

Журнал физической химии, 2020, T. 94, № 8, стр. 1269-1278
Фениламинильные катионы в жидко- и газофазных фотопревращениях протонированного азобензола
Ю. А. Михеев a, *, Ю. А. Ершов b
a Российская академия наук, Институт биохимической физики им. Н.М. Эмануэля
Москва, Россия
b Московский государственный технический университет им. Н.Э. Баумана
Москва, Россия
* E-mail: mik@sky.chph.ras.ru
Поступила в редакцию 20.09.2019
После доработки 20.09.2019
Принята к публикации 12.11.2019
Аннотация
Проведен анализ данных по спектроскопии фотовозбуждения транс → цис-изомеризации и фрагментации протонированного транс-азобензола (ABH+) в атмосфере инертных газов. Результаты газофазной изомеризации сопоставлены с результатами жидкофазной реакции в водной серной кислоте, ведущей к образованию циклического бензо[c]циннолина. Циклизация отсутствует при газофазной t → c-изомеризации в среде разреженного азота при T = 300 K вследствие высокой скорости обратной c → t-изомеризации. Показано, что превращения в обеих фазах идут с участием катионов фениламинильного типа, являющихся хромогенами t- и c-ABH+. Фотофрагментация t-ABH+ в атмосфере криогенно охлажденного гелия (T = 40 K) идет с образованием осколочных катионов фенила и фенилдиазония тоже с участием фениламинильных катионов. Предложены механизмы фотофрагментации, включающие промежуточные акты с участием атомов H, образующихся путем диссоциации группы NH. Быстрая атака атомов H по фенильному кольцу, связанному с NH-группой, ведет к разрывам C–N-связи, исключая возможность t → c-изомеризации в криогенной атмосфере гелия.
Азобензол (AB) не относится к технологическим красителям, однако является химическим базисом азобензольных красителей и важен как объект научных исследований по взаимосвязи химического строения и цветности органических соединений [1]. Он и его замещенные соединения способны обратимо претерпевать t ↔ c-изомеризацию под действием UV- и VIS-света, причем с ультравысокой скоростью в субпикосекундном режиме. В литературе это рассматривается как важное свойство в плане применения замещенных соединений AB в качестве молекулярных фотопереключателей (в том числе в биологических зондах), в механических и оптических материалах, фотофармакологии [2].
Актуальность научного исследования фотореакций производных t-AB стимулировала авторов [2, 3] на проведение экспериментов с протонированным t-азобензолом (t-ABH+) в газовой фазе. Важность полученных экспериментальных результатов [2, 3] существенно нивелируется теоретической трактовкой, основанной на устаревших представлениях о природе цветности (и электронного строения) AB и его производных. Целью настоящей работы является качественно новый анализ данных [2, 3] с учетом того, что ключевым хромогеном азобензола является не катион азония, а катион фениламинильного типа (PhAT) [4–9].
Авторы [2] использовали методику разделения исходных транс- и образующихся при фотовоздействии на t-ABH+цис-катионов ABH+, основанную на разной скорости дрейфа катионов в аппаратурной трубке через зону разреженного буферного газа N2. Дрейфующие катионы создавали, распыляя совместный раствор t-ABH+ (10–5 М) и уксусной кислоты (1%) в метаноле с помощью электрического распылителя (electrospray source) и инжектируя порции пылевидной смеси в зону дрейфа импульсами длительностью 100 мкс с частотой 20 Гц. Движение таких порций (packets) через зону дрейфа обеспечивалось электрическим полем (E = 44 В/см), причем на половине пути дрейфа эти порции подвергали воздействию монохроматического излучения с регулируемой длиной волны в интервале 30–520 нм. Импульсы излучения имели энергию светового потока ≈1 мДж/(см2 импульс). С целью получения разностных масс-спектров, импульсы повторялись с частотой 10 Гц, облучая каждый второй катионный пакет. Сепарирование порций по скорости дрейфа шло вследствие различия величины поперечного сечения катионов в актах соударения с молекулами буферного газа N2 (≈6 Toрр). Результат такого различия в уловиях фотовозбуждения светом с λex = 440 нм однозначно показал, что облучение катионов t-ABH+ приводит к образованию более быстрых катионов c-ABH+.
Экспериментальная установка [2] позволяла разделять дрейфующие порции катионов обоих типов, регулировать время подачи на них импульсов облучения и регистрировать время их подсчета (arrival time distribution, ATD). Были получены разностные спектры ATD и, кроме того, спектр фотовозбуждения t-ABH+ по зависимости интегрального сигнала ATD быстрых катионов c-ABH+ от длины волны возбуждающего света (спектр фотоизомеризационного действия (PISA)).
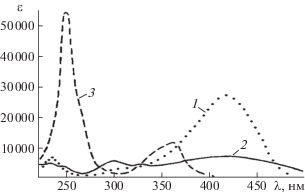
Спектр PISA (полоса в интервале 370–480 нм с максимумом при λm = 430 нм) представлен на рис. 1 в виде кривой 1, характеризующей зависимость выхода катионов с-ABH+ (в произвольных единицах Y). Спектр приписан π → π*-переходу S0 → S1 в катионе азония:
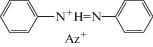
Рис. 1.
Спектр возбуждения фотоизомеризации транс-азобензола в разреженной атмосфере азота. Вертикальные полосы обозначают расположение и интенсивность расчетных электронных уровней S1, S2, S3. Данные [2]. Дополнительные пояснения в тексте.
На рис. 1 авторы [2] нанесли также вертикальные линии S1, S2, S3, характеризующие длины волн и относительные интенсивности вертикальных электронных переходов, рассчитанные для Az+ по аутентичным компьютерным методикам.
Согласно данным [2], выход с-ABH+ при действии импульсов света с λex = 440 нм на t-ABH+ составил ≈85%. Потеря 15% приписана расходованию t-ABH+ в актах фотофрагментации. Фотофрагментация t-ABH+ в газовой фазе установлена в [3].
Авторы [3] исследовали фотопревращение t‑ABH+ в газовой фазе, тоже опираясь на традиционный постулат, что спектр фотовозбуждения t-ABH+ принадлежит катионам азония Az+. В отличие от работы [2], процесс проводили при низкой температуре ≈40 K охлажденного гелия, регистрируя образование продуктов фрагментации с помощью времяпролетного (TOF) масс-спектрометра (спектроскопия фотофрагментационого действия, PFA).
Катионы t-ABH+ продуцировали в электрораспылительном источнике из раствора t-AB (концентрация 0.1 мМ в смеси вода/метанол = 1 : 1) с небольшим количеством уксусной кислоты. Созданные в источнике катионы инжектировали в ловушку, охлажденную импульсом криогенного гелия. На охлаждение катионов в ловушке до Т = = 40 K уходило несколько десятков миллисекунд, после чего подавался лазерный импульс для фотофрагментации катионов t-ABH+. Устройство экспериментальной установки позволяло экстрагировать ионные фрагменты и оставшиеся катионы t-ABH+ из ловушки и вести анализ в TOF масс-спектрометре. Анализ показал, что главными продуктами фотофрагментации катионов t‑ABH+ (отношение масса/заряд – m/q = 183) являются фенильные катионы (Ph+) c m/q = 77, и катионы фенилдиазония (C6H5N$_{2}^{ + }$, benzenediazonium, BD+ c m/q = 105).
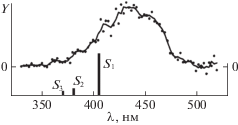
Спектры UV-VIS PFS катионов t-ABH+ представляют собой зависимость интенсивности сигналов катионов-фрагментов от волнового числа ${{\nu }_{{{\text{ex}}}}}$ возбуждающего лазерного излучения. При этом интенсивность сигнала BD+ много ниже, чем сигнала Ph+. Фотофрагментационные UV‑VIS PFS спектры катионов t-ABH+, представленные в [3] в произвольных единицах, приведены на рис. 2. Спектры 1, 2 (рис. 2) характеризуют зависимости выхода осколочных катионов Ph+ (m/q = 77) от волнового числа νex. Спектр 2 передает с увеличенным разрешением участок (выделен кружком) в самом начале полосы UV-PFS-спектра 1. Авторы [3] установили, что фотофрагментация катионов t-ABH+ на Ph+ в интервале 20 800–21 100 см–1 соответствует низкочастотной вибрационной прогрессии с активной частотой 41 см–1, начинающейся с νex = 20 820 см–1 и νex = = 20 842 см–1. Вместе с тем, на основании очень низкой интенсивности этой прогрессии отмечают, что полоса νex = 20 820 см–1 не соотносится в явном виде с электронным 0–0 переходом.
Рис. 2.
Спектры возбуждения фотофрагментации t‑ABH+, полученные по выходу катионов Ph+ c m/q = = 77 (кривые 1, 2) и фенилдиазония с m/q = 105 (кривая 3). Данные [3]. Дополнительные пояснения в тексте.
Отмеченную особенность авторы [3] связывают с значительным отличием геометрии возбужденного S1-состояния азоний катиона t-ABH+ от геометрии основного состояния. Их квантово-химические расчеты показали, что для основного состояния (S0) катиона азония характерна лишь слабая скрученность ароматических колец относительно азогруппы из-за стерических препятствий между кислотным атомом H и смежным фенильным кольцом. В то же время, для возбужденного S1-состояния расчеты показали большую скрученность азоний катиона по N–N-связи, достигающую 96° у диэдрального угла –CN–NC– (ситуация типа изомеризации из планарного состояния в кресловидное). Несмотря на это, начинающуюся с νex = 20 820 см–1 и имеющую νm ≈ ≈ 23 200 см–1 (≈430 нм) полосу (рис. 2, спектр 1), авторы [3] все же приписывают переходу S0 → S1. Последний рассматривается как переход электрона в азоний-катионе с высшей занятой связывающей молекулярной орбитали π-HOMO на антисвязывающую π*-LUMO с расчетной силой осциллятора f = 0.91. Учитывая сильное искажение геометрии азоний-катиона t-ABH+ в S1-состоянии, авторы [3] не исключают наличия t → c-изомеризации. Вместе с тем, подчеркивают, что выход цис-изомеров может быть очень низким из-за быстрой обратной изомеризации кресловидного состояния в планарное при движении возбужденных катионов ABH+ по координате реакции в зоне конического пересечения поверхности S1 с поверхностью S0.
Авторы [3] отмечают, что основное количество осколочных катионов BD+ (phN≡N:+, m/q = 105) образуется в интервале волновых чисел νex от 24 390 до 33 330 см–1 (рис. 2, спектр 3). Именно этим катионам они уделяют главное внимание при рассмотрении механизма фотофрагментации катионов t-ABH+ (конкретного механизма образования катионов Ph+ они не касаются).
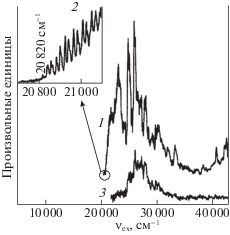
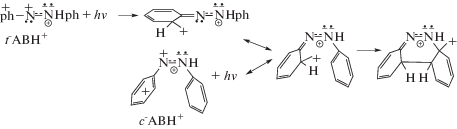
Наблюдаемые в указанном интервале νex на рис. 2 две полосы с близкими значениями νm = 24 534 (407 нм) и 25 648 см–1 (384 нм) отнесены в [3] к переходам с переносом заряда (CT) S0 → S2 (“HOMO – 1” → LUMO) и S0 → S3 (“HOMO – 2” → → LUMO) соответственно. Дана схема 1 актов фрагментации t-ABH+ → BD+ + бензол (B) в возбужденных состояниях S2 и S3:
Согласно схеме 1, фотовозбуждение катиона азония t-ABH+ вызывает перенос электрона с π-орбиталей колец 1, 2 на протонированный атом азота азогруппы. По мнению авторов [3], эти переходы, осуществляемые то с одного, то с другого кольца, приводят к нейтрализации положительного заряда на группе HN+ и последующим гомолитическим разрывам связи NH, миграции образующихся радикалов H к кольцам и, в конечном итоге, образованию бензола (B) и фенилдиазония (BD+). Считается, что именно планарное состояние катиона азония t-ABH+ способствует переносам π-электронов на пустую π-орбиталь атома N+, созданную присоединенным протоном.
Резюмируя обзор данных работ [2, 3] следует отметить, что использованные в них теоретические построения базировались на представлении о катионе азония как основного состояния сопряженной кислоты AB. Между тем, недавно показано, что формулы строения протонированных форм t-ABH+ включают в себя катионы PhAT [4], которые входят как важные хромогенные элементы также в молекулы AB и ридимеры азобензольных красителей [5–9].
Электронная формула катиона PhAT
Существование катиона PhAT установлено в работе [4]. Согласно [4], вносимый в азогруппу AB положительный заряд протона не фиксируется в том состоянии, как определяет формула азоний-катиона и как это представляют авторы [2, 3]. Так, согласно схеме 3 [3], электрон переносится с того или другого кольца на свободную π-орбиталь атома N+, получившего положительный заряд от присоединенного протона. Тем самым вносится путаница в суть реакции протонирования молекул t-AB. В действительности в ходе протонирования сначала один электрон переходит из неподеленной sp2-электронной пары азота молекулы AB на атомную s-орбиталь протона. Затем s-электрон образовавшегося атома водорода и оставшийся sp2-электрон азота образуют σ-связь. При этом положительный заряд протона, локализующийся на pz-орбитали азота образовавшейся группы HN, поляризует азосвязь. Адекватная электронная формула строения ABH+ имеет вид [4]:
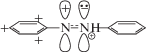
Согласно схеме 2, увеличение положительного заряда на азоте группы HN (отмечено знаком (+) под атомом N) вызывает притяжение обоих π-электронов π-связи так, что они находятся основное время на pz-орбитали этой группы. Таким образом, pz-орбиталь второго атома N, передающая электрон на pz-орбиталь группы NH, приобретает индуцированный положительный заряд и возникает π-катион N+. Этот π-катион N+ поляризует сопряженное с ним фенильное кольцо так, что π‑электроны кольца получают возможность делокализоваться из бензольного электронного секстета на π-катион N+ за счет сопряжения, частично повышая на нем электронную плотность и понижая его положительный заряд. При этом сохраняется sp2-гибридизация атомов азота и плоское строение t-ABH+.
Интенсивный желто-оранжевый цвет t-ABH+, отражающий наличие VIS-полосы с λmax ≈ 430–440 нм, обязан именно катиону PhAT (Ph+N+, схема 2), принимающему на себя роль хромогена подобно тому, как это происходит в бензильных и фениламинильных катионах [4]. Все указанные катионы имеют не только связывающие высшие молекулярные орбитали (HOMO) и нижние антисвязывающие (LAMO) молекулярные орбитали, но еще и вакантные несвязывающие орбитали (NBMО) с нулевой энергией. Именно на NBMO переходит возбуждаемый VIS-светом электрон с занятой HOМО, приводя к появлению в спектрах указанных катионов VIS-полос, сходных по форме и положению [4, 10, 11].
Отмеченное сходство электронных переходов позволило воспользоваться в [4] принципом аналогии с переходом бензильного катиона из основного состояния в возбужденное состояние, описанное в [12], для демонстрации механизма фотоциклизации t-ABH+ с участием образующегося при этом с-ABH+.
Жидкофазная фотоизомеризация с циклизацией t-ABH+
Согласно [12], переход бензильного катиона в первичное возбужденное состояние (соответствует переходу электрона с HOMO на NBMО) влечет за собой изомеризацию фенильного кольца в хиноидное кольцо с локализацией положительного заряда на атоме углерода либо в пара-, либо в орто-положениях кольца. При этом находящийся в кольце атом С, получающий полный положительный заряд, может изменить свое ароматическое валентное sp2-состояние на sp3-состояние:
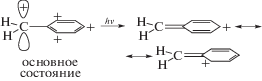
Учет такой ситуации при фотовозбуждении катионов t-ABH+ позволил в [4] объяснить механизм жидкофазной t → c фотоизомеризации катионов t-ABH+, которая, как установлено в [13, 14] ведет к образованию циклического протонированного катиона безо[c]циннолина. Выводы работы [4] позволяют лучше понять экспериментальные данные [2] по газофазной t → c фотоизомеризации катионов t-ABH+.
В жидкой фазе катионы t-ABH+ и c-ABH+ образуются в достаточно сильных кислотах и, согласно данным [13, 15], имеют неодинаковые UV-VIS спектры. Например, свежеприготовленный раствор c-ABH+ (растворитель – этанол/70% HClO4, 1 : 4 об.) отличается низкой интенсивностью VIS-полосы с λm = 418 нм (ε418 = 7460 л/(моль см)) от раствора t-ABH+ (λm = 421 нм, ε421 = 29 300 л/(моль см)) в том же растворителе [15]. Кроме того, c-ABH+ имеет низкую устойчивость и постепенно изомеризуется в t-ABH+. Изомеризация в указанном выше растворителе завершается быстро на открытом свету и медленно (много дней) в отсутствие освещения [15].
В сильно кислой среде 22 н. H2SO4 + 10% этанола реакция c → t изомеризации в отсутствие света заканчивается в течение нескольких дней [13]. В итоге, спектр цис-формы переходит в спектр транс-изомера. В то же время действие света лампы накаливания на растворы t-ABH+ и c-ABH+ в данной сернокислой среде вызывает их относительно быстрое обесцвечивание, сопровождающееся образованием протонированного бензо[c]циннолина [13, 14]. На начальном этапе освещения раствора t-ABH+ (концентрация 2 × × 10–5 М) оптическая плотность VIS-полосы (с λm = 420 нм) быстро снижается из-за частичного фотопревращения в c-ABH+.
На таком же начальном этапе освещения раствор c-ABH+ (тоже 2 × 10–5 М) резко увеличивает оптическую плотность вследствие частичного превращения в t-ABH+. В том и другом случае относительно быстро устанавливается фотодинамическое равновесие между обоими изомерами (их VIS-полосы лежат в одной и той же области спектра (рис. 3, кривые 1, 2)). При этом оптические плотности обоих растворов становятся почти равными за короткое время, и далее их снижение идет с одной скоростью. Одновременное относительно медленное расходование обоих изомеров приводит к образованию протонированного бензо[c]циннолина (рис. 3, кривая 3). Время полупревращения до фоторавновесия примерно в 100 раз меньше, чем время образования продукта циклизации.
Рис. 3.
Спектры протонированных транс-азобензола (1), цис-азобензола (2), бензо[c]циннолина (3). Данные [13].
Механизм данной фотореакции раскрыт в [4] с учетом функционирования катионов PhAT в t‑ABH+ и c-ABH+ (схема 3). Катионы PhAT способны в результате фотовозбуждения превращаться в орто-хиноидные структуры. Одновременно происходит сильное ослабление двойных связей N=N, обеспечивая вращение фенильного и хиноидного колец вокруг ослабленной азосвязи, а также способность орто-хиноидного карбкатиона атаковать незаряженное фенильное кольцо, реализуя реакцию циклизации:
Образующийся в качестве промежуточного соединения протонированный циклический диазин с двумя орто-хиноидными циклами затем окисляется серной кислотой с образованием протонированного бензо[c]циннолина (спектр 3, рис. 3):
При этом серная кислота восстанавливается до сернистой кислоты.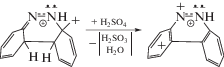
Согласно изложенному выше, традиционную (неадекватную) идею о принципиальной роли катионов азония в фотохимии протонированного азобензола следует заменить адекватной моделью с ключевой ролью катионов PhAT.
Особенности газофазной t → c фотоизомеризации
В [2] не зарегистрирован спектр PISA, соответствующий катиону c-ABH+ (а также циклическому катиону с двумя хиноидными кольцами (схема 3)). Данный факт объясняется высокой скоростью c → t-конверсии [2]. Его можно связать с весьма низкой плотностью газа N2 (8 Торр) и отсутствием клеточного эффекта. В жидкой фазе, благодаря клеточному эффекту [16], встречающиеся фенильное и заряженное орто-хиноидное кольца катионов ABH+ (схема 3), имеют возможность для многократных соударений, ведущих к циклизации по схеме 3. Наоборот, низкая плотность газовой среды N2 не способна оказать сильного торможения торсионному движению колец.
Следует отметить еще одну важную особенность газофазных опытов [2, 3], а именно, усиление основности AB относительно жидкофазной основности. Действительно, для обратимого протонирования (t-AB + H+ ↔ t-ABH+) в жидкой фазе необходима достаточно высокая кислотность [4, 5, 13, 15], которая недостижима с применением очень низкой концентрации уксусной кислоты, используемой в опытах [2, 3]. Между тем, эти опыты однозначно демонстрируют факт протонирования AB. Данный результат можно связать с удачным сочетанием электронного строения катиона t-ABH+, содержащего хромоген PhAT, и использованного в опытах электрораспылительного ионного источника (electrospray ion source).
В электрораспылителе происходит впрыскивание порций раствора через нагретый капилляр в условиях значительного перепада давления в объем с низким давлением ≈1 мБар, где происходит удаление растворителя. При движении в нагретом капилляре раствор находится под действием высоковольтного напряжения (несколько кВ), в результате чего образуются сильно заряженные капли [17, 18], распыляющиеся в объеме и теряющие растворитель. В условиях высокого напряжения образующаяся соль (ацетат AB), не способная к электролитической диссоциации в паровом состоянии, теряет анион ацетата. Это можно объяснить тем, что в катионе t-ABH+ группа NH практически не имеет заряда, так как положительный заряд делокализован в катионе PhAT при условии компенсации зарядов на атоме N (схема 2). По этой причине кулоновское притяжение аниона AcO– к группе NH сильно ослаблено и AcO– легко отщепляется под действием сильного электрического поля. Затем освободившиеся катионы t-ABH+ перемещаются в другой отсек установки с давлением около 10–3 мБар и далее направляются в устройство для фотовозбуждения и сепарирования катионов в разреженном азоте под действием электрического поля.
Особенности газофазной криогенной фотофрагментации
Ключевая роль катионов PhAT в электронно-оптических свойствах ABH+, AB и аминоазобензольных красителей [4–9], свидетельствует о неадекватности теоретических представлений работ [2, 3], базирующихся на модели азоний-катиона t-ABH+. Действительно, образование t-ABH+ идет с присоединением H+ к азоту азогруппы за счет использования пары sp2 электронов на образование ковалентной связи N–H. При этом π-связь азогруппы сильно поляризуется вследствие локализации π-электронов на азоте группы NH и связанной с этим практической компенсацией положительного заряда протона. По этой причине наличие катиона PhAT в t-ABH+ (схема 2) исключает актуальность схемы 1, представленной в [3]. Согласно схеме 1, фрагментация должна идти вслед за актами переноса электрона с того или иного фенильного кольца на pz-орбиталь азота, присоединившего протон (NH+). Между тем, данная pz-орбиталь в основном состоянии t-ABH+ занята двумя электронами (схема 2), и здесь нет места для третьего электрона.
В работе [3] не отмечен наблюдаемый на спектрах UV-VIS PFS (рис. 2, спектры 1, 3) факт, что акты образования фрагментов BD+ идут не только при возбуждении t‑ABH+ в CT-полосах S0 → S2 и S0 → S3, но и в полосе S0 → S1 (νm = 23 200 см–1, λm = 430 нм). Согласно рис. 2, переход S0 → S1 создает интенсивный сигнал m/q = 77 от катионов ph+ (спектр 1), а также значительно менее интенсивный, но все же наблюдаемый сигнал BD+ (спектр 3), который становится значительно сильнее в CT-полосах. То есть в [3] не учтен тот факт, что оба продукта фрагментации – Ph+ и BD+ – образуются параллельно в интервале νex от 21 000 до 33 330 см–1.
Перед обсуждением механизмов фрагментации следует отметить, что в спектре UV-VIS PFS полоса t‑ABH+, связанная с переходом S0 → S1 (рис. 2, спектр 1), имеет λm = 430 нм, как в спектре PISA (рис. 1). Это батохромное смещение в газовой фазе относительно жидкофазной полосы λm = = 420 нм (рис. 3, спектр 1) свидетельствует о наличии достаточно высокой колебательной энергии у катионов t-ABH+, вступающих в фотоизомеризацию при T ≈ 300 K [2] и фрагментацию при T ≈ 40 K [3]. Здесь дело в том, что поглощаемая радиация обладает двойственной ролью, наиболее выраженной в опытах работы [3].
В работе [3] для создания температуры ниже 70 K используют холодный гелий с давлением 0.1–1.0 мТорр. В такой криогенной атмосфере гелия фотовозбуждаемые примесные молекулы испытывают резонансное рамановское рассеяние и получают при переходе в основное состояние сильное колебательное возбуждение, которое теряется значительно медленнее, чем в разреженном азоте [18]. Это позволяет понять тот факт, что UV-VIS PFS-спектр осколочных катионов Ph+ в криогенной гелиевой ловушке [3] на начальном участке полосы S0 → S1 в (рис. 2, интервал 20 800–21 100 см–1) носит характер низкочастотной вибрационной прогрессии с активной частотой 41 см–1. Наблюдаемую прогрессию можно связать с наличием резонансного рамановского рассеяния, индуцирующего высокую скорость генерирования горячих колебательных $S_{0}^{{\text{v}}}$ состояний t‑ABH+. (Для сопоставления отметим, что даже в жидком гексане молекулы AB, перешедшие на $S_{0}^{{\text{v}}}$-уровень с уровня S1, обладают рамановскими антистоксовскими полосами ν = 1440 см–1, имеющими время жизни ∼16 пс [19].) С учетом того, что время акта рассеяния света (порядка фемтасекунды) значительно короче времени лазерного импульса, следует ожидать, что каждый лазерный импульс в [3] (длительность наносекунды) играл двойственную роль, особенно заметную в начале полосы перехода S0 → S1. Действительно, он генерировал $S_{0}^{{\text{v}}}$ состояния t-ABH+ за счет резонансного рамановского рассеяния и сам же зондировал их по полосам UV-VIS PFS осколочных катионов Ph+ (схема “pump + probe ← puls”).
Ранее двойственная роль фотовозбуждения отмечена для протонированных аминоазобензола и диметиламиноазобензола [20]. Действие света на эти соединения вызывает, с одной стороны, акты их депротонирования с образованием хиноидных форм, с другой стороны, свидетельствует об этих актах по интенсивным рамановским полосам образующихся хиноидных форм.
Наличием значительного колебательного возбуждения у катионов t-ABH+ в $S_{0}^{{\text{v}}}$-состоянии можно объяснить также и батохромное смещение спектральной полосы PISA (λm = 430 нм, рис. 1) относительно жидкофазной полосы (λm = 420 нм, рис. 3, спектр 1). Не исключено также, что образующиеся из t-ABH+ катионы с-ABH+ тоже сохраняют колебательное возбуждение, увеличивающее скорость c → t релаксации в разреженном азоте в опытах [2].
Механизм фрагментации в полосе S0 → S1
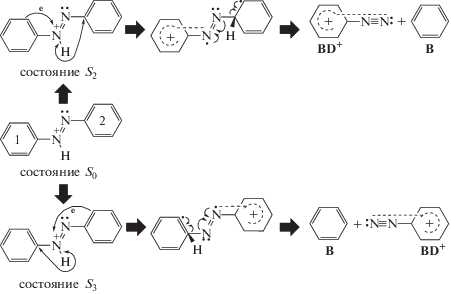
Хромогеном, ответственным за поглощение света в полосе S0 → S1 с νm ≈ 23 200 см–1 (≈430 нм) в t-ABH+ является катион PhAT [4]. Это в принципе исключает актуальность схемы 1. Адекватному механизму образования фенил-катионов Ph+ при переходе S0 → S1 соответствует схема 4:
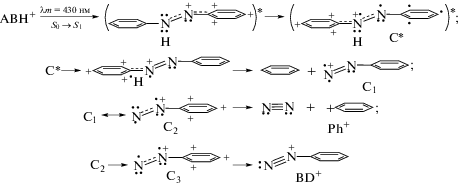
Согласно схеме 4, переход S0 → S1 в ABH+ обусловлен возбуждением катиона PhAT путем перехода электрона с занятой HOМО на NBMO, что создает вакансию на орбитали HOМО, и эта орбиталь становится акцептором для pz-электрона азогруппы HN••. Протекающий после фотовозбуждения неоптический перенос электрона с HN•• изменяет поляризацию ABH+, создавая франк-кондоновское (FK) состояние С* с катионом PhAT на противоположном кольце ABH+. Места бывшего положительного заряда в FK-С* занимает распределенный по азоту и фенильному кольцу акцептированный электрон. Неоптический перенос электрона реализуется в моменты расположения колец в одной плоскости, необходимой для сопряжения pz-орбиталей ослабленной осциллирующей азосвязи.
Дальнейшее превращение состояния FK-С* идет с восстановлением азосвязи N=N путем рекомбинации ее π-электронов, оно сопровождается выделением энергии и элиминированием свободного радикала ${{{\text{H}}}^{ \bullet }}$. Радикал ${{{\text{H}}}^{ \bullet }}$ атакует соседнее фенильное кольцо по углероду с нулевым дефицитом электронной плотности, замещая в нем фрагмент С1, который затем изомеризуется в C2.
Состояние C2 превращается двумя путями, распадаясь в одном из них на молекулу азота N2 и фенил-катион Ph+. По другому пути в нем с малой вероятностью идет переход в крайнем азоте одного из двух π-электронов на sp2-орбиталь с образованием e-изомера C3, который изомеризуется в фенилдиазоний BD+. Вероятность перехода C2 → C3 невелика в силу ортогональности орбиталей π и sp2, однако она все же существует вследствие колебательного возбуждения. Описанная ситуация объясняет факт, хотя малого и не отмеченного в [3], но заметного на рис. 2 сигнала катионов BD+ в полосе фрагментации S0 → S1.
В отличие от фотовозбуждения S0 → S1, фотоперенос электронов в полосах, обозначенных в [3] как CT1 и CT2, идущий тоже при участии катионов PhAT, ярче проявляет параллельный характер реакций образования BD+ и Ph+. Это можно объяснить возникновением FK-состояний с более высокой колебательной (v) энергией. Для CT1 данную ситуацию можно представить схемой 5.
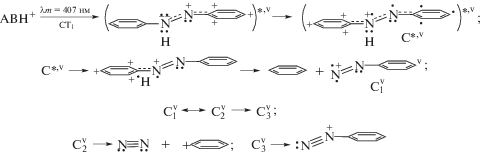
Согласно схеме 5 перенос p-электрона с NH‑группы идет в том же направлении, что и неоптический перенос в схеме 4, но уже после фотовозбуждения катиона PhAT с орбитали HOMO-1 на NBMO. Образующееся более горячее колебательное состояние C*,v диссоциирует, как в схеме 4, на бензол и катион ${\text{C}}_{1}^{{\text{v}}}$. Превращения ${\text{C}}_{1}^{{\text{v}}}$ идут, как в схеме 4, но уже с более высоким выходом BD+, что обусловлено более высокой скоростью перехода ${\text{C}}_{2}^{{\text{v}}}$ → ${\text{C}}_{3}^{{\text{v}}}$ относительно скорости перехода С2 → С3.
Следует отметить, что неодинаковое поведение катионов С2 (${\text{C}}_{2}^{{\text{v}}}$) и С3 (${\text{C}}_{3}^{{\text{v}}}$) обусловлено неодинаковым числом электронов на sp2-орбиталях их азогрупп. Слабые азосвязи в ${\text{C}}_{2}^{{\text{v}}}$ и ${\text{C}}_{3}^{{\text{v}}}$ (С2 и С3 в схеме 4) периодически обеспечивают цис-ориентацию обеих sp2-орбиталей за счет ротационных колебаний. В цис-ориентации, соседние sp2-орбитали образуют две локальные молекулярные орбитали: связывающую МО (BMO) и антисвязывающую МО (ABMO) [21]. В катионе С2 (${\text{C}}_{2}^{{\text{v}}}$) на sp2-ABMO имеется один электрон, что снижает силы электронного отталкивания между sp2-орбиталями двух азотов; при этом энергия связи между атомами N=N выше, чем у фрагментов С3 и ${\text{C}}_{3}^{{\text{v}}}$ (последние имеют по два электрона на BMO и ABMO, и энергия связывания в них компенсируется энергией разрыхления). По этой причине достающаяся катиону С2 энергия (от рекомбинации π-электронов на группе N=N в C*,v) обеспечивает преимущество для разрыва связи N–Ph с образованием N2 и фенилкатиона. В отличие от этого, наличие двух электронов на обеих sp2-орбиталях катионов С3 и ${\text{C}}_{3}^{{\text{v}}}$ не способствует энергетическому выигрышу, оставляя им возможность изомеризоваться в катион BD+.
Результат возбуждения t-ABH+ в полосе CT2, можно представить схемой 6.
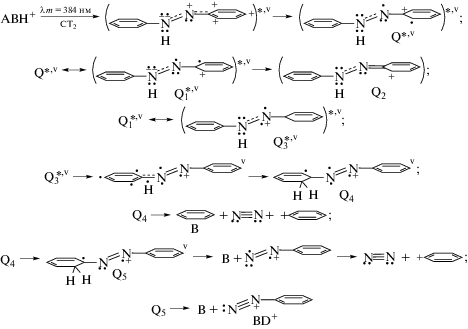
На схеме 6 перенос электрона осуществляется из кольца катиона PhAT на имеющую дефицит электронной плотности pz-орбиталь катиона N+ поляризованной азогруппы. Он идет с pz-орбитали соседнего кольцевого атома углерода, так как на этой орбитали нет электронного дефицита [10, c. 329]. Возникающее при этом возбужденное состояние Q*,v изомеризуется в состояние ${\text{Q}}{{_{1}^{*}}^{{,{\text{v}}}}}$, реагирующее в жидкой и газовой фазах разными путями. В жидкой фазе из него возникает орто-хиноидная структура Q2, имеющая ослабленную азосвязь и участвующая в реакции фотоциклизации по схеме 3. Наоборот, в весьма разреженной криогенной атмосфере гелия, структура ${\text{Q}}{{_{1}^{*}}^{{,{\text{v}}}}}$ сохраняет колебательное возбуждение значительно дольше и претерпевает e-таутомеризацию путем переноса электрона с sp2-орбитали азота на орбиталь орто-углерода кольца с образованием ${\text{Q}}{{_{3}^{*}}^{{,{\text{v}}}}}$.
В состоянии ${\text{Q}}{{_{3}^{*}}^{{,{\text{v}}}}}$ происходит восстановление азосвязи и элиминирование радикала ${{{\text{H}}}^{ \bullet }}$, который присоединяется к соседнему фенильному кольцу по орто-углероду с повышенной электронной плотностью. При этом возникает промежуточное соединение Q4 с циклогексадиенильной структурой и тетраэдрической метиленовой группой. Соединение Q4 в одном случае претерпевает фрагментацию с переносом атома H и восстановлением ароматичности кольца, завершающуюся распадом на молекулы бензола, азота и катиона фенила. В другом случае, на азоте, соседнем с циклогексадиенильным кольцом, идет с низкой вероятностью перенос электрона с pz-орбитали на sp2-орбиталь, ведущий к образованию катиона Q5. Далее катион Q5 распадается либо на молекулу азота и катион фенила, либо на бензол и катион фенилдиазония BD+.
Схемы 4–6, построенные на основании данных работы [3], адекватно и более полно раскрывают механизмы параллельных реакций фотофрагментации t-ABH+ при относительно низком выходе BD+. Они позволяют также объяснить значительный процент потери катионов t-ABH+ (15%) в опытах по газофазной t → c фотоизомеризации [2]. В свою очередь, отсутствие t → c фотоизомеризации в опытах [3] по газофазной фотофрагментации можно связать с тем, что более высокая колебательная температура катионов t‑ABH+, фотоиндуцированных в разреженной криогенной атмосфере гелия [3, 18], обеспечивала более высокую конкурентную способность фрагментации с участием весьма подвижных радикалов ${{{\text{H}}}^{ \bullet }}$ (схемы 4–6).
В дополнение к изложенному следует отметить, что на спектрах рис. 2 имеется еще ряд пиков с низкой интенсивностью, свидетельствующих о появлении осколков Ph+ и BD+ в интервале более высоких частот возбуждающего излучения. Они свидетельствуют о наличии дополнительных актов фотовозбуждения ABH+ с участием катиона фениламинильного типа PhAT. К таковым можно отнести, например, акты фотопереноса электрона в PhAT с sp2-орбитали азота N+ на π*-уровень NBMO (n → π*-переход [22]), а также внутримолекулярного переноса энергии от возбужденного фенильного кольца анилинового участка (PhNH-) на катион PhAT [23].
В заключение можно отметить, что показанное на основании экспериментальных данных работ [2, 3] фундаментальное значение электронных свойств катиона PhAT позволяет:
– дать адекватные механизмы реакций фотоизомеризации и фотофрагментации t-ABH+, возбуждаемых в главных полосах поглощения радиации;
– сделать вывод, что этот результат способствует расширению границ понимания фотоники t-ABH+;
– сделать вывод о неэффективности традиционной модели катиона азония в теоретических работах.
Список литературы
Гордон П., Грегори П. Органическая химия красителей. М.: Мир, 1987. 344 с.
Scholz M.S., Bull J.N., Coughlan J.A. et al. // J. Phys. Chem. A. 2017. V. 121. № 34. P. 6413.
Féraud G., Dedonder-Lardeux C., Jouvet C., Marceca E. // Ibid. 2016. V. 120. № . P. 3897.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Журн. физ. химии. 2015. Т. 89. № 2. С. 243.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Там же. 2015. Т. 89. № 11. С. 1773.
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 6. С. 946.
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 7. С. 1111.
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 11. С. 1746.
Михеев Ю.А., Ершов Ю.А. // Там же. 2018. Т. 92. № 10. С. 1552.
Стрейтвизер Э. Теория молекулярных орбит для химиков-органиков. М.: Мир, 1965. 435 с.
Хигаси К., Баба Х., Рембаум А. Квантовая органическая химия. М.: Мир, 1967. С. 127.
Grace J.A., Symons M.C.R. // J. Chem. Soc. 1959. P. 958.
Lewis G.E. // J. Org. Chem. 1960. V. 25. № 12. P. 2193.
Lewis G.E. // Tetrahedron Letters. 1960. № 9. P. 12.
Gerson F., Heilbronner E., van Veen A., Wepster B.M. // Helvetica Chim. Acta. 1960. V. 43. P. 1889.
Колдин Е. Быстрые реакции в растворе. М.: Мир, 1966. С. 282.
Andersen J.U., Hvelplund P., Nielsen S.B. et al. // Rev. Sci. Instruments. 2002. V. 73. № 3. P. 1284.
Wang Xue-Bin, Wang Lai-Sheng // Rev. Sci. Instruments. 2008. V. 79. № 7. P. 073108.
Fujino T., Tahara T. // J. Phys. Chem. A. 2000. V. 104. № 18. P. 4203.
Михеев Ю.А., Ершов Ю.А. // Журн. физ. химии. 2019. Т. 93. № 2. С. 313.
Mason S.F. // J. Chem. Soc. 1959. P. 1240.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Журн. физ. химии. 2018. Т. 92. № 8. С. 1251.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Там же. 2020. Т. 94. № 1. С. 143.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии