

Журнал физической химии, 2020, T. 94, № 8, стр. 1202-1205
Реакционная способность ароматических и алифатических нитросоединений в триплетном состоянии по отношению к аминам
Д. В. Овсянников a, *, С. В. Зеленцов a
a Национальный исследовательский Нижегородский государственный университет им. Н.И. Лобачевского
603950 Нижний Новгород, Россия
* E-mail: ovsyannikov@chem.unn.ru
Поступила в редакцию 30.09.2019
После доработки 30.09.2019
Принята к публикации 15.10.2019
Аннотация
Методами квантовой химии проведено моделирование реакций фотохимического переноса атомов водорода от молекул аминов к молекулам нитросоединений. В качестве акцепторов атомов водорода использованы нитрометан и нитробензол в триплетном состоянии, в качестве доноров атома водорода – аммиак, метиламин и диметиламин. Найдены структуры реакционных комплексов исходных веществ, переходных состояний и продуктов реакции, энергии активации реакций и изменения стандартных энтальпий реакций. Смоделированы профили поверхностей потенциальной энергии некоторых реакций. Установлено, что все профили достаточно ровные, а энергии активации имеют низкие значения. Распределение электронной плотности в комплексах изучено методом теории Бейдера. Все комплексы имеют характерную кольцевую структуру, что можно считать наличием дополнительного взаимодействия между амином и нитрогруппой в ходе реакции. Предположено, что подобная кольцевая структура – причина низких энергий активации реакций.
Реакция отрыва атома водорода широко известна в фотохимии кетонов, хинонов и нитросоединений [1–5]. Служить донорами водорода могут различные классы химических соединений. Основная особенность подобных реакций – акцептор водорода находится в возбужденном состоянии, чаще всего, в триплетном электронном состоянии. Благодаря близкому расположению энергетических уровней в молекулах нитросоединений очень сильно возрастает вероятность интеркомбинационной конверсии этих молекул [4]. В частности, 1-нитронафталин и 9-нитроантрацен имеют близко расположенные уровни S1 и T2, T3, T4 [4]. Такая конфигурация резко повышает вероятность синглет-триплетного перехода. Известно, что для нитробензола квантовый выход триплетного состояния составляет 0.67 [1]. Теоретические исследования нитробензола методом CASPT2 также показывают наличие близко расположенных уровней различной мультиплетности [6]. Между уровнями S0 и S1 расположены уровни T1, T2, T3 (причем различие между уровнями S1 и T2 составляет всего около 0.1 эВ). При синглет-триплетных переходах в нитросоединениях меняется геометрия. В частности, сообщалось о том, что в нитробензоле нитрогруппа покидает плоскость бензольного кольца в триплетном состоянии [7].
Кроме того, известно, что п-нитроанилин способен инициировать фотополимеризацию акриловых мономеров [8]. Процесс сопровождается эмиссией, отнесенной к триплетному нитросоединению. Интенсивность испускания зависит от концентрации вещества, что подтверждает участие в процессе триплетных частиц [9]. В зависимости от наличия или отсутствия кислорода в системе реакция протекает по разным маршрутам. Аналогичные результаты получены для п‑N,N-диметиламинобензальдегида. Процесс полимеризации развивается очень быстро, и система достигает максимальной конверсии за ∼10 мин в присутствии соединений, содержащих тяжелые атомы [10, 11].
Наиболее широкое исследование фотохимии нитросоединений проведено Гёрнером и сотр.; в результате их работы сделан вывод о том, что перенос атома водорода происходит в две стадии: перед переносом иона водорода должна быть стадия переноса электрона [12–14]. Пик УФ-спектра в области 400–410 нм Гёрнер относит к анион-радикалу нитросоединения. Полоса спектра в области 380 нм отнесена к ArN · O2H. Особое внимание Гёрнер и коллеги уделяют влиянию кислорода и растворителя на ход реакции [12]. В случае, если реакция проводится при насыщении кислородом, то наблюдается тушение триплетного состояния. Образуется синглетный кислород. При этом квантовый выход реакции фоторазложения уменьшается в сотни раз.
Существует альтернативная точка зрения на данную проблему. К примеру, в работе [15] предлагается механизм одновременного переноса атома водорода и заряда. Выводы сделаны на основании времен жизни триплетных состояний.
Цель данной работы – квантово-химическое исследование фотохимического переноса атома водорода в системе нитросоединение–амин. Авторы считают, что реакция должна протекать по более простому механизму – совместному переносу водорода и заряда.
МЕТОДОЛОГИЧЕСКАЯ ЧАСТЬ
Использовали метод B3LYP-D/cc-pVDZ из программного пакета NWChem-6.8 [16] и метод DF-OMP2/cc-pVDZ-RI из программного пакета Psi4 [17]. Приближение Resolution of Identity использовали в целях экономии времени вычисления. Визуализацию результатов проводили при помощи программы Pymol [18], для анализа электронной плотности в рамках теории Бейдера использовали программу Multiwfn [19].
В качестве нитросоединений использовали нитрометан и нитробензол – наиболее простые представители класса, в качестве аминов – аммиак, метиламин и диметиламин.
Оптимизацию геометрий комплексов, участвующих в реакциях, проводили стандартным методом (RFO). Для каждой из оптимизированных геометрий вычисляли частоты колебаний. Для переходного комплекса полагали геометрию, которой соответствует одна колебательная мода вдоль координаты реакции (“отрицательная частота”). Частоты колебаний ниже 100 см–1 не учитывали. К абсолютной энергии комплексов делали поправку на нулевые колебания (ZPE).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Мы считаем, что реакция фотохимического переноса водорода происходит по следующей схеме:
возбуждение нитросоединения
интеркомбинационная конверсия нитросоединения
образование предреакционного комплекса
(3)
$^{3}{\text{RN}}{{{\text{O}}}_{2}} + {\text{NHRR}}{\kern 1pt} ' \to {}^{3}\{ {\text{RN}}{{{\text{O}}}_{2}} + {\text{NHRR}}{\kern 1pt} '\} ,$перенос атома водорода
(4)
$^{3}\{ {\text{RN}}{{{\text{O}}}_{2}} + {\text{NHRR}}{\kern 1pt} {\text{'}}{\kern 1pt} \} \to {}^{3}\{ {\text{RNO}}_{2}^{ \bullet }{\text{H}}\; + \;\centerdot {\text{NRR}}{\kern 1pt} {\text{'}}{\kern 1pt} \} ,$распад постреакционного комплекса
(5)
$^{3}\{ {\text{RNO}}_{2}^{ \bullet }{\text{H}}\; + \;\centerdot {\text{NRR}}{\kern 1pt} {\text{'}}{\kern 1pt} \} \to {\text{RNO}}_{2}^{ \bullet }{\text{H}} + \,\,\centerdot {\text{NRR}}{\kern 1pt} {\text{'}}{\kern 1pt} .$Перенос водорода с участием метиламина и диметиламина происходит по водороду аминогруппы. Кроме реакции отрыва атома водорода от атома азота может наблюдаться отрыв атома водорода от атомов углерода метильных групп аминов (в метиламине и диметиламине). Перенос от метильной группы рассматривался ранее в работе [20]. Изменение стандартных энтальпий реакции при температуре 298.15 K приведены в табл. 1. Результаты получены методами B3LYP-D/cc-pVDZ и DF-OMP2/cc-pVDZ-RI. Несмотря на большие различия, общим является то, что реакции с аммиаком – эндотермические, остальные реакции – экзотермические.
Таблица 1.
Изменение стандартных энтальпий исследуемых реакций при температуре 298.15 K, вычисленных методами B3LYP/cc-pVDZ DF-OMP2/cc-pVDZ-RI, кДж моль–1
| Реагент | CH3NO2 | C6H5NO2 | ||
|---|---|---|---|---|
| B3LYP | OMP2 | B3LYP | OMP2 | |
| NH3 | 14.35 | 28.37 | 8.95 | 23.47 |
| CH3NH2 | –19.45 | 0.88 | –24.89 | –4.06 |
| (CH3)2NH | –42.76 | –18.95 | –48.16 | –23.85 |
В табл. 2 приведены величины энергий активации реакций между исследуемыми аминами и нитросоединениями в триплетном состоянии, вычисленные методами B3LYP-D/cc-pVDZ и DF-OMP2/cc-pVDZ-RI. Использование метода DF-OMP2/cc-pVDZ-RI к отрицательным значениям энергии активации, в отличие от B3LYP-D/cc-pVDZ, не приводит.
Таблица 2.
Энергии активации реакций указанных веществ, вычисленные методами B3LYP/cc-pVDZ и DF-OMP2/cc-pVDZ, кДж моль–1
| Реагент | CH3NO2 | C6H5NO2 | ||
|---|---|---|---|---|
| B3LYP | OMP2 | B3LYP | OMP2 | |
| NH3 | –0.42 | 41.61 | –3.26 | 7.53 |
| CH3NH2 | –6.31 | 26.31 | –7.33 | 2.50 |
| (CH3)2NH | 4.55 | 29.13 | –27.00 | 1.05 |
Известно [9], что метод B3LYP имеет тенденцию неверно передавать абсолютные высоты барьеров активации реакций переноса водорода. Мы считаем, что этот эффект возникает при удлинении связей, а также при разрыве и образовании новых связей, как и в исследуемой системе. Известно [21], что феномен отрицательных энергий активации часто имеет место при переносе атома водорода в системах, содержащих амины. В работах многих авторов продемонстрировано, что метод B3LYP сильно занижает величины энергий активации реакций переноса водорода [21–25].
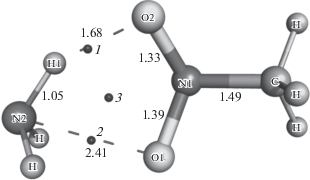
Таким образом, установлено, что, несмотря на поправку Гримме, метод B3LYP занижает энергии активации исследуемых реакций. Мы считаем, что это происходит вследствие сложности системы нитросоединение – амин. Данный вывод можно сделать, если рассмотреть переходные комплексы реакций методом Бейдера [26]. Метод позволяет локализовать критические точки электронной плотности в молекуле. Эти точки классифицируются следующим образом: (3, –3) – точка атома, (3, –1) – точка связи, (3, +1) – кольцевая точка. Последний тип возникает в случае, если образуется “провал” электронной плотности. Пример такой конфигурации – центр бензольного кольца. На рис. 1 изображена топология предреакционного комплекса нитрометана с аммиаком. Точка 1 является точкой связи типа (3, –1). Она соединяет реагирующие молекулы в предреакционный комплекс. Точка 2 возникает между атомом азота аммиака N2 и кислородом нитрогруппы O1. Расстояние между ними составляет 2.41 Å. Благодаря этой точке между молекулами возникает кольцевая точка 3 типа (3, +1). Точки 2 и 3 – свидетельство дополнительного взаимодействия между молекулами. Поэтому барьеры активации оказываются очень низкими.
Остается открытым вопрос об отрицательных барьерах активации, предсказанных методами DFT. Были восстановлены некоторые профили ППЭ реакций с помощью метода B3LYP-D/cc-pVDZ по алгоритму Intrinsic Reaction Coordinate. В случае абсолютных энергий без добавки поправки на нулевые колебания, высота барьеров для всех реакций оказывается положительной. Активация реакции аммиака с нитрометаном происходит при 44.93 кДж моль–1, аммиака с нитробензолом – при 6.02 кДж моль–1. Эти данные очень близки к полученным методом DF-OMP2/cc-pVDZ-RI. Однако, вычисление энергий в полученных точках с добавкой поправки на нулевые колебания приводит к отрицательным барьерам активации. Таким образом, причина появления отрицательных барьеров активации – поправка на нулевые колебания.
Таким образом, в результате проведения квантово-химических вычислений кинетических и термодинамических параметров реакций переноса водорода нитросоединений с аминами в триплетном состоянии установлено, что метод B3LYP-D/cc-pVDZ дает ненадежные результаты, слабо согласующиеся с методом DF-OMP2/cc-pVDZ-RI. Особенно следует отметить, что восстановление профилей ППЭ при помощи алгоритма IRC дает адекватные результаты для метода B3LYP-D. Электронная плотность предреакционного комплекса нитрометана с аммиаком изучена методом Бейдера. Установлено наличие кольцевой структуры, которой соответствует критическая точка типа (3, +1).
Результаты экспериментов получены с использованием узла кластера, содержащего два восьмиядерных процессора Intel Sandy Bridge E5-2660 2.2 GHz, 64GB RAM, работающего под управлением ОС Linux CentOS 7.2 [27].
Список литературы
Trotter W., Testa A.C. // J. Amer. Chem. Soc. 1968. V. 90. № 25. P. 7044.
Rehorek D., Janzen E.G. // J. Photochemistry. 1986. V. 35. № 2. P. 251.
Sundararajan K., Ramakrishnan V., Kuriacose J.C. // Ibid. 1984. V. 27. № 1. P. 61.
Ohtani H. и дp. // Bull. Chem. Soc. Japan. 1980. V. 53. № 1. P. 43.
Hurley R., Testa A.C. // J. Amer. Chem. Soc. 1968. V. 90. № 8. P. 1949.
Giussani A., Worth G.A. // J. Chem. Theory and Comp. 2017. V. 13. № 6. P. 2777.
Arenas J.F. et al. // J. Org. Chem. 2006. V. 71. № 3. P. 983.
Costela A. et al. // J. Photochem. Photobiol. A. 1997. V. 109. № 1. P. 77.
Costela A. et al. // J. Polym. Sci., Part A: Polym. Chem. 1997. V. 35. № 17. P. 3801.
Zelentsov S.V. et al. // High Energy Chemistry. 2004. V. 38. № 2. P. 115.
Zelentsov S.V., Simdyanov I.V., Kuznetsov M.V. // High Energy Chemistry. 2005. V. 39. № 5. P. 309.
Görner H. // J. Chem. Soc.Perkin Trans. II. 2002. № 10. P. 1778.
Görner H. // J. Photochem. Photobiol., A. 2008. V. 195. № 3. P. 235.
Biczók L., Görner H. // Chem. Phys. 2012. V. 392. № 1. P. 10.
Abd El Latif F.M. et al. // J. Photochem. Photobiol., A. 1999. V. 121. № 2. P. 111.
Valiev M. et al. // Comput. Phys. Commun. 2010. V. 181. № 9. P. 1477.
Parrish R.M. et al. // Journal of Chemical Theory and Computation. 2017. V. 13. № 7. P. 3185.
Schrödinger L. Pymol. 2015.
Lu T., Chen F. // J. Comput. Chem. 2012. V. 33. № 5. P. 580.
Овсянников Д.В., Зеленцов С.В. // Химия высоких энергий. 2019. Т. 53. № 2. С. 95.
Diamanti A. et al. // Ind. Eng. Chem. Res. 2017. V. 56. № 4. P. 815.
Joly J.-F., Miller R.E. // Ibid. 2018. V. 57. № 3. P. 876.
Kobayashi Y., Kamiya M., Hirao K. // Chem. Phys. Lett. 2000. V. 319. № 5–6. P. 695.
Lynch B.J., Truhlar D.G. // J. Phys. Chem. A. 2001. V. 105. № 13. P. 2936.
Jursic B.S. // THEOCHEM. 1998. V. 428. № 1–3. P. 49.
Bader R.F.W. Atoms in Molecules: A Quantum Theory: International Series of Monographs on Chemistry. Atoms in Molecules. Oxford, New York: Oxford University Press, 1994. 458 c.
Суперкомпьютер ННГУ “Лобачевский”: http://hpc-educ.unn.ru/ru/ресурсы
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии