

Журнал физической химии, 2020, T. 94, № 8, стр. 1153-1160
Озонолитическая делигнификация соломы пшеницы
Е. М. Бенько a, *, Д. Г. Чухчин b, Н. А. Мамлеева a, А. Н. Харланов a, В. В. Лунин a
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
b Северный (Арктический) федеральный университет имени М.В. Ломоносова
Москва, Россия
* E-mail: benko_elena@mail.ru
Поступила в редакцию 11.10.2019
После доработки 11.10.2019
Принята к публикации 12.11.2019
Аннотация
Исследована динамика делигнификации пшеничной соломы при обработке озоном. Определена зависимость содержания остаточного лигнина от удельного поглощения озона. Установлено, что на продолжительном этапе озонирования озон расходуется преимущественно на реакцию с лигнином. С помощью метода ВЭЖХ получены кинетические профили водорастворимых продуктов озонирования. Проанализированы ИК-спектры образцов соломы различной степени делигнификации. Полученные результаты подтверждают озонолитическую деструкцию лигнина и указывают на частичное окисление полисахаридов при длительном озонировании. Установлена связь между содержанием лигнина и реакционной способностью предобработанной озоном соломы в процессах ферментативного гидролиза в сахара. Оптимальный расход озона составляет 2 мол. экв. О3 на фенилпропановую структурную единицу (ФПЕ) содержащегося в соломе лигнина и соответствует содержанию остаточного лигнина ∼10%.
Делигнификация лигноцеллюлозного растительного сырья является необходимым этапом его предобработки в биотехнологии получения моносахаридов и спиртов. Согласно литературным данным лигнин затрудняет доступ ферментов к целлюлозе, сдерживает ее набухание, а также адсорбирует часть целлюлолитических ферментов, выводя их из зоны реакции [1, 2]. Деструкция лигнина с помощью озона рассматривается в качестве перспективного альтернативного метода делигнификации благодаря высокой активности и селективности озона по отношению к лигнину и экологической безопасности процесса [3].
В предыдущей работе [4] нами исследована озонная предобработка соломы пшеницы для последующего ферментативного гидролиза в сахара. Показано значительное увеличение степени конверсии растительных образцов после озонирования и установлена зависимость выхода восстанавливающих сахаров от количества поглощенного озона. На основании полученных результатов сделано предположение о том, что в процессе обработки лигноцеллюлозного композита озон расходуется преимущественно на реакцию с лигнином. Задача настоящей работы состояла в изучении динамики удаления лигнина из соломы пшеницы в процессе обработки озоном.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исследовали образцы пшеничной соломы (Tritcum L.), предоставленные и охарактеризованные Алтайским государственным университетом (Барнаул, Россия). Состав: лигнин – 20%, целлюлоза – 40%, гемицеллюлозы – 24%; размер частиц 0.6–1.0 мм.
Для озонирования использовали образцы с влажностью ∼100%. Для их приготовления к навеске (0.5–1.5 г) воздушно сухой соломы (Wабс 5–7%) добавляли необходимое количество дистиллированной воды и выдерживали в течение нескольких суток в закрытой емкости.
Озонирование проводили в реакторе с неподвижным слоем при начальной концентрации озона 50–60 мг/л, объемной скорости газового потока 10 л/ч, комнатной температуре, как описано в работе [4]. Из кинетических кривых зависимости концентрации озона на выходе из реактора рассчитывали удельный расход озона [4].
Образцы соломы после озонирования заливали водой (20 мл/г), выдерживали 2 ч, после центрифугирования водный раствор анализировали на содержание органических кислот и других продуктов озонирования. Твердый образец промывали водой, сушили при 105oC до постоянного веса, определяли потерю массы и использовали для определения содержания остаточного лигнина.
Содержание лигнина определяли по стандартной методике [5] с помощью двухстадийного гидролиза серной кислотой. Образец (0.1 г в расчете на абсолютно сухой материал – АСМ) выдерживали в 1 мл 72% H2SO4 при комнатной температуре в течение 1 ч, затем добавляя 28 мл воды, и нагревали в автоклаве при 120°C в течение 1 ч. После охлаждения раствор фильтровали, остаток промывали, высушивали и взвешивали, определяя кислотонерастворимый лигнин (КНР). Фильтрат разбавляли и анализировали содержание в нем кислоторастворимого лигнина (КРЛ) по поглощению при λ = 205 нм (ε = 110 г/(л см)) [5].
Органические кислоты определяли с помощью ВЭЖХ на хроматографе Agilent 1100. Рабочие параметры: УФ-детектор (λ = 190 нм), колонка Rezex ROA-Organic acids, подвижная фаза 0.05 М H2SO4, температура 65°C, скорость потока 0.5 мл/мин. Содержание восстанавливающих сахаров в водной фракции анализировали по методу Шомоди-Нельсона [6].
Для получения ИК-спектров предварительно промытые и высушенные на воздухе образцы соломы дополнительно измельчали, смешивали с КВr в соотношении: 1:850 и прессовали при давлении 6000 кгс/см2 в таблетки массой 300 мг. ИК-спектры регистрировали на ИК-Фурье спектрометре Equinox 55/S (Bruker) с разрешением 4 см–1 (32 скан.). Базисную линию вычитали из экспериментального спектра с помощью программы OPUS 6.0 (Bruker), суммарный сложный спектр аппроксимировался гауссовскими компонентами.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Содержание лигнина в озонированных образцах
На рис. 1 приведены зависимости количества поглощенного озона и степень делигнификации соломы от времени озонирования. Содержание общего (ОЛ), кислотонерастворимого (КНРЛ) и кислоторастворимого (КРЛ) лигнина как функция количества озона, поглощенного образцами в результате обработки, показана на рис. 2.
Рис. 1.
Зависимости количества поглощенного озона (1) и степени делигнификации (2) пшеничной соломы от времени озонирования.
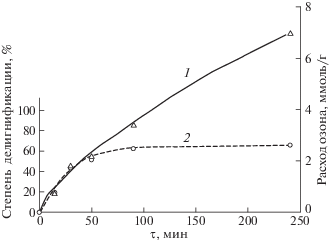
Рис. 2.
Зависимости содержания общего (ОЛ), кислотонерастворимого (КНРЛ) и кислоторастворимого лигнина (КРЛ) от удельного поглощения озона.
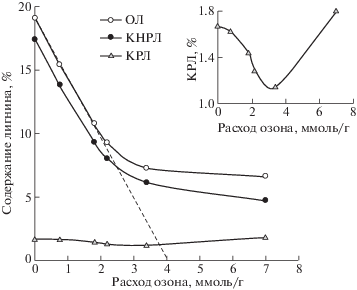
За 50 мин озонирования при расходе озона 2.2 ммоль/г содержания лигнина в соломе падает более чем на 50%: ОЛ с 19.1 до 9.3%, КНРЛ с 17.5 до 8.0%. В дальнейшем скорость делигнификации уменьшается, и для снижения содержания лигнина на 70% (до 6–4%) требуется 4 ч озонирования при расходе озона 7 ммоль/г. После 1.5 ч обработки степень делигнификации практически не меняется, а скорость поглощения озона остается достаточно высокой, т.е. озон расходуется не на реакцию с лигнином (рис. 1).
Как следует из приведенных данных, на начальном этапе озонирования наблюдается линейное падение содержания лигнина с ростом расхода озона. Лигнин является наиболее реакционноспособным компонентом растительной биомассы. Известно, что реакции молекулярного озона с лигнином, имеющим ароматическую структуру, протекают в 103–106 раз быстрее, чем с углеводами [7, 8].
Если предположить, что сначала озон реагирует исключительно с лигнином, можно оценить стехиометрическое соотношение реагентов (озона и лигнина). В 1 г исследуемой соломы содержится ∼0.2 г (20%) лигнина. Считая молекулярную массу С9-фенилпропановой субъединицы (ФПЕ) лигнина соломы величиной порядка 170–200 [9] получаем, что содержание ароматических субъединиц в соломе составляет 1.0–1.2 ммоль/г. Согласно схеме озонолиза [10] на деструкцию ароматического кольца до алифатических кислот требуется 3 молекулы озона. Таким образом, на полное разрушение ароматики в исследуемом образце потребуется 3.0–3.6 ммоль озона (3О3/ФПЕ). Эта оценка удовлетворительно согласуется с экспериментальным результатом, представленным на рис. 3, где пунктиром показана гипотетическая зависимость убыли лигнина при условии, что озон взаимодействует исключительно с фенилпропановыми структурными субъединицами лигнина.
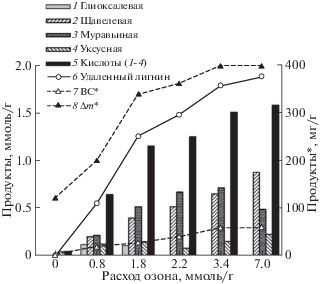
Рис. 3.
Выход продуктов озонирования при различном расходе озона: 1 – глиоксалевая, 2 – щавелевая, 3 – муравьиная, 4 – уксусная кислоты, 5 – сумма идентифицированных веществ (1–4), 6 – количество удаленного лигнина (в пересчете на содержание непредельных связей); 7 – общее содержания водорастворимых продуктов озонирования.
Аналогичный подход использован в работе [9], где установлено, что при озонировании пшеничной соломы с начальным содержанием лигнина 10% зависимость количества остаточного лигнина от расхода озона линейна в области от 0 до 1 экв. О3/ФПЕ. В [11] для образцов хвойной древесины (японский кедр) такая зависимость сохраняет линейность до значений расхода озона 3 экв. О3/ФПЕ.
Согласно результатам, приведенным на рис. 2, при озонировании соломы пшеницы первые 2 экв. О3/ФПЕ (∼2.5 ммоль О3/г) расходуются практически только на взаимодействие с лигнином. По мере разрушения лигнина могут происходить структурные изменения, например, слипание целюлозных фибрилл и сминание клеточных стенок [12], модификация лигнина с образованием полимерных продуктов его конденсации [13–15] и иные процессы, затрудняющие доступ озона к остаточному лигнину. При этих условиях озон расходуется на реакции с другими компонентами растительной биомассы и промежуточными продуктами озонолиза лигнина. Нельзя исключить и реакции озона с лигнином, не приводящие к его деструкции и удалению из лигноцеллюлозной матрицы, например, модификация ароматических структур кислородсодержащими заместителями или окислительная деструкция эфирных связей [16–18].
По данным различных авторов степень делигнификации соломы пшеницы при больших дозах озона достигает, в среднем, 50–70% [9, 19–22], в отдельных случаях – 90% [23, 24].
На фоне уменьшения значения общего и кислотонерастворимого лигнина обычно наблюдается незначительное повышение содержания КРЛ, что связывают с переходом части КНРЛ в растворимую форму в результате окисления [25].
В наших экспериментах количество кислоторастворимого лигнина во всех образцах не превышает 2%, что близко к пределу ошибок определения, поэтому его изменения не обсуждаются. Тем не менее, следует отметить, что зависимость содержания КРЛ от расхода озона (рис. 2a) имеет четкую закономерность, которая в рамках изложенных представлений согласуется с данными по изменению КНРЛ и ОЛ (рис. 2).
Водорастворимые продукты озонирования
Известно, что основным направлением реакции молекулярного озона с лигнином является электрофильная атака ароматических структурных субъединиц с образованием алифатических кислот, кетокислот и альдегидов [10, 16]. В зависимости от условий возможны также радикальные реакции озона с лигнином и полисахаридами, приводящие к окислению функциональных групп и расщеплению эфирных и гликозидных связей [26, 27]. Реакции полисахаридов с молекулярным озоном не должны вносить заметный вклад из-за сравнительно маленьких констант скорости [8].
В водорастворимой фракции продуктов озонирования пшеничной соломы были обнаружены глиоксалевая, щавелевая, муравьиная, уксусная кислоты. Зависимость выхода этих веществ от количества поглощенного озона приведена на рис. 3.
Глиоксалевая кислота в небольших количествах зафиксирована в интервале расхода озона до 2 ммоль/г. Содержание муравьиной кислоты проходит через максимум, при расходе озона около 3 ммоль/г, что коррелирует с профилем делигнификации образца соломы (рис. 2). Количество уксусной и щавелевой кислот, устойчивых к действию озона, с увеличением времени озонирования возрастает.
На рис. 3 также приведены результаты оценки количества кислот, которое может образоваться из удаленного в процессе озонирования лигнина (кривая 6). Расчет проведен в предположении, что количество молей образовавшихся кислот пропорционально количеству ФПЕ прореагировавшего лигнина в соотношении 3 : 1, что соответствует схеме озонолиза [10]. Как видно, оно сопоставимо с количеством идентифицированных веществ (1–4), из чего можно заключить, что щавелевая, уксусная и муравьиная кислоты являются основными продуктами деструкции лигнина. Суммарная масса идентифицированных продуктов при максимальном расходе озона составляет 120 мг/г.
На рис. 3 также приведены данные по количеству удаленного в процессе озонирования лигнина. Для оценки соотношений реагентов и продуктов представлено мольное содержание двойных связей в удаленном лигнине. Как видно, оно сопоставимо с количеством идентифицированных веществ (1–4), из чего можно заключить, что щавелевая, уксусная и муравьиная кислоты являются основными продуктами деструкции лигнина. Следует, однако, учитывать, что эти же кислоты образуются, в конечном итоге, при озонолизе полисахаридов [28]. Суммарная масса идентифицированных продуктов при максимальном расходе озона составляет 120 мг/г.
В водной фракции с помощью химического анализа определены редуцирующие сахара. Их содержание возрастает от 2 мг/г для необработанной соломы до 60 мг/г для расхода озона 3 ммоль/г, и при дальнейшем озонировании до 7 ммоль/г не изменяется (кривая 7). Такой же характер имеет зависимость общей потери массы образца после озонирования и промывания водой, которая в пределе достигает 400 мг/г (кривая 8). Это свидетельствует о том, что образование водорастворимых редуцирующих сахаров, как и других водорастворимых продуктов проходит на стадии деструкции лигнина, сопровождающейся разрушением лигноуглеводной матрицы и частичной деполимеризацией полисахаридов.
Теоретически низкомолекулярные карбоновые кислоты, как и другие водорастворимые продукты могут образоваться при окислении озоном целлюлозы и гемицеллюлоз [28]. Однако эти реакции протекают очень медленно [7, 8] и в условиях нашего эксперимента не проявляются. Этот факт подтверждается насыщением на кривой потери массы образца (кривая 8) в интервале расхода озона от 3 до 7 ммоль/г, когда условия для окисления полисахаридов наиболее благоприятны.
Согласно литературным данным в продуктах озонировании растительной биомассы помимо щавелевой, уксусной и муравьиной кислот могут содержаться молочная, левулиновая, малоновая, глюконовая кислоты, а также ксилит, бутаналь и др. [25, 29]. В [30] при озонировании багассы зафиксированы такие летучие продукты как СО, CO2, Н2О2, НСООН метанол.
В [31] при озонировании пшеничной соломы идентифицировали промежуточные продукты деструкции лигнина ароматической природы, такие как п-гидроксибензальдегид, ванилин, ванилиновая, капроновая, п-гидроксибензойная кислота, а также продукты окисления сахаров с ненасыщенными связями (фурфурол и его производные). В наших экспериментальных условиях, как и в работе [25] каких-либо водорастворимых ароматических соединений, имеющих заметное поглощение в УФ-области спектра, не обнаружено.
ИК-спектры озонированной соломы
На рис. 4 представлены ИК-спектры поглощения образцов соломы пшеницы при различных значениях удельного расхода озона.
Рис. 4.
ИК-спектры поглощения образцов озонированной соломы (а–в). Расход озона, ммоль /г: 0 (1), 0.8 (2), 2.0 (3), 3.2 (4), 6.5 (5).
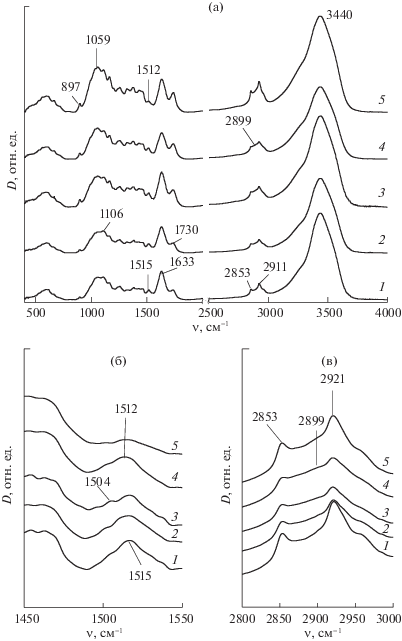
Интенсивные полосы поглощения в спектре исходного образца в области 1000–1200 см–1 обусловлены валентными колебаниями групп С–О, присутствующих в структуре углеводов и лигнина. Полосы при 700–900 см–1 обусловлены внеплоскостными колебаниями СН-связей и пиранозных звеньев целлюлозы [32]. В области 1300–1400 см–1 наблюдаются деформационные колебания групп СН связей [33]. Полосы поглощения 1473 и 1380 см–1 относят к симметричным и асимметричным деформационным С–Н-колебаниям, 1327 см–1 к С–С- и С–О-скелетным колебаниям [32]. Полосы поглощения, соответствующие С–С-валентным колебаниям в ароматических структурах лигнина соломы наблюдаются по данным разных авторов [33–35] при 1595, 1510–1514 и 1430 см–1.
Из рис. 4 видно, что обработка озоном приводит к изменениям оптической плотности многих полос поглощения. За изменением ИК-полос поглощения удобно проследить с помощью табл. 1, где приведены значения Dν/$D_{\nu }^{0}$, которые представляют собой оптическую плотность Dν в спектре озонированного образца при волновом числе ν, нормированное к $D_{\nu }^{0}$ (оптическая плотность при том же ν в спектре исходной соломы).
Таблица 1.
Оптическая плотность полосы поглощения в спектре озонированного образца соломы при волновом числе ν (Dν), нормированная к оптической плотности полосы в спектре не озонированного образца ($D_{\nu }^{0}$)
| (Dν)/($D_{\nu }^{0}$) | Отнесение полосы | ||||||
|---|---|---|---|---|---|---|---|
| № образца | 1 | 2 | 3 | 4 | 5 | ||
| Расход озона, ммоль/г | 0 | 0.8 | 2.0 | 3.2 | 7.0 | ||
| Волновое число, см–1 | 1512–1515 | 1.0 | 0.9 | 0.8 | 0.8 | 0.6 | С–C-колебания ароматического кольца |
| 1059 | 1.0 | 1.0 | 1.3 | 1.4 | 2.0 | C–O-валентные колебания в целлюлозе и гемицеллюлозе | |
| 1730 | 1.0 | 1.0 | 1.4 | 1.9 | 2.0 | C=O-валентные колебания в неконъюгированных карбоксильных группах лигнина и гемицеллюлоз | |
| 2853 | 1.0 | 1.0 | 1.2 | 1.7 | 2.0 | C–H-валентные колебания | |
| 2921 | 1.0 | 0.8 | 1.0 | 1.5 | 1.9 | ||
| 3440 | 1.0 | 1.0 | 1.0 | 1.1 | 1.1 | O–H-валентные колебания (связанные) | |
На рис. 4a представлены ИК-спектры в области валентных С–С-колебаний лигнина. Видно, что увеличение удельного поглощения озона сопровождается смещением положения максимума поглощения ароматических структур и изменением контура полосы поглощения. Это можно объяснить тем, что лигнин соломы пшеницы построен из субъединиц трех типов: п-гидроксифенильных (Н), гваяцильных (G) и сиренгильных (S) [36]. Их реакционная способность по отношению к озону значительно отличается, убывая в ряду S–G–H [37]. В связи с этим, в процессе озонирования происходит не только разрушение ароматики, но и изменение относительного содержания ароматических структур разного типа, имеющих различные спектральные характеристики. Это отражается на интенсивности и положении полос ИК-спектра. Приведенные в табл. 1 значения 2Dν/$D_{\nu }^{0}$ в максимуме полосы поглощения ароматики (1512–1515 см–1) уменьшаются по мере увеличения расхода озона.
Полосы 1595 и 1430 см–1, где также наблюдаются С–С-валентные колебания ароматических структур в данном случае не рассматриваются из-за наложения первой с полосой поглощения деформационных ОН-колебаний (∼1625 см–1), а при 1430 см–1 валентные С–С-колебания ароматики комбинируются также с деформационными С–Н-колебаниями в ОСН3 группах лигнина и СН- и СН2-группах углеводов [38].
Полоса поглощения 1730 см–1 относится к С=О-валентным колебаниям карбоксильных групп кислот и сложных эфиров, а также карбонильных групп, характерных для гемицеллюлоз, входящих в состав растительного сырья [33, 38]. Значение D1730/$D_{{1730}}^{0}$ возрастает при увеличении количества поглощенного озона и становится вдвое больше, чем в исходном образце при удельном поглощении озона 7 ммоль/г (образец 5).
Из табл. 1 видно заметное увеличение D1059/$D_{{1059}}^{0}$ С–О-валентных колебаний. Возрастание поглощения в этой области спектра может быть связано с увеличением содержания карбоксильных групп, а также c возрастанием содержания углеводов в составе образца озонированной соломы.
Валентные С–Н-колебания в спектре исходной соломы характеризуются максимумами при 2921 и 2853 см–1 (рис. 4). Полоса С–Н-колебаний представляет собой суперпозицию полос, относящихся к С–Н-колебаниям в метильных и метиленовых группах лигнина, а также валентных С‒Н в метиленовых и метиновых группировках целлюлозы (2899 см–1) [32]. В этой области спектра также поглощают алифатические группы гемицеллюлоз и воскообразных веществ, входящих в состав озонированного биоматериала [33, 39]. В спектрах озонированных образцов положение максимумов С–Н валентных колебаний сохраняется. Значения D2921/$D_{{2921}}^{0}$ и D2853/$D_{{2853}}^{0}$ возрастают по мере увеличения расхода озона (рис. 4a). Контур спектра искажается, возрастает поглощение при 2899 см–1 (рис 4в), что свидетельствует об увеличении содержания целлюлозы в озонированных образцах, и согласуется с отмеченным увеличением D1059/$D_{{1059}}^{0}$.
Спектр образца 5 (рис. 4), которому соответствует наибольший расход озона (7.0 ммоль/г), характеризуется существенно более высокими значениями D2921/$D_{{2921}}^{0}$ и D2853/$D_{{2853}}^{0}$. Одним из возможных объяснений этого результата может быть накопление при высоких расходах озона нерастворимых в воде продуктов озонирования с алифатической углеводородной цепью. Образование подобных соединений наблюдали при делигнификации древесины под действием озона [39].
Полоса 3440 см–1 относится к валентным колебаниям О–Н-групп, связанных водородной связью. Значение D3440/$D_{{3440}}^{0}$ спектра озонированных образцов имеет тенденцию к увеличению (табл. 1).
Таким образом, анализ ИК-спектров показывает, что обработка озоном соломы пшеницы приводит к удалению части лигнина и увеличению содержания целлюлозы. При высоких значениях удельного поглощения озона наблюдается значительное возрастание содержания карбоксильных групп, возможным источником которых является непосредственное окисление углеводов и алифатических структур лигнина биомассы. Кроме того, нельзя исключить и предлагаемое авторами [40], наличие взаимосвязи между процессами деструкции ароматических систем и углеводов за счет промежуточных продуктов окисления лигнина (феноксильные радикалы, ${\text{О}}{{{\text{Н}}}^{ \bullet }}$-радикалы, озониды).
Даже при высоком значении поглощения озона озона (7.0 ммоль/г) в ИК-спектрах озонированных образцов интенсивность полос поглощения, характерных для ароматического кольца лигнина, остается достаточно высокой. Это результат согласуется с данными определения остаточного лигнина, подтверждая вывод о том, что часть ароматических структур недоступна озону.
Влияние делигнификации на ферментативный гидролиз соломы
В нашей предыдущей работе [4] определена зависимость выхода восстанавливающих сахаров от расхода озона при ферментативном гидролизе обработанной озоном пшеничной соломы: ВС48(О3). Для ферментативного гидролиза образцов использовали коммерческий препарат целлюлазного комплекса Trichoderma Viride “Целловиридин Г20Х”. Показано, что выход восстанавливающих сахаров после озонирования возрастает, достигается максимума (∼412 мг/г) при расходе озона 2.5–3.0 ммоль О3/г биомассы, и при дальнейшем увеличении расхода озона снижается [4].
В настоящей работе получены данные о зависимости содержания остаточного лигнина от расхода озона: ОЛ(О3). Сопоставив эти результаты, можно установить зависимость выхода продуктов ферментативного гидролиза от содержания остаточного лигнина. Зависимость степени конверсии полисахаридов при ферментативном гидролизе предобработанной озоном пшеничной соломы от содержания остаточного лигнина представлена на рис. 5.
Рис. 5.
Зависимость степени конверсии полисахаридов предобработанной озоном пшеничной соломы от содержания остаточного лигнина (с учетом результатов работы [4]).
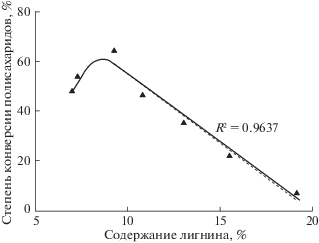
При малых дозах озона степень конверсии полисахаридов возрастает пропорционально количеству удаленного лигнина. Аналогичную тенденцию наблюдали при ферментации озонированной соломы в [19]. Полученный результат находится в соответствии с представлениями о том, что содержащийся в лигноцеллюлозной биомассе лигнин является основным препятствием для ферментов в процессе гидролиза целлюлозы [1, 2]. При содержании остаточного лигнина менее 10% выход продуктов ферментативного гидролиза уменьшается. Можно предположить, что вызванная удалением лигнина деструкция лигноуглеводной матрицы, приводит к блокировке внутренней поверхности и затрудняет доступ ферментов к целлюлозе.
Таким образом, проведенные исследования указывают на понятные (с точки зрения стехиометрии реакции озона с лигнином) корреляции между величиной удельного расхода озона, содержанием остаточного лигнина и выходом восстанавливающих сахаров при ферментативном гидролизе. Согласно полученным результатам, на начальном этапе озонирования озон расходуется преимущественно на реакцию с лигнином. При удалении более 60% лигнина скорость делигнификации резко падает, возможно, из-за структурных изменений, приводящих к тому, что остаточный лигнин становится недоступен озону. Выход сахаров в ферментативной реакции соответствует профилю делигнификации: вначале возрастает пропорционально количеству удаленного лигнина, а затем падает. Оптимальный расход озона в процессе предобработки пшеничной соломы соответствует 2 моль.экв. О3/ФПЕ содержащегося в соломе лигнина (∼10 мас. %).
Работа выполнена с использованием оборудования ЦКП химического факультета МГУ и ЦКП НО “Арктика” (САФУ) при финансовой поддержке Минобрнауки РФ.
Список литературы
Chang V.S., Holtzapple M.T. // Appl. Biochem. Biotechnol. 2000. V. 84. P. 5.
Neely W.C. // Biotechnol. Bioeng. 1984. V. 26. P. 59.
Travaini R., Martín-Juárez J., Lorenzo-Hernando A., Bolado-Rodríguez S. // Bioresour. Technol. 2016. V. 199. P. 2.
Ben’ko E.M., Chukhchin D.G., Lunin V.V. // Russ. J. Phys. Chem A. 2017. V. 91. P. 2092.
Aldaeus F., Sjöholm E. COST Action FP0901 Round Robins of lignin samples Part 1: Lignin content. Version 3 (2011) (2010-12-14). Innventia Report No.: IR 108. fredrik.aldaeus@innventia.com.
Somogyi M. // J. Biol. Chem. 1952. V. 200. P. 245.
Hoigne J. Hand book of ozone technology and Applications. Ann. Aiber Science Publisher. M. I. 1982. V. 1.
Nompex P., Dore M. // Ozone Sci. Eng. 1991. V. 13. P. 265.
Binder A., Pelloni L., Fiechter A. // Eur. J. Appl. Microbiol. Biotechnol. 1980. V. 11. P. 1.
Bailey P.S. Ozonation in Organic Chemistry. V. II. New York: Acad Press, 1982.
Sugimoto T., Magara K., Hosoya S. et al. // Holzforshung. 2009. V. 63. P. 537.
Wong K.K.Y., Deverell K.F., Mackie K.L. et al. // Biotechnol. Bioeng. 1988. V. 31. P. 447.
Bule M.V., Gao A.H., Hiscox B., Chen S. // J. Agric. Food Chem. 2013. V. 61. P. 3916.
Kobayashi M., Asano T., Kajiyama M., Tomita B. // J. Wood Sci. 2005. V. 51. P. 348.
Kádár Z., Schultz-Jensen N., Jensen J.S. et al. // Biomass Bioenergy. 2015. V. 81. P. 26.
Sarkanen K.V., Islam A., Anderson C. in Methods in Lignin Chemistry. Lin S.Y., Dence C.W., Eds. Berlin: Springer-Verlag, 1992. P. 387–406.
Ferron B., Croué J.P., Dore M. // Ozone: Sci. Eng. 1995. V. 17. P. 687.
Bertaud F., Croué J.P., Legube B. // Ozone: Sci. Eng. 2001. V. 23. P. 139.
García-Cubero M.T., Palacín L.G., González-Benito G. et al. // Bioresour. Technol. 2012. V. 107. P. 229.
Garcia-Cubero M.T., Gonzalez-Benito G., Indacoechea I., Coca M., Bolado S. // Bioresour. Technol. 2009. V. 100. P. 1608.
Wu J., Upreti S., Ein-Mozaffari F. // Int. J. Hydrogen Energy. 2013. V. 38. P. 10270.
Bhattarai S., Bottenus D., Ivory C.F. et al. // Bioresource Technol. 2015. V. 196. P. 78.
Shultz-Jensen N., Kádár Z., Thomsen A.B. et al. // Appl. Biochem. Biotechnol. 2011. V. 165. P. 1010.
Baig K.S., Wu J., Turcotte G., Doan H.D. // Energy and Environment. 2015. V. 26. P. 303.
Travaini R., Otero M.D.M., Coca M. et al. // Bioresour. Technol. 2013. V. 133. P. 332.
Criegee R. // Angew. Chem. Int. 1975. Ed. 14. P. 745.
Ragnar M., Eriksson T., Reitberger T. // Holzforschung. 1999. V. 53. P. 292.
Marcq O., Barbe J.M. // Carbohydrate Research. 2009. V. 344. P. 1303.
Adarme O.F.H., Baêta B.E.L., Diego Roberto Sousa Lima D.R.S. et al. // Ind. Crops. Products. 2017. V. 109. P. 288.
Souza-Corrêa J.A., Ridenti M.A., Olivera C.S. et al. // J. Phys. Chem. B. 2013. V. 117. P. 3110.
Schultz-Jensen N., Kádár Z., Thomsen A. et al. // J. Biochem. Biotechnol. 2011. V. 65. P. 1010.
Ciolacu D., Ciolacu F., Popa V. // Cellulose Chem. Technol. 2011. V. 45. P. 13.
Kristensen J.B., Thygesen L.G., Felby C. et al. // Biotechnol. Biofuels. 2008. V. 1. P. 3.
Sun X.F., Xu F., Sun R.C. et al. // Carbohydrate Research. 2005. V. 340. P. 97.
Stewart D., Wilson H.M., Hendra P.J., Morrison I.M. // J. Agricult. Food Chem. 1995. V. 43. P. 2219.
Río J.C., Rencoret J., Prinsen P. et al. // J. Agric. Food Chem. 2012. V. 60. P. 5922.
Kaneko H., Hosoya S., Iiyama K., Nakano J. // J. Wood Chem. Technol. 1983. V. 3. P. 399.
Smith L. Applied Infrared Spectroscopy. Fundamentals, techniques, and analytical problem-solving. Wiley, John Sons., 1967.
Mamleeva N.A., Babayeva N.A., Kharlanov A.N., Lunin V.V. // Russ. J. Phys. Chem. A. 2019. V. 93. P. 37.
Olkkonen C., Tylli Y.I., Forsskåhl I. et al. // Holzforschung. 2000. V. 54. P. 397.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии