего. При рассмотрении приведенного ряда растворителей видно, что скорость накопления катиона ферроцения ${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}}$ падает симбатно увеличению диэлектрической проницаемости растворителя. Это свидетельствует о существенном вкладе неспецифической сольватации в стабилизацию карбокатиона. Ее можно оценить по уравнению
где ΔG – изменение свободной энергии при переносе иона из газовой фазы в диэлектрическую среду или та же самая свободная энергия сольватации [1]. Из приведенного уравнения видно, что стабилизация карбокатиона будет наиболее существенна в растворителях с наиболее высоким значением ε, и именно поэтому в диоксане, который характеризуется самым низким значением диэлектрической проницаемости (ε = 2.4) среди используемых растворителей, скорость редокс-изомерии самая высокая, независимо от природы протонируемой ферроценилкарбоновой кислоты. Специфическая сольватация карбокатиона должна влиять прежде всего на его стационарную концентрацию. В случае ДМФА и ДМСО (S), молекулы которых могут существовать в виде резонансных структур с полностью разделенными отрицательными и положительными зарядами [12], предельно низкая концентрация карбокатиона связана, скорее всего, с образованием донорно-акцепторного комплекса [FcCH2S]+, в котором положительный заряд локализован уже на одном из атомов молекулы растворителя. Этанол, как и вода, может вступать в прямое взаимодействие с карбокатионом, давая соответствующий эфир. Ацетонитрил, являющийся n-донором электронов, может стабилизировать карбокатион не только за счет неспецифической сольватации, но и за счет специфической.Результаты изучения кинетики накопления катиона ферроцения при протонировании I и III хлорной кислотой указывают на существенное влияние соотношения концентраций металлокомплекса на скорость накопления катиона ферроцения, что проявляется в значительном отличии зависимостей ${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}}$ = f([МК]0) и ${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}}$ = f([IV]0) друг от друга (рис. 4). При небольших концентрациях реагентов скорость реакции линейно растет с ростом их концентрации, что свидетельствует о первом порядке процесса по каждому из реагентов:
(5)
${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}} = {{k}_{{{\text{эфф}}}}}{{[{\text{FcCOOH}}]}_{0}}{{[{{{\text{H}}}^{ + }}]}_{0}}.$Рис. 4.
Зависимости скорости накопления катиона ферроцения при протонировании соединений I (1, 2) и III (3, 4) от концентрации металлокомплекса (1, 3) и кислоты IV (2, 4) в диоксане; $С_{{{\text{IV}}}}^{0}$ = 0.001 М (1, 3); $~С_{{{\text{II}},{\text{\;III}}}}^{0}$ = 0.001 М (2, 4).
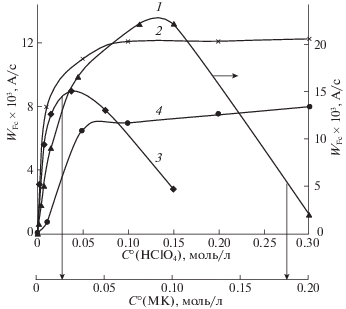
Наличие экстремума на зависимостях ${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}} = f(\left[ {{\text{МК}}{{]}_{0}}} \right)$ свидетельствует о том, что увеличение концентрации металлокомплекса относительно заданной концентрации кислоты IV приводит к ингибированию процесса редокс-изомерии карбокатиона, которое может быть результатом частичной или полной его дезактивации за счет образования донорно-акцепторных комплексов типа FcC+O · FcCOOH со свободной молекулой МК, если оно взято в избытке. Естественно, что концентрация комплекса возрастает с ростом концентрации МК.
Стабилизация ферроценилкарбениевого катиона за счет координации с исходным производным ферроцена ранее была отмечена в работах [7, 8].
Рассмотрим схемы 1 и 2 процесса протонирования III, анализ которых позволил бы объяснить экспериментально установленные зависимости ${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}} = f({{{\text{[}}{{{\text{H}}}^{ + }}]}_{0}})$ и ${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}} = f({{{\text{[MK}}]}_{0}})$:
Схема 1
(1.1)
${\text{FcCOOH}} + {{{\text{H}}}^{ + }}\;\overset K \leftrightarrows \;{\text{Fc}}\mathop {\text{C}}\limits^ + {\text{O}} + {{{\text{H}}}_{2}}{\text{O,}}$(1.2)
${\text{Fc}}\mathop {\text{C}}\limits^ + {\text{O}}\;\xrightarrow{{{{k}_{2}}}}\;{\text{F}}{{{\text{c}}}^{ + }}{\dot {C}O}.$В соответствии со схемой 1 , скорость накопления катиона ферроцения будет равна:
(6)
${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}} = {{k}_{2}}[{\text{Fc}}\mathop {\text{C}}\limits^ + {\text{O}}].$(7)
$[{\text{Fc}}\mathop {\text{C}}\limits^ + {\text{O}}] = \frac{{K{{{[{\text{FcCOOH}}]}}_{0}}[{{{\text{H}}}^{ + }}]}}{{[{{{\text{H}}}_{2}}{\text{O}}] + K[{{{\text{H}}}^{ + }}]}}.$(8)
${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}} = \frac{{{{k}_{2}}K{{{[{\text{FeCOOH}}]}}_{0}}[{{{\text{H}}}^{ + }}]}}{{[{{{\text{H}}}_{2}}{\text{O}}] + K[{{{\text{H}}}^{ + }}]}}.$При малых концентрациях кислоты уравнение (8) трансформируется в уравнение, которое соответствует первому порядку процесса по металлокомплексу и кислоте и минус первому порядку по воде:
(9)
${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}} = \frac{{{{k}_{2}}K{{{[{\text{FeCOOH}}]}}_{0}}[{{{\text{H}}}^{ + }}]}}{{[{{{\text{H}}}_{2}}{\text{O}}]}}.$(10)
${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}} = {{k}_{2}}{{[{\text{FcCOOH}}]}_{o}} = {{W}_{{\max {\text{F}}{{{\text{c}}}^{ + }}}}}.$Схема 2 отличается от схемы 1 наличием равновесной стадии образования комплекса FcC+O · FcCOOH при избытке соединения III.
Схема 2
(2.1)
${\text{FcCOOH}} + {{{\text{H}}}^{ + }}\;\overset K \leftrightarrows \;{\text{Fc}}\mathop {\text{C}}\limits^ + {\text{O}} + {{{\text{H}}}_{2}}{\text{O,}}$(2.2)
${\text{Fc}}\mathop {\text{C}}\limits^ + {\text{O}} + {\text{FcCOOH}}\;\overset {{{K}_{a}}} \leftrightarrows \;{\text{Fc}}\mathop {\text{C}}\limits^ + {\text{O}} \cdot {\text{FcCOOH,}}$(2.3)
${\text{Fc}}\mathop {\text{C}}\limits^ + {\text{O}}\;\xrightarrow{{{{k}_{2}}}}\;{\text{F}}{{{\text{c}}}^{ + }}{\text{CO}},$При [FcCOOH]o$ \gg $ [H+]o выражение материального баланса по концентрации кислоты Н+ имеет вид:
(11)
$\begin{gathered} {{[{{{\text{H}}}^{ + }}]}_{o}} = [{{{\text{H}}}^{ + }}] + [{\text{Fc}}\mathop {\text{C}}\limits^ + {\text{O}}] + [{\text{Fc}}\mathop {\text{C}}\limits^ + {\text{O}} \cdot {\text{FcCOOH}}] = \\ = \;[{{{\text{H}}}^{ + }}] + [{\text{Fc}}\mathop {\text{C}}\limits^ + {\text{O}}] + {{K}_{a}}[{\text{Fc}}\mathop {\text{C}}\limits^ + {\text{O}}][{\text{FcCOOH}}]. \\ \end{gathered} $Из выражения для константы равновесия K можно найти выражение для равновесной концентрации FсC+O и далее скорость реакции:
(12)
$\begin{gathered} \text{[}{\text{Fc}}\mathop {\text{C}}\limits^ + {\text{O}}] = \\ = \frac{{K{{{[{{{\text{H}}}^{ + }}]}}_{o}}[{\text{FcCOOH}}]}}{{[{{{\text{H}}}_{2}}{\text{O}}] + K[{\text{FcCOOH}}] + K{{K}_{a}}{{{[{\text{FcCOOH}}]}}^{2}}}}, \\ \end{gathered} $(13)
${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}} = \frac{{{{k}_{2}}K{{{[{{{\text{H}}}^{ + }}]}}_{o}}[{\text{FcCOOH}}]}}{{[{{{\text{H}}}_{2}}{\text{O}}]\, + \,K[{\text{FcCOOH}}]\, + \,K{{K}_{a}}{{{[{\text{FcCOOH}}]}}^{2}}}}.$(14)
${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}} = \frac{{{{k}_{2}}K{{{[{{{\text{H}}}^{ + }}]}}_{o}}[{\text{FcCOOH}}]}}{{[{{{\text{H}}}_{2}}{\text{O}}] + K[{\text{FcCOOH}}]}}.$(15)
${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}} = \frac{{{{k}_{2}}{{{[{{{\text{H}}}^{ + }}]}}_{o}}}}{{1 + {{K}_{a}}[{\text{FcCOOH}}]}}.$Установленные зависимости для ${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}}$ от концентрации III аналогичны зависимостям для скорости ферментативной реакции от концентрации субстрата в условиях субстратного ингибирования (схема 3 ) [13].
Схема 3
Результатом субстратного ингибирования является экстремальная зависимость начальной скорости реакции от концентрации [S]0, описываемая уравнением:
(16)
$W = \frac{{\left( {{{k}_{2}} + \frac{{\beta {{k}_{2}}{{{[{\text{S}}]}}_{0}}}}{{{{K}_{s}}}}} \right){{{[{\text{E}}]}}_{0}}{{{[{\text{S}}]}}_{0}}}}{{K + {{{[{\text{S}}]}}_{0}} + \frac{{{{{[{\text{S}}]}}_{0}}}}{{{{K}_{s}}}}}},$Согласно этим данным, асимметричный вид экстремальной зависимости W = f([S]0) свидетельствует о том, что субстратный комплекс ES2 не полностью теряет ферментативную активность, а лишь становится менее реакционноспособным. Поскольку зависимость ${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}} = f({{{\text{[III}}]}_{0}})$ является асимметричной, то это указывает на то, что карбокатион в составе комплекса FcC+O · FcCOOH способен к редокс-изомерии, хотя и с меньшей скоростью. Если в схеме 2 учесть это обстоятельство, то, в соответствии с [13], она должна быть дополнена уравнением реакции:
(2.4)
${\text{Fc}}\mathop {\text{C}}\limits^ + {\text{O}} \cdot {\text{FcCOOH}}\;\xrightarrow{{\beta {{k}_{2}}}}\;{\text{F}}{{{\text{c}}}^{ + }}{\dot {C}O} + {\text{FcCOOH}}{\text{.}}$(17)
${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}}\, = \,\frac{{({{k}_{2}}\, + \,\beta {{k}_{2}}{{K}_{a}}[{\text{FcCOOH}}])K{{{[{{{\text{H}}}^{ + }}]}}_{0}}[{\text{FcCOOH}}]}}{{[{{{\text{H}}}_{2}}{\text{O}}]\, + \,K[{\text{FcCOOH}}]\, + \,K{{K}_{a}}{{{[{\text{FcCOOH}}]}}^{2}}}}{\kern 1pt} .$Основное отличие в протонировании I и III связано с возможностью фрагментации ацилиевого иона FcCH2C+O, что уже отмечалось выше. Это позволяет считать, что основной канал образования катиона ферроцения при протонировании I связан с редокс-изомерией ${\text{FcCH}}_{2}^{ + }$, и исключить из рассмотрения редокс-изомерию ацилиевого иона (схема 4 ). Не исключено, что редокс-изомерия карбокатиона FcCH2C+O вообще затруднена вследствие удаленности вакантной р-орбитали карбокатиона от высших заполненных d-орбиталей металлокомплекса, локализованных на атоме железа.
Схема 4
(4.1)
${\text{FcC}}{{{\text{H}}}_{2}}{\text{COOH}} + {{{\text{H}}}^{ + }}\;\overset K \leftrightarrows \;{\text{FcC}}{{{\text{H}}}_{2}}{{{\text{C}}}^{ + }}{\text{O}} + {{{\text{H}}}_{2}}{\text{O,}}$(4.2)
${\text{FcC}}{{{\text{H}}}_{2}}{{{\text{C}}}^{ + }}{\text{O}}\;\xrightarrow{{{{k}_{2}}}}\;{\text{CO}} + {\text{Fc}}{{{\text{C}}}^{ + }}{{{\text{H}}}_{2}}\;\xrightarrow{{{{k}_{3}}}}\;{\text{F}}{{{\text{c}}}^{ + }}{\dot {C}}{{{\text{H}}}_{2}},$(18)
${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}} = {{k}_{3}}[{\text{Fc}}\mathop {\text{C}}\limits^ + {{{\text{H}}}_{2}}].$Примем, что в условиях, когда [H+]0 > > [FcCH2COOH]0,
(19)
$\begin{gathered} {{[{\text{FcC}}{{{\text{H}}}_{2}}{\text{COOH}}]}_{0}} = \\ = \;[{\text{FcC}}{{{\text{H}}}_{2}}{\text{COOH}}] + [{\text{FcC}}{{{\text{H}}}_{2}}{{{\text{C}}}^{ + }}{\text{O}}] + [{\text{Fc}}{{{\text{C}}}^{ + }}{{{\text{H}}}_{2}}]. \\ \end{gathered} $(20)
$\frac{{d[{\text{Fc}}{{{\text{C}}}^{ + }}{{{\text{H}}}_{2}}]}}{{dt}} = {{k}_{2}}[{\text{FcC}}{{{\text{H}}}_{2}}{{{\text{C}}}^{ + }}{\text{O}}] - {{k}_{3}}[{\text{Fc}}{{{\text{C}}}^{ + }}{{{\text{H}}}_{2}}] = 0,$(21)
$[{\text{FcC}}{{{\text{H}}}_{2}}{{{\text{C}}}^{ + }}{\text{O}}] = \frac{{{{{[{\text{FcC}}{{{\text{H}}}_{2}}{\text{COOH}}]}}_{0}} - [{\text{Fc}}{{{\text{C}}}^{ + }}{{{\text{H}}}_{2}}]}}{{\frac{{[{{{\text{H}}}_{2}}{\text{O}}]}}{{[{{{\text{H}}}^{ + }}]K}} + 1}}.$Подставляя соотношение (21) в выражение (20), найдем концентрацию [FcC+H2] и далее скорость реакции:
(22)
$[{\text{Fc}}{{{\text{C}}}^{ + }}{{{\text{H}}}_{2}}] = \frac{{\frac{{{{k}_{2}}}}{{{{k}_{2}} + {{k}_{3}}}}K{{{[{\text{FcC}}{{{\text{H}}}_{2}}{\text{COOH}}]}}_{0}}[{{{\text{H}}}^{ + }}]}}{{\frac{{{{k}_{2}}}}{{{{k}_{2}} + {{k}_{3}}}}[{{{\text{H}}}_{2}}{\text{O}}] + K[{{{\text{H}}}^{ + }}]}},$(23)
${{W}_{{{\text{F}}{{{\text{c}}}^{ + }}}}} = \frac{{\frac{{{{k}_{3}}{{k}_{2}}}}{{{{k}_{2}} + {{k}_{3}}}}K{{{[{\text{FcC}}{{{\text{H}}}_{2}}{\text{COOH}}]}}_{0}}[{{{\text{H}}}^{ + }}]}}{{\frac{{{{k}_{2}}}}{{{{k}_{2}} + {{k}_{3}}}}[{{{\text{H}}}_{2}}{\text{O}}] + K[{{{\text{H}}}^{ + }}]}}.$Список литературы
Бетел Д., Голд В. Карбониевые ионы. М.: Мир, 1970. С. 344.
Методы элементоорганической химии. Железоорганические соединения / Под редакцией А.Н. Несмеянова, К.А. Кочеткова. М.: Наука, 1983. С. 544.
Rinehart K.L., Jr., Michejda C.J., Kittle P.A. // J. Amer. Chem. Soc. 1959. V. 81. P. 3162. https://doi.org/10.1021/ja01521a082
Weinmayr V. // Ibid. 1955. V. 77. P. 3009. https://doi.org/10.1021/ja01616a026
Фомин В.М., Зайцева К.С., Шарова М.Н., Климова М.Н. // Журн. общ. химии. 2016. Т. 86. Вып. 5. С. 815.
Фомин В.М., Кочеткова Е.С., Ключевский К.В. // Там же. 2018. Т. 88. Вып. 5. С. 805.
Фомин В.М., Широков А.Е. // Там же. 2009. Т. 89. Вып. 5. С. 1782. https://doi.org/10.1134/S1070363209050119
Фомин В.М., Широков А.Е. // Журн. общ. химии. 2008. Т. 78. Вып. 7. С. 1125.
Фомин В.М., Широков А.Е. // Там же. 2012. Т. 82. № 6. С. 921.
Fomin V.M., Orlova E.A., Zaitseva K.S. // Russ. Y. Gen. Chem. 2014. V. 84. № 4. P. 645. https://doi.org/10.1134/S1070363214040215
Несмеянов А.Н., Шульпин Г.Б., Рыбинская М.Г., Петровский П.В. // Докл. АН СССР. 1974. Т. 215. № 3. С. 599.
Матье Ж., Панико Р. Курс теоретических основ органической химии. М.: Мир. 1975. С. 528.
Березин И.В., Мартинек К. Основы физической химии ферментативного катализа. М.: Высш. школа, 1977. С. 235.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии