

Журнал эволюционной биохимии и физиологии, 2023, T. 59, № 3, стр. 198-206
Противосудорожная эффективность ингибирования синаптических и внесинаптических ГАМК-транспортеров в предотвращении гипербарических кислородных судорог
О. С. Алексеева 1, *, Б. А. Герда 1, А. С. Жиляева 1, И. Т. Демченко 1
1 Федеральное государственное бюджетное учреждение науки
Институт эволюционной физиологии и биохимии им. И.М. Сеченова Российской академии наук
Санкт-Петербург, Россия
* E-mail: osa72@inbox.ru
Поступила в редакцию 10.02.2023
После доработки 13.04.2023
Принята к публикации 18.04.2023
- EDN: YGRLBE
- DOI: 10.31857/S0044452923030026
Аннотация
Гипербарический кислород (ГБО2) подавляет ГАМК-ергическую нейропередачу в мозге, что может приводить к развитию судорожного синдрома, известного как “кислородная эпилепсия”. Дефицит ГАМК-ергической передачи в ГБО2, возникающий за счет снижения уровня синаптического ГАМК, может быть компенсирован путем ингибирования нейрональных и глиальных ГАМК-транспортеров (GAT). В настоящем исследовании проводилась сравнительная оценка противосудорожного действия тиагабина, ингибитора GAT-1, и SNAP 5114, ингибитора GAT-2/3. Противосудорожные эффекты оценивались после введения препаратов в латеральный мозговой желудочек крыс за 30 мин до начала гипербарической кислородной экспозиции при давлении 5 АТА. В отдельных опытах измерялась концентрация ГАМК в стриатуме крыс при дыхании кислородом под давлением 5 АТА после ингибирования GAT с помощью тиагабина или SNAP 5114. Основными результатами выполненных исследований являются: (1) ингибирование GAT-1 или GAT-2/3 предотвращало развитие “кислородной эпилепсии” у крыс; (2) среди двух используемых ингибиторов, TGB оказался более эффективным в предотвращении кислородных судорог по сравнению со SNAP 5114; (3) совместное использование TGB и SNAP 5114 вызывало аддитивный противосудорожный эффект; (4) кислородные судороги появлялись при снижении ГАМК в головном мозге на 30–40% от исходного уровня; (5) ингибирование GAT с помощью тиагабина увеличивало содержание внеклеточного ГАМК в 2.9 раза и в 1.7 раза при использовании SNAP 5114. Таким образом, ингибирование GAT-1 или GAT-2/3 повышает внеклеточную концентрацию ГАМК до уровня, достаточного для восстановления нарушенной в ГБО2 тормозной нейропередачи, и предотвращает развитие гипербарических кислородных судорог.
ВВЕДЕНИЕ
Кислород под давлением (гипербарический кислород или ГБO2) используется в клинике при лечении заболеваний методом гипербарической оксигенации и для дыхания водолазами при осуществлении их профессиональной деятельности. Основным лимитирующим фактором использования кислорода под давлением выше 2 АТА (атмосфер абсолютных) является его нейротоксическое действие, проявляющееся в виде пароксизмальной активности на ЭЭГ и тонико-клонических судорог, известных как “кислородная эпилепсия” [1].
Принято считать, что “кислородная эпилепсия” может развиваться в результате снижения ГАМК-опосредованной тормозной нейропередачи в ЦНС [2–4]. Ослабление ГАМК-ергической нейротрансмиссии в ГБО2 связано с ингибированием синтеза медиатора и снижением его доступности к рецепторам [5]. Причиной снижения внутримозговой ГАМК при ГБО2 является инактивация глутаматдекарбоксилазы (GAD), катализирующей синтез медиатора в нервных клетках [6–8]. Механизм снижения активности GAD при экстремальной гипероксии происходит за счет S-нитрозилирования фермента – ковалентного присоединения группы оксида азота (NO) к тиоловым остаткам цистеина в белке [9].
ГАМК является основным тормозным нейротрансмиттером в мозге млекопитающих. После высвобождения из пресинаптических окончаний и активации рецепторов излишек ГАМК быстро удаляется из синаптического пространства с помощью ГАМК-транспортеров (GAT), которые прерывают синаптическую передачу [10, 11], регулируют спилловер ГАМК на соседние синапсы [12] и поддерживают гомеостаз медиатора для предотвращения чрезмерной тонической активации синаптических и экстрасинаптических рецепторов ГАМК [13]. На сегодняшний день описано четыре различных транспортера ГАМК: GAT-1, GAT-2, GAT-3 и транспортер бетаина/ГАМК типа 1 (BGT-1). Все они являются членами большого семейства 12-трансмембранных транспортеров [14]. GAT-1, транспортер обратного захвата медиатора в пресинаптический нейрон, был первым клонированным GAT [15]. Белковая последовательность GAT-1 крысы [15], мыши [16] и человека [17] демонстрирует высокую степень гомологии и почти идентичные фармакологические свойства. Согласно имеющимся морфологическим данным, GAT-2/3, скорее всего, расположены в астроглии и, частично, в синапсе в непосредственной близости к GAT-1 [18]. Клонированные GAT-2 и GAT-3 обнаруживают более высокую степень аминокислотной идентичности друг с другом (67% идентичности) и с BGT-1 (68 и 65% идентичности для GAT-2 и GAT-3 соответственно), чем с GAT-1 (~52% идентичности аминокислот). Аминокислотная последовательность GAT-3 у человека, крысы и мыши практически идентична лишь с несколькими заменами [10]. Транспортеры ГАМК способны создавать 105-кратный градиент между внутри- и внеклеточной концентрацией ГАМК [19].
Регулирование содержания синаптического ГАМК с участием GAT предполагает возможность повышения доступности медиатора для активации рецепторов путем использования фармакологических веществ, ингибирующих функцию транспортеров обратного захвата медиатора. Блокада GAT увеличивает синаптическую доступность ГАМК, тем самым облегчая активацию синаптических и внесинаптических ГАМК-рецепторов. Для этой цели широко применяются фармакологические вещества с селективным и неселективным ингибированием транспортеров ГАМК, такие как тиагабин, селективный ингибитор GAT-1, и SNAP 5114, неселективный ингибитор GAT-2 и GAT-3. Тиагабин содержит лиофильную группу, позволяющую препарату беспрепятственно проникать в головной мозг и оказывать противосудорожное действие. Благодаря этому свойству TGB нашел широкое применение в клинике для лечения эпилепсии. SNAP 5114 является неселективным ингибитором GAT-2 и GAT-3 с более высоким сродством к GAT-3, чем GAT-2 (IC50 ~ 5 и 20 мкМ соответственно). Учитывая, что GAT-3 локализован преимущественно в глии, а GAT-2 в слоях лептоменинкса мозга, SNAP 5114 обычно используется в качестве неселективного блокатора GAT-2/3 в исследованиях регуляции GAT-опосредованной синаптической передачи в центральной нервной системе [20].
Можно предположить, что тиагабин и SNAP 5114, блокируя функцию GAT, повышают содержание ГАМК в синаптическом и внесинаптическом пространстве и оказывают выраженное противосудорожное действие. Действительно, тиагабин и SNAP 5114 продемонстрируют противосудорожное действие на разных моделях экспериментальной эпилепсии [18, 20, 21]. Тиагабин ослабляет развитие судорог и на модели “кислородной эпилепсии” [3, 5], однако противосудорожная потенция SNAP 5114 еще не тестировалась при развитии гипербарических кислородных судорог. Кроме того, динамика ГАМК в мозге при ингибировании GAT с помощью тиагабина или SNAP 5114 в условиях гипербарической гипероксии не изучалась.
Цель работы состояла в сравнительной оценке противосудорожного эффекта ингибирования cинаптических и внесинаптических ГАМК-транспортеров при развитии гипербарических кислородных судорог. Для этого изучали противосудорожные эффекты тиагабина и SNAP 5114, а также их совместное действие в условиях гипероксической экспозиции. Кроме того, мы оценивали временной профиль изменений ГАМК в головном мозге после ингибирования GAT-1 с помощью тиагабина и GAT-2/3 с помощью SNAP 5114 у крыс, подвергшихся воздействию кислорода под давлением 5 АТА.
МЕТОДЫ ИССЛЕДОВАНИЯ
В работе использовали крыс линии Вистар массой 278–311 г, полученных из питомника лабораторных животных “Рапполово” (Всеволожский район, Ленинградская область). Протокол опытов одобрен Комиссией по биоэтике ИЭФБ РАН (протокол № 1-12/2022 от 27.01.2022). Было проведено две серии экспериментов.
В первой серии опытов за неделю до проведения экспериментов каждому животному под наркозом (нембутал 50 мг/кг, внутрибрюшинно) вводили металлическую канюлю в боковой желудочек мозга по стереотаксическим координатам [22]: AP = = ‒1.2 мм, LM = ± 2.3 мм, V = 3.5 мм. Канюлю фиксировали акриловым стоматологическим цементом и двумя анкерными винтами, установленными билатерально в височной области черепа. Животном давали восстановиться после операции в течение 5–7 дней. Перед опытом с использованием гипербарического кислорода в боковой желудочек через имплантированную канюлю с помощью микрошприца вводили ингибиторы GAT (Sigma Aldrich, USA) – тиагабин или SNAP 5114. Контрольным животным вводили искусственный ликвор (CSF). Ингибиторы GAT растворяли в искусственном ликворе и вводили в объеме 10 мкл. Содержание препаратов в вводимом объеме CSF составляло: тиагабин – 0.05 мг, SNAP 5114–0.5 мг. Животным контрольной группы вводили искусственный ликвор в объеме 10 мкл.
После введения препаратов крыс размещали в барокамере объемом 100 л по 2 особи в каждом эксперименте. Давление кислорода в камере повышали до 5 АТА со скоростью 1 АТА/мин. Температуру в камере поддерживали в пределах 23–25°С, относительную влажность – около 60%, содержание СО2 – не более 0.05%. Во время экспозиции в ГБО2 проводили непрерывную видеосъемку поведения животных. Гипербарическая экспозиция продолжалась до появления генерализованных клонико-тонических судорог или максимум 90 мин. Время декомпрессии составляло 8 мин. Для оценки противосудорожных эффектов препаратов отмечали время (в минутах) появления специфических двигательных нарушений от начала экспозиции 5 АТА, соответствующих известной шкале состояний судорожного синдрома [23]. При отсутствии генерализованных судорог латентный период принимали за 90 мин.
В опытах на бодрствующих животных было использовано 42 крысы, разделенных на 4 группы. За 30 мин до гипербарического воздействия животным контрольной группы 1 (n = 12) в мозговой желудочек вводили CSF, животным группы 2 (n = 12) вводили TGB, крысам группы 3 (n = 10) – SNAP 5114 и крысам группы 4 (n = 8) вводили комбинацию TGB + SNAP 5114. Экспозиция животных в барокамере продолжалась до появления судорог, но не более 90 мин.
Во второй серии опытов измеряли содержание внеклеточной ГАМК в мозге крыс при ингибировании GAT-1 и GAT-3 с помощью TGB и SNAP 5114. Для этого наркотизированным животным (уретан 750 мг/кг + хлоралоза 250 мг/кг) в стриатум (стереотаксические координаты: А = + 1.0 мм, LМ ± 2.5 мм, D = 5.8 мм) вводили микродиализные зонды (CMA/11, CMA/Microdialysis AB, Sweden). Во время экспозиции в ГБО2 зонды перфузировали искусственным ликвором со скоростью 1.0 мкл/мин, содержащим TGB (30 мкмоль/л) или SNAP 5114 (100 мкмоль/л), а пробы диализата автоматически отбирали каждые 15 мин (CMA 142 Microfraction Collector, AB, Sweden). Концентрацию ГАМК в пробах измеряли с помощью высокопроизводительной жидкостной хроматографии (HPLC) с электрохимической детекцией медиатора (ESA model 5100A), как было ранее подробно описано [24]. ГАМК в образцах количественно определяли в мкмоль/л, используя внешние стандарты. В данной серии опытов у крыс также регистрировали ЭЭГ. Для этого наркотизированным животным ввертывали билатерально в теменную часть черепа до соприкосновения с поверхностью сенсомоторной коры два винта из нержавеющей стали, каждый диаметром 2 мм. При регистрации и анализе ЭЭГ использовали аппаратно-программный комплекс iWorx LX 228 c программным обеспечением LabScribe 2 (iWorx System, Dover, США). Записи биоэлектрической активности использовали для определения латентного периода судорожной активности мозга, которая проявлялась в виде ЭЭГ- спайков.
Для измерения внеклеточной ГАМК было использовано 25 животных, разделенных на 5 групп. ГАМК измеряли у крыс при дыхании атмосферным воздухом с введенным TGB (группа 1, n = 5) или SNAP 5114 (группа 2, n = 5). У крыс групп 1 и 2 измеряли также контрольные значения ГАМК в стриатуме в период постоянной перфузии микродиализных зондов искусственным ликвором до введения ингибиторов. Крыс групп 3-5 подвергали воздействию гипербарического кислорода 5 АТА. Во время экспозиции в ГБО2 животным в стриатум через микродиализные зонды вводили CSF (группа 3, n = 5), TGB (группа 4, n = 5) или SNAP 5114 (группа 5, n = 5). Пробы диализата собирали каждые 15 мин во время ГБО2-экспозиции для измерения ГАМК после декомпрессии с помощью HPLC.
Статистическую обработку полученных результатов проводили с использованием программы SigmaPlot 13.0 (Systat Software, Inc., San Jose, CA, США) и GraphPad Prism 9 (GraphPad Software, Inc., США) с использованием критерия Колмогорова–Смирнова для оценки нормальности распределения. Оценку равенства дисперсий проводили с помощью F-критерия Фишера. Однофакторный дисперсионный анализ ANOVA применяли для выявления отличий латентного периода судорожных реакций в ГБО2 у животных при введении ингибиторов GAT по сравнению с контролем (искусственный ликвор). Двухфакторный дисперсионный анализ ANOVA применяли для сравнения изменений мозговой ГАМК у крыс двух экспериментальных групп. Оценку значимости различий между группами проводили с использованием двухвыборочного t-теста Стьюдента и пост-хок теста Тьюки, внутригрупповые сравнения зависимых признаков оценивали с помощью парного t-теста Стьюдента c поправкой Бонферрони для множественных сравнений. Данные представлены в виде М ± SEM, при этом в качестве статистически значимых различий принимали значения p < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
У контрольной группы бодрствующих крыс, находящихся в барокамере под давлением кислорода 5 АТА, были выявлены характерные двигательные нарушения, соответствующие известной шкале состояний судорожного синдрома [23]. В течение первых 10 мин воздействия ГБО2 животные оставались неподвижными. После этого у них проявились интенсивный груминг, легкое дрожание головы и передних лап, которое у части животных переходило во встряхивание всего тела, известное как симптом “мокрой собаки” (стадия 1). На стадии 2 наблюдались повторяющиеся локальные подергивания мышц мордочки, головы и передних конечностей, а также дрожь всего тела. Миоклонии продолжались от 5 до 15 с и могли повторяться. На стадии 3 у животных наблюдались ритмичные сокращения мышц всего тела продолжительностью 5–15 с, некоторые крысы вставали на задние лапы и двигались назад. На 4-й стадии у животных присутствовали генерализованные клонические или тонические конвульсии. Приступы сопровождались тахикардией и гипервентиляцией.
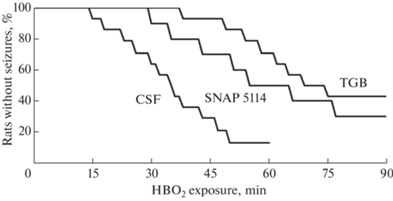
Начальные стадии судорожной активности (стадии 1–3) отчетливо проявлялись также у животных после введения GAT-ингибиторов. Генерализованные судороги (стадия 4) не выявлялись у 40% крыс, получавших TGB, и у 25% крыс после введения SNAP 5114 в течение 90 мин ГБО2-экспозиции (рис. 1).
Рис. 1.
Зависимость появления судорожных реакций у бодрствующих крыс в кислородной среде под давлением 5 АТА с введенными GAT-ингибиторами от времени гипероксической экспозиции.
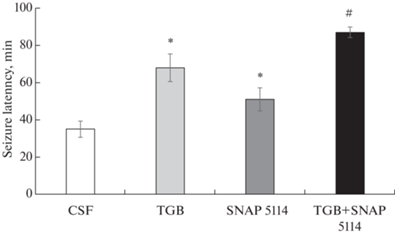
Латентное время появления четвертой стадии судорожной активности у крыс, получавших GAT-ингибиторы, было достоверно большим, чем у контрольной группы, причем TGB вызывал более выраженное противосудорожное действие, чем SNAP 5114 (рис. 2). Совместное введение TGB+SNAP 5114 аддитивно усиливало противосудорожное действие ингибиторов и латентное время появления судорог было еще больше, чем после введения каждого из препаратов в отдельности (рис. 2).
Рис. 2.
Средние значения латентного периода появления судорог у крыс под давлением кислорода 5 АТА после введения в мозговой желудочек искусственного ликвора (CSF), тиагабина (TGB) и SNAP 5114. * p < 0.05 по сравнению с CSF, # p < 0.05 по сравнению с TGB и SNAP 5114.
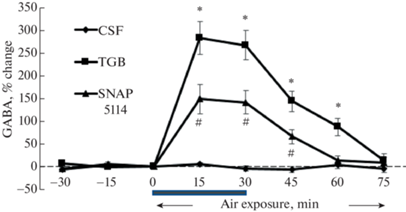
Концентрация ГАМК в стриатуме крыс при дыхании атмосферным воздухом составила 0.059 ± ± 0.011 мкмоль/л. После введения GAT-ингибиторов уровень ГАМК повышался и пиковые значения медиатора были максимальными после введения TGB по сравнению с SNAP 5114 (F(2.21) = 3.349, p = 0.0347) (рис. 3). Двухфакторный анализ ANOVA с повторными измерениями (в конфигурации “mixed model ANOVA”) с пост-хок тестами Тьюки показал достоверность различий между контрольной (ГБО2 5 АТА) и обеими опытными группами с введением препаратов (p < 0.05). Достоверных различий между экспериментальными группами не было выявлено (p > 0.05).
Рис. 3.
Временной профиль изменения концентрации ГАМК в стриатуме крыс при дыхании атмосферным воздухом и 30‑минутном введении в стриатум через микродиализный зонд искусственного ликвора (CSF), тиагабина (TGB) или SNAP 5114. *,#p < 0.05 по сравнению с CSF.
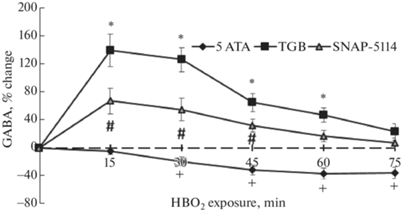
У наркотизированных животных, подвергшихся воздействию кислорода под давлением 5 АТА, двигательных нарушений не наблюдалось, но эпилептиформная активность на ЭЭГ появлялась через 69 ± 5.3 мин. Кислород под давлением 5 АТА вызывал прогрессирующее снижение уровня медиатора в стриатуме контрольных животных, которое в конце кислородной экспозиции достигало 32 ± 4.4% от начального значения (рис. 4). Микродиализное введение TGB в стриатум повышало уровень ГАМК в этом отделе мозга перед началом ГБО2-экспозиции, а в период гипероксического воздействия уровень медиатора постепенно снижался и к концу экспозиции оставался выше, чем у контрольных животных (F(2.15) = 8.726, p = 0.003) (рис. 4). После внутримозгового введения SNAP 5114 уровень ГАМК в стриатуме повышался, но на меньшую величину, чем после введения TGB и снижение под давлением кислорода 5 АТА было более выражено, чем у крыс, которым вводился ингибитор GAT-1 (рис. 4), причем двухфакторный ANOVA с пост-хок тестами Тьюки показал достоверность различий между контрольной (CSF) и обеими опытными группами с введением препаратов (p < 0.05), но не между группами с введением SNAP 5114 или TGB (p > 0.05).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В результате проведенных исследований получено несколько новых данных: (1) ингибирование GAT-1 или GAT-2/3 с помощью TGB или SNAP 5114 соответственно предотвращало развитие гипербарических кислородных судорог; (2) совместное ингибирование GAT-1 и GAT-2/3 вызывало аддитивный противосудорожный эффект; (3) при ингибировании GAT-1 или GAT-3 уровень внеклеточного ГАМК в стриатуме повышался; (4) в условиях гипероксии 5 АТА ингибирование GAT-1 или GAT-2/3 компенсировало понижение внеклеточного ГАМК, наблюдаемого в ГБО2 без введения препаратов.
Причастность ГАМК-ергической системы к развитию кислородных судорог давно установлена и первым доказательством этому являлось снижение содержания тормозного медиатора в мозге животных, подвергшихся воздействию гипербарического кислорода [6–8, 25]. Позднее были получены данные о том, что судорожные разряды на ЭЭГ появлялись, когда внеклеточный ГАМК в мозге понижается у наркотизированных крыс на 37% [24] и на 28% у бодрствующих животных [26]. Скорость снижения внеклеточного медиатора зависела от парциального давления вдыхаемого кислорода. Так, после первых 30 минут дыхания кислородом под давлением 6 АТА снижение внеклеточного ГАМК было в 1.5 раза больше, чем при 5 АТА [5].
Уменьшение внеклеточной ГАМК в ГБО2 связано с понижением активности GAD – фермента, обеспечивающего синтез тормозного медиатора в мозге. Ферментативная активность GAD быстро снижается в ГБО2, о чем свидетельствует показанное нами появление судорог уже через 30–50 мин после начала гипероксического воздействия при 5 АТА, а при 6 АТА менее чем через 30 мин кислородной экспозиции [27, 28]. Для сравнения, в нормальных условиях ингибирование синтеза ГАМК с помощью 3-меркаптопропионовой кислоты понижало уровень ГАМК в мозжечке морских свинок на 39% через 15 мин, в гипоталамусе на 27% через 30 мин и в коре головного мозга на 43% через 90 мин [29]. Потеря каталитической активности GAD связана с посттрансляционной модификацией ее молекулярной структуры под действием различных редокс-молекул [30–32]. Во время гипероксической экспозиции образование окислительно-восстановительных молекул достигает чрезмерных уровней, способных модифицировать структуру GAD посредством окисления или нитрозилирования аминокислотных остатков [33–36]. Наши исследования показали, что механизм инактивации GAD состоит в S-нитрозилировании белка по цистеину путем присоединения группы оксида азота (NO) к его тиоловым остаткам [9]. Валидность такого механизма подавления каталитической активности GAD с участием NO подтверждается экспериментами, в которых ингибирование NO-синтаз (NOS) в мозге крыс с помощью L-NAME защищало животных от развития кислородных судорог [27], а у мышей с нокаутом нейрональной NOS наблюдался более длительный латентный период начала судорог, чем у обычных животных [37].
Экспериментальные данные настоящей работы показывают, что раздельное ингибирование транспортеров GAT-1 или GAT-2/3 восстанавливает нарушенную в экстремальной гипероксии ГАМК-нейропередачу. Доказательством этому может служить следующее. Во-первых, у крыс с предварительно введенными в мозговой желудочек TGB или SNAP 5114 развитие кислородных судорог не проявлялось, что может свидетельствовать о восстановлении ГАМК-передачи за счет увеличения концентрации тормозного медиатора в межклеточном пространстве. Во-вторых, у крыс при дыхании воздухом ингибирование GAT-1 с помощью TGB увеличивало концентрацию ГАМК в стриатуме в 2.9 раза, а внутримозговое введение SNAP 5114 повышало уровень медиатора в 1.7 раза (рис. 1). Эти данные согласуются с другими исследованиями, в которых показано, что внутримозговое введение антагонистов GAT-1 с помощью микродиализа повышало внеклеточную концентрацию ГАМК в мозге грызунов в 1.5–4 раза по сравнению с исходным уровнем [38]. Точно так же применение SNAP 5114 в дозе 100 мкМ увеличивало уровень ГАМК на 250% [21]. В-третьих, в данной работе после ингибирования транспортеров ГАМК уровень мозговой ГАМК при 5 АТА кислорода оставался повышенным, тогда как у контрольных животных концентрация медиатора в стриатуме снижалась на 37 ± 5.4%. Принимая во внимание, что внутримозговой микродиализ позволяет оценивать уровень ГАМК, на 50–70% состоящий из синаптического нейротрансмиттера [39], и некоторой части ГАМК, высвобождаемой из глиальных клеток [40], можно полагать, что ингибирование ГАМК-транспортеров восстанавливает ГАМК-ергическую функцию в ГБО2 за счет усиления фазической синаптической и тонической экстрасинаптической передач. Используемые в работе TGB и SNAP 5114 нацелены на GAT, локализованные в пресинаптической мембране и в астроцитах. Противосудорожная аддитивность действия двух ингибиторов, вероятно, является следствием ингибирования как синаптических, так и внесинаптических ГАМК-транспортеров.
Вместе с тем имеющиеся данные пока не дают прямого ответа на вопрос, какая из двух ГАМК-ергических нейропередач, синаптическая или экстрасинаптическая, угнетается больше в условиях гипероксического воздействия. Показанное в нашей работе увеличение межклеточной ГАМК в стриатуме или в гиппокампе крыс у Kersante и соавт. [20] при ингибировании GAT-1 и более эффективное противосудорожное действие TGB по сравнению с SNAP 5114 косвенно указывают на разный вклад синаптической и внесинаптической нейропередач в снижение ГАМК-опосредованной нейротрансмиссии в ГБО2. В то же время зависимость между содержанием медиатора в синаптической щели и в прилежащем межклеточном пространстве позволяет предположить, что внесинаптическая ГАМК-ергическая передача также может подавляться при ГБО2. Это предположение основано на известных данных о наличии в головном мозге тонической экстрасинаптической ГАМК-опосредованной нейротрансмиссии через ГАМКА-рецепторы, локализованные на теле нейронов [13].
Выраженный противосудорожный эффект применяемых препаратов свидетельствует о том, что ГБО2 не подавляет функцию транспортеров ГАМК. В противном случае, вводимые в головной мозг антагонисты ГАМК транспортеров не вызывали бы столь выраженного противосудорожного действия. Среди двух ингибиторов GAT, использованных в данной работе, TGB оказался более эффективным в предотвращении кислородных судорог по сравнению с SNAP 5114. Тиагабин является селективным ингибитором транспортеров ГАМК, переносящих медиатор преимущественно в пресинаптические нейроны, поэтому при их ингибировании синаптический уровень медиатора повышается, обеспечивая противосудорожный эффект за счет усиления тормозных постсинаптических потенциалов [41].
Дефицит ГАМК-ергической функции при ГБО2, возникающий из-за ингибирования синтеза ГАМК, может быть преодолен повышением уровня ГАМК в синаптическом пространстве и во внесинаптической среде. Проверка этой гипотезы показала, что ингибиторы транспортера ГАМК, вводимые в мозговой желудочек, повышают уровень ГАМК в головном мозге и предотвращают развитие кислородных судорог. Следовательно, блокирование механизмов синаптического клиренса ГАМК может компенсировать снижение синтеза ГАМК в ГБО2, повысить концентрацию ГАМК до уровня, достаточного для обеспечения тормозной нейропередачи, и тем самым предотвратить развитие кислородного судорожного синдрома.
Список литературы
Зальцман ГЛ (1968) Стадии развития кислородной эпилепсии и функциональное состояние нервной системы. В кн. Гипербарические эпилепсия и наркоз. Л. Наука. [Zaltsman GL (1968) Stages of formation of oxygen epilepsy and the functional state of the centres of the nervous system. In book: Hyperbaric epilepsy and narcosis. Ed. Zaltsman G.L. Leningrad. Nauka. (In Russ)].
Demchenko IT, Zhilyaev SY, Moskvin AN, Krivchenko AI, Piantadosi CA, Allen BW (2017) Antiepileptic drugs prevent seizures in hyperbaric oxygen: A novel model of epileptiform activity. Brain Res 1657: 347–354. https://doi.org/10.1016/j.brainres.2016.12.032
Demchenko IT, Zhilyaev SY, Alekseeva OS, Krivchenko AI, Piantadosi CA, Gasier HG (2019) Increased Antiseizure Effectiveness with Tiagabine Combined with Sodium Channel Antagonists in Mice Exposed to Hyperbaric Oxygen. Neurotox Res 36 (4): 788–795. https://doi.org/10.1007/s12640-019-00063-5
Ciarlone GE, Hinojo CM, Stavitzski NM, Dean JB (2019) CNS function and dysfunction during exposure to hyperbaric oxygen in operational and clinical settings. Redox Biol 27: 101–159. https://doi.org/10.1016/j.redox.2019.101159
Demchenko IT, Zhilyaev SY, Platonova TF, Alekseeva OS, Nikitina ER (2021) Inhibition of GABA-Transaminase and GABA-Transporters in the Brain with Vigabatrin and Tiagabin Prevents Seizure Development in Rats Breathing Hyperbaric Oxygen. J Evol Biochem Physiol 57 (5): 1101–1109. https://doi.org/10.1134/S0022093021050112
Щербакова ГВ (1962) Активность глютаматдекарбоксилазы и содержание γ-аминомасляной кислоты в мозге крыс при разных функциональных состояниях, вызванных повышенным давлением кислорода. ДАН СССР 146(5): 1213–1215. [Shcherbakova GV Glutamate decarboxylase activity and γ-aminobutyric acid content in rat brain at different functional states caused by high oxygen pressure. DAN USSR 146 (5): 1213–1215. (In Russ)].
Wood JD, Watson WJ (1964) The effect of oxygen on glutamic acid decarboxylase and gamma-aminobutyric acid-alpha-ketoglutaric acid transaminase activities in rat brain homogenates. Can J Physiol Pharmacol 42: 277–279. https://doi.org/10.1139/y64-032
Mialon P, Gibey R, Bigot JC, Barthelemy L (1992) Changes in striatal and cortical amino acid and ammonia levels of rat brain after one hyperbaric oxygen-induced seizure. Aviat Space Environ Med 63 (4): 287–291.
Gasier HG, Demchenko IT, Tatro LG, Piantadosi CA (2017) S-nitrosylation of GAD65 is implicated in decreased GAD activity and oxygen-induced seizures. Neurosci Lett 653: 283–287. https://doi.org/10.1016/j.neulet.2017.05.067
Borden LA, Dhar TG, Smith KE, Branchek TA, Gluchowski C, Weinshank RL (1994) Cloning of the human homologue of the GABA transporter GAT-3 and identification of a novel inhibitor with selectivity for this site. Recept Channels 2 (3): 207–213.
Richerson GB, Wu Y (2003) Dynamic equilibrium of neurotransmitter transporters: not just for reuptake any more. J Neurophysiol 90 (3): 1363–1374. https://doi.org/10.1152/jn.00317.2003
Overstreet LS, Westbrook GL (2003) Synapse density regulates independence at unitary inhibitory synapses. J Neurosci 23 (7): 2618–2626. https://doi.org/10.1523/JNEUROSCI.23-07-02618.2003
Semyanov A, Walker MC, Kullmann DM, Silver RA (2004) Tonically active GABA A receptors: modulating gain and maintaining the tone. Trends Neurosci 27 (5): 262–269. https://doi.org/10.1016/j.tins.2004.03.005
Böck MC, Höfner G, Wanner KT (2020) N-Substituted Nipecotic Acids as (S)-SNAP-5114 Analogues with Modified Lipophilic Domains. Chem Med Chem 15 (9): 756–771. https://doi.org/10.1002/cmdc.201900719
Guastella J, Nelson N, Nelson H, Czyzyk L, Keynan S, Miedel MC, Davidson N, Lester HA, Kanner BI (1990) Cloning and expression of a rat brain GABA transporter. Science 249 (4974): 1303–1306. https://doi.org/10.1126/science.1975955
Liu QR, López-Corcuera B, Mandiyan S, Nelson H, Nelson N (1993) Molecular characterization of four pharmacologically distinct gamma-aminobutyric acid transporters in mouse brain. J Biol Chem 268 (3): 2106–2112.
Nelson H, Mandiyan S, Nelson N (1990) Cloning of the human brain GABA transporter. FEBS Lett 269 (1): 181–184. https://doi.org/10.1016/0014-5793(90)81149-i
Jin XT, Galvan A, Wichmann T, Smith Y (2011) Localization and Function of GABA Transporters GAT-1 and GAT-3 in the Basal Ganglia. Front Syst Neurosci 5: 63. https://doi.org/10.3389/fnsys.2011.00063
Schousboe A (1981) Transport and metabolism of glutamate and GABA in neurons are glial cells. Int Rev Neurobiol 22: 1–45. https://doi.org/10.1016/s0074-7742(08)60289-5
Kersanté F, Rowley SC, Pavlov I, Gutièrrez-Mecinas M, Semyanov A, Reul JM, Walker MC, Linthorst AC (2013) A functional role for both-aminobutyric acid (GABA) transporter-1 and GABA transporter-3 in the modulation of extracellular GABA and GABAergic tonic conduc tances in the rat hippocampus. J Physiol 591 (10): 2429–2441. https://doi.org/10.1113/jphysiol.2012.246298
Dalby NO (2000) GABA-level increasing and anticonvulsant effects of three different GABA uptake inhibitors. Neuropharmacology 39 (12): 2399–2407. https://doi.org/10.1016/s0028-3908(00)00075-7
Paxinos G, Watson C (2005) The Rat Brain in Stereotaxic Coordinates. Boston, MA: Elsevier.
Racine RJ (1972) Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32 (3): 281–294. https://doi.org/10.1016/0013-4694(72)90177-0
Demchenko IT, Piantadosi CA (2006) Nitric oxide amplifies the excitatory to inhibitory neurotransmitter imbalance accelerating oxygen seizures. Undersea Hyperb Med 33(3): 169–174.
Faiman MD, Nolan RJ, Baxter CF, Dodd DE (1977) Brain gamma-aminobutyric acid, glutamic acid decarboxylase, glutamate, and ammonia in mice during hyperbaric oxygenation. J Neurochem 28 (4): 861–865. https://doi.org/10.1111/j.1471-4159.1977.tb10640.x
Zhang S, Takeda Y, Hagioka S, Takata K, Aoe H, Naka-tsuka H, Yokoyama M, Morita K (2005) Measurement of GABA and glutamate in vivo levels with high sensitivity and frequency. Brain Res Brain Res Protoc 14 (2): 61–66. https://doi.org/10.1016/j.brainresprot.2004.03.005
Demchenko IT, Boso AE, O’Neill TJ, Bennett PB, Piantadosi CA (2000) Nitric oxide and cerebral blood flow responses to hyperbaric oxygen. J Appl Physiol (1985) 88 (4): 1381–1389. https://doi.org/10.1152/jappl.2000.88.4.1381
Dean JB, Mulkey DK, Garcia AJ 3rd, Putnam RW, Henderson RA 3rd (2003) Neuronal sensitivity to hyperoxia, hypercapnia, and inert gases at hyperbaric pressures. J Appl Physiol (1985) 95 (3): 883–909. https://doi.org/10.1152/japplphysiol.00920.2002
Alsip NL, DiMicco JA (1992) Time course of effects of 3-mercaptopropionic acid on GABA levels in different brain regions in guinea pigs: possible relationship with associated cardiovascular changes. Neurochem Res 17 (5): 443–448. https://doi.org/10.1007/BF00969890
Dickinson BC, Chang CJ (2011) Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol 7 (8): 504–511. https://doi.org/10.1038/nchembio.607
Corcoran A, Cotter TG (2013) Redox regulation of protein kinases. FEBS J 280 (9): 1944–1965. https://doi.org/10.1111/febs.12224
Go YM, Jones DP (2013) The redox proteome. J Biol Chem 288 (37): 26512–26520. https://doi.org/10.1074/jbc.R113.464131
Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J (1992) S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A 89 (1): 444–448. https://doi.org/10.1073/pnas.89.1.444
Chung HS, Wang SB, Venkatraman V, Murray CI, Van Eyk JE (2013) Cysteine oxidative posttranslational modifications: emerging regulation in the cardiovascular system. Circ Res 112 (2): 382–392. https://doi.org/10.1161/CIRCRESAHA.112.268680
Gould N, Doulias PT, Tenopoulou M, Raju K, Ischiropoulos H (2013) Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J Biol Chem 288 (37): 26473–26479. https://doi.org/10.1074/jbc.R113.460261
Kovacs I, Lindermayr C (2013) Nitric oxide-based protein modification: formation and site-specificity of protein S-nitrosylation. Front Plant Sci 4: 137.https://doi.org/10.3389/fpls.2013.00137
Atochin DN, Demchenko IT, Astern J, Boso AE, Piantadosi CA, Huang PL (2003) Contributions of endothelial and neuronal nitric oxide synthases to cerebrovascular responses to hyperoxia. J Cereb Blood Flow Metab 23 (10): 1219–1226. https://doi.org/10.1097/01.WCB.0000089601.87125.E4
Fink-Jensen A, Suzdak PD, Swedberg MDB, Judge ME, Hansen L, Nielsen PG (1992) The γ-aminobutyric acid (GABA) uptake inhibitor, tiagabine, increases extracellular brain levels of GABA in awake rats. Eur J Pharmacol 220 (2–3): 197–201. https://doi.org/10.1016/0014-2999(92)90748-s
Van der Zeyden M, Oldenziel WH, Rea K, Cremers TI, Westerink BH (2008) Microdialysis of GABA and glutamate: analysis, interpretation and comparison with microsensors. Pharmacol Biochem Behav 90 (2): 135–147. https://doi.org/10.1016/j.pbb.2007.09.004
Del Arco A, Segovia G, Fuxe R, Mora F (2003) Changes in dialysate concentrations of glutamate and GABA in the brain: An index of volume transmission mediated actions? J Neurochem 85: 23–33. https://doi.org/10.1046/j.1471-4159.2003.01692
Keros S, Hablitz JJ (2005) Subtype-specific GABA transporter antagonists synergistically modulate phasic and tonic GABAA conductances in rat neocortex. J Neurophysiol 94 (3): 2073–2085. https://doi.org/10.1152/jn.00520.2005
Дополнительные материалы отсутствуют.
Инструменты
Журнал эволюционной биохимии и физиологии