

Журнал аналитической химии, 2020, T. 75, № 4, стр. 338-347
Применение неподвижной фазы на основе пористого графитизированного углерода для определения продуктов трансформации 1,1-диметилгидразина методом жидкостной хроматографии–масс-спектрометрии
Н. В. Ульяновский a, *, Д. С. Косяков a, М. С. Попов a, И. И. Пиковской a, О. Ю. Хорошев a
a Северный (Арктический) федеральный университет имени М.В. Ломоносова, Центр коллективного пользования научным оборудованием “Арктика”
163002 Архангельск, Набережная Северной Двины, 17, Россия
* E-mail: n.ulyanovsky@narfu.ru
Поступила в редакцию 06.12.2018
После доработки 14.03.2019
Принята к публикации 06.10.2019
Аннотация
Предложен подход к одновременному определению пяти продуктов трансформации ракетного топлива на основе 1,1-диметилгидразина (1-метил-1Н-1,2,4-триазола, N-нитрозодиметиламина, N,N-диметилформамида, диметилгидразонов формальдегида и ацетальдегида), основанный на сочетании ВЭ-ЖХ на пористом графитизированном углероде (Hypercarb) с масс-спектрометрическим и тандемным масс-спектрометрическим детектированием. Пределы обнаружения 0.6–7 мкг/л. Установлено, что неподвижная фаза на основе пористого графитизированного углерода обеспечивает эффективное хроматографическое удерживание и разделение азотсодержащих продуктов трансформации 1,1-диметилгидразина за счет реализации смешанного механизма, включающего как гидрофобные, так и индукционные взаимодействия с аналитами. Показано отсутствие матричных эффектов при анализе природных вод и метанольного экстракта торфяной болотной почвы.
Экологическое сопровождение ракетно-космической деятельности помимо контроля содержания высокотоксичного ракетного топлива на основе 1,1-диметилгидразина (несимметричный диметилгидразин, НДМГ) в объектах окружающей среды неотъемлемо связано с определением широкого круга опасных продуктов, образующихся в ходе его окислительной деградации. Среди них в первую очередь выделяют алкиламины, N,N-диметилгидразоны формальдегида (ДМГФ) и ацетальдегида (ДМГА), 1,1,4,4-тетраметил-2-тетразен, N-нитрозодиметиламин (НДМА), диметилгидразид муравьиной кислоты (ДМГМК), N,N-диметилформамид (ДМФА), 1-метил-1Н-1,2,4-триазол (МТ) и некоторые другие соединения [1–3]. Благодаря высокому содержанию ДМГФ, ДМГА, ДМФА, НДМА, ДМГМК и МТ в загрязненных ракетным топливом почвах [4–6], данные соединения могут выступать в качестве своеобразных маркеров [3] загрязнения в течение длительного времени даже при полном отсутствии исходного НДМГ. Разработка экспрессных, селективных и высокочувствительных методов их определения является актуальной задачей.
Для определения продуктов трансформации НДМГ в объектах окружающей среды наибольшее применение нашли методы газовой хроматографии [7–10] и высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием (ВЭЖХ–МС) [5, 6, 11], обеспечивающие высокую селективность и чувствительность анализа. Метод ВЭЖХ–МС наиболее предпочтителен, так как позволяет одновременно анализировать как экстракты почв, так и различные водные объекты без дополнительных стадий пробоподготовки и дериватизации. Ключевой проблемой для определения продуктов трансформации НДМГ методом ВЭЖХ является достижение оптимального удерживания сильно различающихся по свойствам соединений для обеспечения высокой селективности анализа и снижения помех со стороны матрицы.
Учитывая высокую полярность многих продуктов трансформации НДМГ, применение обращенно-фазовой хроматографии для их определения ограничено. Описаны [12, 13] лишь отдельные примеры определения N-нитрозодиметиламина, который является наиболее опасным соединением из образующихся в ходе деградации НДМГ в окружающей среде, в различных природных объектах с разделением на октадецильных сорбентах. В случае традиционно используемой при определении гидразинов ионной хроматографии (ИХ) с разделением на сульфокатионообменной неподвижной фазе [6, 14] затруднительно определять неионогенные аналиты (НДМА, ДМФА), а также МТ, который не способен протонироваться в значительной степени при значении рН, используемом для разделения подвижной фазы. Похожая ситуация наблюдается при разделении в режиме гидрофильной хроматографии (HILIC) и ион-парной хроматографии (ИПХ) [11, 15]. В последнем случае, помимо слабого удерживания многих аналитов, существенно снижается чувствительность масс-спектрометрического детектирования за счет применения высоких концентраций буферных растворов и ион-парных реагентов.
Решением проблемы является применение неподвижных фаз со смешанным механизмом удерживания, способных эффективно разделять азотсодержащие продукты трансформации НДМГ, обеспечивая хорошее отделение от матрицы. Наиболее перспективным в этом плане представляется неподвижная фаза на основе пористого графитизированного углерода (ПГУ). Этот сорбент обладает такими преимуществами, как возможность работать со 100%-ными водными подвижными фазами, устойчивость в широких диапазонах рН и температур [16]. Благодаря эффекту “полярного удерживания” [16, 17] ПГУ демонстрирует значительное увеличение коэффициентов удерживания (k) полярных и ионных аналитов по сравнению с октадецильными неподвижными фазами. В работах [18, 19] изучено удерживание азотсодержащих гетероциклических соединений различных классов (1,2,4-триазола, имидазола и др.) на неподвижной фазе Hypercarb и показана перспективность применения ПГУ для хроматографического разделения указанных аналитов. При этом установлено, что гидрофобность и дисперсионные взаимодействия, хотя и оказывают влияние на разделение, не определяют порядок элюирования. Удерживание исследуемых соединений определялось преимущественно взаимодействием полярных групп аналитов с поверхностью ПГУ с образованием на ней индуцированных зарядов. Помимо ограниченного круга азотсодержащих гетероцикличеких соединений, имеются сведения о применении неподвижной фазы на основе ПГУ для хроматографического определения нитрозаминов (в частности, НДМА) [20].
Цель настоящего исследования – оценка возможности применения неподвижной фазы на основе ПГУ для определения важнейших продуктов трансформации НДМГ в объектах окружающей среды методом ВЭЖХ–МС. В качестве аналитов выбраны пять соединений, для которых характерно слабое удерживание как в обращенно-фазовом, так и в ионообменном режимах хроматографии: МТ, НДМА, ДМФА, диметилгидразоны формальдегида и ацетальдегида.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты и материалы. 1-Метил-1H-1,2,4-триазол (98%, Fluorochem, Великобритания), N,N-диметилформамид (ГСО, раствор с концентрацией 1 г/л, Экоаналитика, Россия), ДМФА (99.8%, Lab-Scan, Польша) использовали для приготовления градуировочных и рабочих растворов. Для синтеза ДМГФ и ДМГА использовали формальдегид (37%-ный водный раствор), ацетальдегид (≥99.5%), 1,1-диметилгидразин (99%), хлорид кальция (≥96.0%) и гидроксид натрия (≥98%) (Sigma-Aldrich, Германия). Для приготовления буферных растворов, растворов аналитов и подвижной фазы использовали ацетонитрил (0 сорт, Криохром, Россия), метанол (HPLC grade, J.T.Baker, США), формиат аммония (10 М раствор, Sigma-Aldrich, Швейцария), муравьиную кислоту (98%, Sigma-Aldrich, Германия), а также ультрачистую воду с удельным сопротивлением 18.2 MΩ см, полученную с применением системы Milli-Q (Millipore, Франция).
Диметилгидразоны формальдегида и ацетальдегида синтезировали по известным методикам [21], при этом выход целевых продуктов составил 72 и 79%, соответственно. Подтверждали структуру и проверяли чистоту препаратов методами ЯМР-спектрометрии, ИК-спектроскопии и газовой хромато-масс-спектрометрии.
Структурные формулы исследуемых продуктов трансформации 1,1-диметилгидразина представлены на схеме 1 .
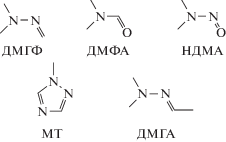
Схема 1 . Структурные формулы исследуемых соединений.
Приготовление растворов. Исходные растворы исследуемых соединений (1000 мг/л), за исключением НДМА, готовили по точной навеске и хранили в холодильнике при 4°С не более недели. Рабочие растворы аналитов с концентрациями 0.001–10 мг/л получали путем последовательных разбавлений исходного раствора водой. Рабочие растворы готовили непосредственно перед анализом и хранили при 4°С в течение не более 24 ч.
Объекты исследования и подготовка проб. В качестве реальных природных объектов для апробации разработанных подходов выбрали три образца природных вод: воды из р. Северная Двина (I), грунтовой воды из скважины в Архангельской области (II), незагрязненной поверхностной воды торфяного болота, отобранной в районе падения отработанных частей ракет-носителей в 2014 г. в Архангельской области (III) и образец загрязненной ракетным топливом торфяной болотной почвы (IV), отобранной в 2014 г. в Архангельской области в месте падения первой ступени ракеты-носителя Циклон-3. Влажность образца почвы составляла 86%, величина pH водной вытяжки 3.7, pH солевой вытяжки 2.8, зольность 2.6%. Валовое содержание НДМГ, определенное методом ИХ с амперометрическим детектированием после отгонки с паром из щелочной среды [14], составило 158 ± 26 мг/кг абсолютно сухого образца.
В день отбора после доставки в лабораторию образец почвы тщательно перемешивали, замораживали в герметичных полиэтиленовых контейнерах и сохраняли при –18°С без доступа воздуха. Непосредственно перед проведением анализа содержимое контейнера размораживали и снова перемешивали до однородности. Экстракцию почвы по Сокслету проводили с использованием автоматической системы экстракции B-811 (BUCHI, Швейцария) [5]. Образец почвы массой 10 г помещали в патрон из фильтровальной бумаги и экстрагировали метанолом в течение 7 ч. Полученный экстракт (170 мл) упаривали в токе азота (99.99%) до объема 1 мл, разбавляли в 10 раз водой для предотвращения размывания хроматографических пиков при вводе образца в растворителе с высокой элюирующей силой, фильтровали через нейлоновый фильтр с размером пор 0.22 мкм и 1 мл фильтрата помещали в автоматический дозатор для последующего анализа. Поскольку при пробоподготовке из 10 г почвы получали 10 мл анализируемого раствора, содержание аналитов в почвах (мг/кг) соответствовало их концентрации в анализируемых растворах (мг/л).
Хромато-масс-спектрометрический анализ. Хроматографическое разделение проводили с использованием системы для ВЭЖХ LC-30 Nexera, состоящей из двух насосов LC-30AD, дегазатора DGU-20A5R, автоматического дозатора SIL-30AC и термостата колонок CTO-20A (Shimadzu, Япония). Для детектирования в режиме мониторинга выделенных ионов (МВИ) использовали одноквадрупольный масс-спектрометрический детектор LCMS-2020 (Shimadzu, Япония). Детектирование в режиме мониторинга заданных реакций (МЗР) проводили с использованием трехквадрупольного тандемного масс-спектрометрического детектора LCMS-8040 (Shimadzu, Япония). Для управления приборами, сбора и обработки данных использовали программное обеспечение LabSolutions LCMS (Shimadzu, Япония). Разделение аналитов проводили на колонке 150 × 3 мм (размер частиц 3 мкм) с пористым графитизированным углеродным сорбентом Hypercarb (Thermo, США) в изократическом режиме. Хроматографическую колонку термостатировали при 40°C. Расход подвижной фазы составлял 0.3 мл/мин, объем вводимой пробы 20 мкл.
Ввиду высокого содержания воды в подвижной фазе при разделении аналитов на ПГУ затруднено распыление элюента в режиме ионизации электрораспылением, поэтому масс-спектрометрический анализ проводили в условиях химической ионизации при атмосферном давлении (ХИ-АД) при положительной полярности, которая способствует лучшей ионизации аналитов в данных условиях. Использовали одинаковые параметры ионного источника для обоих масс-спектрометрических детекторов, обеспечивающие максимальную интенсивность ионов аналитов в масс-спектрах, установленные в результате предварительных экспериментов: поток распыляющего и осушающего газов (N2 1.5 и 10 л/мин соответственно, температуры источника, нагревательного блока и линии десольватации 450, 250 и 200°C соответственно. Напряжение на игле коронного разряда 4.5 кВ. Параметры масс-спектрометрического детектирования представлены в табл. 1.
Таблица 1.
Параметры детектирования исследуемых аналитов
| Аналит | Молекулярная масса, Да | Ион-предшественник*, m/z |
Ион-продукт, m/z | Энергия соударений, эВ | Время накопления, с |
|---|---|---|---|---|---|
| ДМФА | 73 | 74 | 46 | 18 | 0.2 |
| 42** | 26 | ||||
| НДМА | 74 | 75 | 43 | 19 | 0.2 |
| 58** | 17 | ||||
| ДМГФ | 72 | 73 | 44 | 13 | 0.2 |
| 45** | 16 | ||||
| ДМГА | 86 | 87 | 44 | 14 | 0.2 |
| 45** | 15 | ||||
| МТ | 83 | 84 | 57 | 22 | 0.2 |
| 42** | 21 |
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Хроматографическое удерживание и разделение аналитов на неподвижной фазе на основе пористого графитизированного углерода. Учитывая смешанный механизм удерживания на ПГУ, важнейшими факторами, определяющими взаимодействия аналитов с неподвижной фазой, являются природа и содержание органического растворителя в подвижной фазе, температура, рН и ионная сила среды.
Элюирование чистой водой обеспечивает близкое к оптимальному удерживание всех исследуемых аналитов – величины k для них лежат в диапазоне от 2 до 6. Добавление в подвижную фазу органического растворителя (ацетонитрила или метанола) приводит к значительному снижению факторов удерживания (рис. 1), при этом наибольшее влияние состав подвижной фазы оказывает на поведение МТ, характеризующегося наибольшей величиной k в водной среде. Примечательно, что при использовании в качестве органического сорастворителя ацетонитрила наблюдается инверсия времен удерживания для пар МТ–ДМГА (3% CH3CN) и МТ–ДМГФ (8% CH3CN), в то время как в случае водно-метанольного элюента порядок выхода аналитов остается неизменным независимо от состава подвижной фазы. Это означает дифференцированное изменение вкладов различных типов взаимодействий в удерживание МТ и гидразонов (или смену преобладающего механизма удерживания) при добавлении даже небольших количеств ацетонитрила в подвижную фазу. Так, причиной наблюдаемого явления может служить конкуренция МТ с высокоосновными молекулами ацетонитрила или комплексами ацетонитрил–вода [22] при реализации удерживания за счет взаимодействий неподеленных электронных пар сорбата с поверхностью ПГУ, важность которых отмечалась ранее [23, 24]. В случае полярных молекул гидразонов, обладающих способной к протонированию аминогруппой, во взаимодействиях с ПГУ ведущую роль должны играть индукционные эффекты.
Рис. 1.
Зависимость фактора удерживания (k) от типа органического модификатора и его содержания в подвижной фазе. 1 – МТ, 2 – ДМГА, 3 – ДМГФ, 4 – НДМА, 5 – ДМФА. Расход подвижной фазы 0.3 мл/мин, pH 6.5.
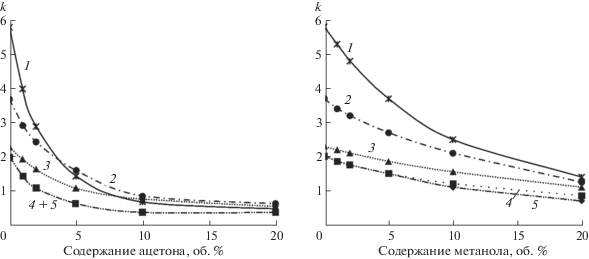
Добавление в подвижную фазу органических растворителей, несмотря на нежелательное снижение величин k аналитов, благоприятно сказывается как на ширине и форме хроматографических пиков, так и на чувствительности масс-спектрометрического детектирования. Компромиссное использование для элюирования 5%-ного водного раствора метанола позволяет повысить интенсивность сигнала в 2–3 раза при сохранении хорошего разрешения пиков МТ и ДМГА и приемлемых величинах факторов удерживания всех аналитов (1.5–4). В то же время добиться полного разделения НДМА и ДМФА не удалось в изучаемом диапазоне составов элюента.
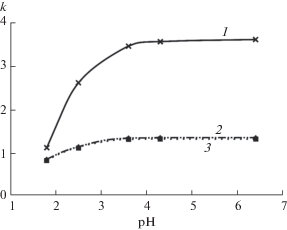
Варьирование величины рН элюента в диапазоне от 1.8 до 6.5 выявило нестабильность диметилгидразонов при рН < 5 – наблюдали ухудшение воспроизводимости площадей хроматографических пиков и их существенное уменьшение при снижении рН. Данное наблюдение согласуется с литературными данными [25]. Для МТ, НДМА и ДМФА величины k снижаются при повышении кислотности элюента (рис. 2), что может быть обусловлено несколькими факторами. Во-первых, происходит протонирование МТ, для сопряженной кислоты (протонированной формы) которого величины pKa лежат в диапазоне 3–3.5 [26]. Это способствует уменьшению гидрофобных взаимодействий между неподвижной фазой и аналитами и приводит к значительному снижению времен удерживания. Во-вторых, для создания низких величин pH (2–3) требуется добавка существенного количества муравьиной кислоты, что приводит к увеличению доли органического компонента в подвижной фазе и соответствующему изменению ее элюирующей силы (по аналогии с рассмотренным выше действием небольших добавок ацетонитрила и метанола). Этим фактором можно объяснить снижение k для ДМФА и НДМА, которые не протонируются в указанном диапазоне pH. Наилучшие результаты с учетом необходимости обеспечения стабильности диметилгидразонов достигаются в диапазоне pH 6–6.5, что приводит к необходимости контроля этой величины, особенно при анализе экстрактов торфяных болотных почв, имеющих кислую реакцию среды.
Рис. 2.
Зависимость факторов удерживания МТ (1), НДМА (2) и ДМФА (3) от рН подвижной фазы. Подвижная фаза: вода–метанол (95 : 5, по объему), расход 0.3 мл/мин.
Для создания достаточной буферной емкости подвижной фазы при заданном уровне рН использовали добавки формиата аммония, при этом концентрацию соли варьировали в диапазоне 0–50 мМ. Установлено, что факторы удерживания аналитов практически не зависят от концентрации формиата аммония во всем исследуемом диапазоне, что в свою очередь свидетельствует о незначительной роли межионных взаимодействий между аналитами в подвижной фазе и пористым графитизированным углеродным сорбентом. Повышение концентрации буферной соли более 20 мМ вызывает существенное подавление ионизации аналитов в источнике ионов масс-спектрометра.
Варьирование температуры хроматографической колонки в диапазоне 30–50°С не привело к заметным изменениям удерживания аналитов, что свидетельствуют о небольших значениях теплот адсорбции аналитов на неподвижной фазе. По-видимому, существенное изменение удерживания и селективности может быть достигнуто лишь в условиях субкритического состояния подвижной фазы, в первую очередь, путем снижения полярности растворителя.
Суммируя полученные данные по влиянию состава подвижной фазы, величины pH и ионной силы, можно предположить, что основной вклад в удерживание исследуемых продуктов трансформации НДМГ вносят гидрофобные и индукционные (образование наведенных диполей на поверхности графита) взаимодействия с неподвижной фазой, вклады которых определяются составом подвижной фазы, рН и природой аналитов. Благодаря такому смешанному механизму, азотсодержащие соединения хорошо удерживаются на неподвижной фазе на основе ПГУ, который можно рассматривать в качестве альтернативы применявшимся ранее для определения продуктов трансформации НДМГ обращенно-фазовым и ионообменным сорбентам, а также полярным неподвижным фазам в режиме HILIC.
В качестве оптимальной подвижной фазы для разделения выбранных аналитов можно рекомендовать 10 мМ водный раствор формиата аммония (рН 6.4), содержащий 5 об.% метанола. Пример хроматограммы модельной смеси аналитов в оптимальных условиях представлен на рис. 3.
Рис. 3.
Хроматограмма модельной смеси исследуемых аналитов с концентрацией 1 мг/л каждого компонента в режиме мониторинга выделенных ионов. Подвижная фаза: 10 мМ водный раствор формиата аммония (рН 6.4), содержащий 5 об. % метанола, расход 0.3 мл/мин.

Метрологические характеристики. Анализ модельных растворов аналитов в режимах МВИ и МЗР показал, что градуировочные зависимости линейны в широком диапазоне концентраций, охватывающем не менее трех порядков (табл. 2), с коэффициентами корреляции более 0.99 для каждого компонента. На основе критериев отношения сигнал/шум, равных 3 и 10, рассчитаны пределы обнаружения (сmin) и нижние границы определяемых концентраций (сн) соответственно. Полученные значения подтверждены путем анализа модельного раствора с концентрациями аналитов, близкими к сн. Минимальные пределы обнаружения при детектировании в режиме МВИ достигнуты для МТ (1.5 мкг/л), максимальные – для ДМГФ и ДМГА (13 и 10 мкг/л соответственно). Диметилформамид и нитрозодиметиламин характеризуются величинами cmin на уровне нескольких единиц мкг/л. Переход к детектированию в режиме МЗР позволил повысить чувствительность в 2–5 раз в зависимости от аналита (табл. 2).
Таблица 2.
Метрологические характеристики разработанного подхода
| Аналит | Режим анализа | Линейный диапазон, мкг/л | R2 | сmin, мкг/л | сн, мкг/л |
|---|---|---|---|---|---|
| ДМФА | МВИ | 13–5000 | 0.9987 | 3.8 | 13 |
| МЗР | 3–5000 | 0.9993 | 0.80 | 2.6 | |
| НДМА | МВИ | 19–5000 | 0.9998 | 5.8 | 19 |
| МЗР | 8–5000 | 0.9996 | 2.4 | 7.9 | |
| ДМГФ | МВИ | 43–10 000 | 0.9996 | 13 | 43 |
| МЗР | 23–10 000 | 0.9985 | 7.1 | 23 | |
| ДМГА | МВИ | 33–10 000 | 0.9994 | 10 | 33 |
| МЗР | 17–10 000 | 0.9988 | 5 | 17 | |
| МТ | МВИ | 5–5000 | 0.9963 | 1.5 | 5.0 |
| МЗР | 2–5000 | 0.9991 | 0.60 | 2.0 |
Достигнутые пределы обнаружения для ДМФА, НДМА и МТ в 2–11 раз ниже полученных нами ранее методом ИХ–МС/МС [6], и в 2–5 раз ниже по сравнению с методом HILIC–МС/МС [11]. Такое различие можно объяснить более подходящими для масс-спектрометрического детектирования условиями хроматографического разделения (высокая эффективность, низкая ионная сила подвижной фазы, отделение пиков аналитов от мертвого объема и др.), а также использованием ХИАД. Еще более значительное повышение чувствительности наблюдается при сравнении разработанной методики с результатами, полученными с применением методов ИХ–МС и ИПХ–МС [5, 15]. В последнем случае выигрыш в чувствительности при разделении на неподвижной фазе Hypercarb составляет до 2 порядков.
Разработанный подход значительно уступает по чувствительности (особенно для ДМГФ – в 25 раз) лишь методам, основанным на применении газовой хроматографии – тандемной хромато-масс-спектрометрии [27] (ГХ–МС/МС). Более того, чувствительность метода ГХ–МС/МС можно дополнительно повысить за счет концентрирования продуктов трансформации НДМГ твердофазной микроэкстракцией [9].
Следует отметить, что разработанный подход по чувствительности удовлетворяет действующим нормативам по содержанию НДМА, предельно-допустимая концентрация которого для водных объектов хозяйственно-бытового назначения установлена на уровне 0.01 мг/л [28], а для почв – 0.01 мг/кг. Достигнутые пределы обнаружения ДМФА и МТ на 2–3 порядка ниже установленных в Республике Казахстан нормативов [29].
Валидация разработанного подхода. Правильность определения МТ, НДМА, ДМФА, ДМГФ и ДМГА оценивали методом введено–найдено на двух уровнях концентрации (в 10 и 100 раз выше величины сн для режима МВИ). Вследствие связывания аналитов торфяной болотной почвой эксперименты проводили только с образцами природных вод I–III. Правильность определения (табл. 3) оценивали на основе отклонения получаемых результатов от заданных (n = 3) как относительное извлечение добавки. Видно, что даже при применении режима МВИ для анализа образцов I и II степень извлечения добавки составила 84–100% в зависимости от соединения и уровня концентрации. При переходе к поверхностной воде болота (образец III), характеризующейся повышенным содержанием растворенного органического вещества, возникают проблемы с определением диметилгидразонов. Например, даже после введения добавки ДМГФ высокой концентрации (6580 мкг/л) удается определить не более 10% от заданного содержания. При меньших уровнях вносимых концентраций диметилгидразонов данные аналиты обнаружить не удалось. Этот факт может быть обусловлен разложением гидразонов в кислой среде с высвобождением НДМГ и дальнейшими процессами его связывания и трансформации под действием растворенных гуминовых веществ.
Таблица 3.
Результаты оценки правильности определения аналитов в природных водах методом введено–найдено в режиме мониторинга выделенных ионов (n = 3, P = 0.95)
| Аналит | Введено, мкг/л | Найдено, мкг/л | Степень извлечения добавки, % |
|---|---|---|---|
| Образец 1 | |||
| НДМА | 250 | 240 ± 30 | 96 ± 12 |
| 25 | 23 ± 4 | 92 ± 16 | |
| ДМФА | 1010 | 960 ± 60 | 95 ± 6 |
| 101 | 94 ± 8 | 93 ± 8 | |
| ДМГФ | 6580 | 6220 ± 490 | 95 ± 7 |
| 658 | 644 ± 53 | 98 ± 8 | |
| ДМГА | 2500 | 2240 ± 330 | 90 ± 13 |
| 250 | 210 ± 36 | 84 ± 14 | |
| МТ | 55 | 53 ± 6 | 96 ± 11 |
| 5.5 | 5.0 ± 0.7 | 91 ± 13 | |
| Образец 2 | |||
| НДМА | 250 | 250 ± 27 | 100 ± 11 |
| 25 | 21 ± 4 | 84 ± 16 | |
| ДМФА | 1010 | 921 ± 94 | 91 ± 9 |
| 101 | 90 ± 12 | 89 ± 12 | |
| ДМГФ | 6580 | 6460 ± 530 | 98 ± 8 |
| 658 | 579 ± 80 | 88 ± 14 | |
| ДМГА | 2500 | 2375 ± 290 | 95 ± 12 |
| 250 | 230 ± 26 | 92 ± 10 | |
| МТ | 55 | 54 ± 5 | 98 ± 9 |
| 5.5 | 4.9 ± 0.8 | 89 ± 15 | |
| Образец 3 | |||
| НДМА | 250 | 260 ± 44 | 104 ± 18 |
| 25 | 20 ± 6 | 80 ± 24 | |
| ДМФА | 1010 | 990 ± 110 | 98 ± 11 |
| 101 | 92 ± 15 | 91 ± 15 | |
| ДМГФ | 6580 | 626 ± 60 | 10 ± 1 |
| 658 | – | – | |
| ДМГА | 2500 | – | – |
| 250 | – | – | |
| МТ | 55 | 53 ± 9 | 96 ± 16 |
| 5.5 | 4.8 ± 0.8 | 87 ± 15 | |
Анализ хроматограмм образцов природных вод I–III с введенными в разных концентрациях аналитами и без них показал отсутствие мешающего влияния со стороны матрицы, что свидетельствует о достаточной селективности метода ВЭЖХ–МС при определении продуктов трансформации Н-ДМГ в поверхностных водах. Для оценки влияния потенциальных помех со стороны НДМГ на ионизацию аналитов, который может присутствовать в объектах исследования в значительных концентрациях, сравнивали площади пиков аналитов (концентрация каждого соединения в смеси составляла 5 мг/л), полученные в отсутствие НДМГ в пробе и с добавкой его в концентрации 10 мг/л. Значимых различий в площадях пиков не обнаружено (расхождение составило менее 5% для всех аналитов), что свидетельствует об отсутствии мешающего влияния НДМГ.
Эксперименты по оценке внутрилабораторной воспроизводимости (на протяжении 24 ч при n = 7) на уровне концентраций 30–60 мкг/л показали, что величина относительного стандартного отклонения не превысила 16% для каждого аналита.
Особое внимание уделено надежности предложенного подхода к хроматографическому разделению продуктов трансформации НДМГ, основанного на применении химически стойкой неподвижной фазы ПГУ. Времена удерживания аналитов хорошо воспроизводятся (отклонение менее 1%) на протяжении сотен вводов проб природных вод с проведением анализа в разные дни с различными партиями подвижной фазы. При работе с сильнозагрязненными природным органическим веществом образцами (болотная вода и экстракты почв) полная регенерация хроматографической колонки и восстановление времен удерживания аналитов достигалась промывкой органическими растворителями и растворами щелочей для полного удаления гуминовых кислот и полифенольных соединений. Это невозможно при использовании неподвижных фаз на основе силикагеля.
Анализ почвы. Исследование образца торфяной болотной почвы, отобранной в месте падения отработанной ступени ракеты-носителя, показало применимость разработанных подходов для решения задач экологического мониторинга объектов окружающей среды, подверженных влиянию ракетно-космической деятельности. В экстракте обнаружены три из пяти исследуемых аналитов – НДМА, МТ и ДМФА (рис. 4). Как и следовало ожидать исходя из результатов экспериментов по введению диметилгидразонов в поверхностную воду болота, вследствие нестабильности в кислых средах ДМГФ и ДМГА не могут присутствовать в свободном виде в метанольном экстракте торфа. По-видимому, в сильнокислых почвах, богатых органическим веществом, такие соединения могут присутствовать только в связанных формах, разложение которых может приводить к высвобождению гидразонов в почвенный раствор или экстракт. Соответственно определение этих аналитов в торфяных болотных почвах возможно лишь при использовании более жестких методов извлечения в щелочных средах, например, предложенной нами ранее субкритической экстракцией ацетонитрилом в присутствии гидроксида бария [8].
Рис. 4.
Хроматограмма экстракта почвы, загрязненной ракетным топливом, в режимах мониторинга выделенных ионов (а) и мониторинга заданных реакций (б). Подвижная фаза: 10 мМ водный раствор формиата аммония (рН 6.4), содержащий 5 об. % метанола, расход 0.3 мл/мин.
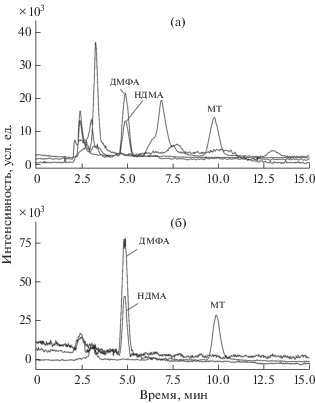
Применение режима МВИ для анализа таких сложных объектов, как экстракты торфяных почв, не обеспечивает достаточной селективности – на полученной хроматограмме (рис. 4а) наблюдаются интенсивные пики изобарных и/или изомерных соединений, особенно в области, близкой к мертвому объему. При тандемном масс-спектрометрическом детектировании матричные эффекты на хроматограмме оказались незначительными (рис. 4б). Тем не менее, применение неподвижной фазы на основе ПГУ позволило практически полностью отделить целевые аналиты от матрицы. Вследствие этого результаты анализа образца IV, полученные в режимах МВИ и МЗР, практически совпали (табл. 4). Такое преимущество ПГУ существенно снижает требования к аппаратурному оформлению анализа и, как следствие, стоимость его выполнения.
Концентрации обнаруженных в экстракте аналитов находятся на уровне нескольких единиц мг/кг, при этом основным продуктом трансформации при использовании выбранного способа экстрагирования из почвы является ДМФА (4.6 мг/кг), присутствующий в почве в количествах, в два раза больших по сравнению с МТ и НДМА (табл. 4).
* * *
Хроматографическое разделение азотсодержащих продуктов трансформации высокотоксичного ракетного топлива на неподвижной фазе на основе ПГУ является перспективной альтернативной имеющимся подходам, применяемым в экологическом сопровождении ракетно-космической деятельности. Они открывают серьезные перспективы для создания способов нецелевого скрининга и одновременного определения широкого круга продуктов трансформации 1,1-диметилгидразина, относящихся к различным классам.
Работа выполнена при финансовой поддержке Российского научного фонда (проект № 17-13-01112) с использованием оборудования Центра коллективного пользования научным оборудованием “Арктика” Северного (Арктического) федерального университета им. М.В. Ломоносова.
Список литературы
Родин И.А., Москвин Д.Н., Смоленков А.Д., Шпигун О.А. Превращения несимметричного диметилгидразина в почвах // Журн. физ. химии. 2008. Т. 82. № 6. С. 1039. (Rodin I.A., Moskvin D.N., Smolenkov A.D., Shpigun O.A. Transformations of asymmetric dimethylhydrazine in soils // Russ. J. Phys. Chem. A. 2008. V. 82. № 6. P. 911.)
Ul'yanovskii N.V., Kosyakov D.S., Pikovskoi I.I., Khabarov Yu.G. Characterisation of oxidation products of 1,1-dimethylhydrazine by high-resolution orbitrap mass spectrometry // Chemosphere. 2017. V. 174. P. 66.
Kenessov B., Alimzhanova M., Sailaukhanuly Y., Baimatova N., Abilev M., Batyrbekova S., Carlsen L., Tulegenov A., Nauryzbayev M. Transformation products of 1,1-dimethylhydrazine and their distribution in soils of fall places of rocket carriers in Central Kazakhstan // Sci. Total Environ. 2012. V. 427–428. P. 78.
Bakaikina N.V., Kenessov B.N., Ul’yanovskii N.V., Kosyakov D.S. Quantification of transformation products of rocket fuel unsymmetrical dimethylhydrazine in soils using SPME and GC-MS // Talanta. 2018. V. 184. P. 332.
Родин И.А., Ананьева И.А., Смоленков А.Д., Шпигун О.А. Определение продуктов окислительной трансформации несимметричного диметилгидразина в почвах методом жидкостной хроматомасс-спектрометрии // Масс-спектрометрия. 2009. Т. 6. № 4. С. 302. (Rodin I.A., Anan’eva I.A., Smolenkov A.D., Shpigun O.A. Determination of the products of the oxidative transformation of unsymmetrical dimethylhydrazine in soils by liquid chromatography/mass spectrometry // J. Analyt. Chem. 2010. V. 65. № 13. P. 1405.)
Kosyakov D.S., Ul’yanovskii N.V., Bogolitsyn K.G., Shpigun O.A. Simultaneous determination of 1,1-dimethylhydrazine and products of its oxidative transformations by liquid chromatography–tandem mass spectrometry // Int. J. Environ. Anal. Chem. 2014. V. 94. № 12. P. 1254.
Смирнов Р.С., Родин И.А., Смоленков А.Д., Шпигун О.А. Хромато-масс-спектрометрическое определение продуктов трансформации несимметричного диметилгидразина в почвах // Журн. аналит. химии. 2010. Т. 65. № 12. С. 1295. (Smir-nov R.S., Rodin I.A., Smolenkov A.D., Shpigun O.A. Determination of the products of the transformation of unsymmetrical dimethylhydrazine in soils using chromatography/mass spectrometry // J. Analyt. Chem. 2010. V. 65. №12. P. 1266.)
Kosyakov D.S., Ul’yanovskii N.V., Pokryshkin S.A., Lakhmanov D.E., Shpigun O.A. Rapid determination of 1,1-dimethylhydrazine transformation products in soil by accelerated solvent extraction coupled with gas chromatography–tandem mass spectrometry // Int. J. Environ. Anal. Chem. 2015. V. 95. № 14. P. 1321.
Bakaikina N.V., Kenessov B., Ul’yanovskii N.V., Kosyakov D.S., Pokryshkin S.A., Derbissalin, M., Zhubatov Z.K. Quantification of Transformation Products of Unsymmetrical Dimethylhydrazine in Water Using SPME and GC-MS // Chromatographia. 2017. V. 80. № 6. P. 931.
Zhubatov Z.K., Kenessov B., Bakaikina N.V., Bimaganbetova A.O., Akynbayev N., Bakhytkyzy I. Fast Determination of 1-methyl-1H-1,2,4-triazole in soils contaminated by rocket fuel using solvent extraction, isotope dilution and GC–MS // Chromatographia. 2016. V. 79. № 7–8. P. 491.
Ul’yanovskii N.V., Kosyakov D.S., Pikovskoi I.I., Shavrina I.S., Shpigun O.A. Determination of 1,1-dimethylhydrazine and its transformation products in soil by zwitterionic hydrophilic interaction liquid chromatography/tandem mass spectrometry // Chromatographia. 2018. V. 81. № 6. P. 891.
Ripollés C., Pitarch E., Sancho J.V., López F.J., Hernández F. Determination of eight nitrosamines in water at the ng L–1 levels by liquid chromatography coupled to atmospheric pressure chemical ionization tandem mass spectrometry // Anal. Chim. Acta. 2011. V. 702. № 1. P. 62.
Plumlee M.H., Lo’pez-Mesasa M., Heidlberger A., Ishida K.P., Reinhard M. N-nitrosodimethylamine (NDMA) removal by reverse osmosis and UV treatment and analysis via LC–MS/MS // Water Res. 2008. V. 42. № 1–2. P. 347.
Smolenkov A.D., Krechetov P.P., Pirogov A.V., Koroleva T.V., Bendryshev A.A., Shpigun O.A., Martynova M.M. Ion chromatography as a tool for the investigation of unsymmetrical hydrazine degradation in soils // Int. J. Environ. Anal. Chem. 2005. V. 85. № 14. P. 1089.
Смоленков А.Д., Родин И.А., Смирнов Р.С., Татаурова О.Г., Шпигун О.А. Применение ионной и ион-парной хроматографии с масс-спектрометрическим детектированием для определения нессиметричного диметилгидразина и продуктов его трансформации // Вестн. Моск. ун-та. Серия 2. Химия. 2012. Т. 53. № 5. С. 312. (Smolenkov A.D., Rodin I.A., Smirnov R.S., Tataurova O.G., Shpigun O.A. Use of ion and ion-pair chromatography with mass spectrometric detection to determine unsymmetrical dimethylhydrazine and its transformation products // Mosc. Univ. Chem. Bull. 2012. V. 67. № 5. P. 229.)
West C., Elfakir C., Lafosse M. Porous graphitic carbon: A versatile stationary phase for liquid chromatography // J. Chromatogr. A. 2010. V. 1217. № 19. P. 3201.
Knox J.H., Kaur B. Structure and performance of porous graphitic carbon in liquid chromatography // J. Chromatogr. A. 1986. V. 352. P. 3.
Polyakova Y.L., Row K.H. Retention of some five-membered heterocyclic compounds on a porous graphitized carbon, Hypercarb // Chromatographia. 2007. V. 65. № 1–2. P. 59.
Polyakova Y.L., Row K.H. HPLC of some polar compounds on a porous graphitized carbon Hypercarb column // J. Liq. Chromatogr. Relat. Technol. 2005. V. 28. № 20. P. 3157.
Ngongang A.D., Duy S.V., Sauv´ S. Analysis of nine N-nitrosamines using liquid chromatography-accurate mass high resolution-mass spectrometry on a Q-Exactive instrument // Anal. Methods. 2015. V. 7. № 14. P. 5748.
Class J.B., Aston J.G., Oakwood T.S. Trimethylhydrazine and Tetramethylhydrazine // J. Am. Chem. Soc. 1953. V. 75. № 12. P. 2937.
Catalán J., Díaz C., García-Blanco F. Characterization of binary solvent mixtures: the water–acetonitrile mixture // Org. Biomol. Chem. 2003. V. 1. № 3. P. 575.
Bapiro T.E., Richards F.M., Jodrell D.I. Understanding the complexity of porous graphitic carbon (PGC) chromatography: modulation of mobile-stationary phase interactions overcomes loss of retention and reduces variability // Anal. Chem. 2016. V. 88. № 12. P. 6190.
Lepont C., Gunatillaka A.D., Poole C.F. Retention characteristics of porous graphitic carbon in reversed-phase liquid chromatography with methanol–water mobile phases // Analyst. 2001. V. 126. P. 1318.
Kalia J., Raines R.T., Hydrolytic stability of hydrazones and oximes // Angew. Chem. Int. Ed. 2008. V. 47. P. 7523.
Zhang S. A reliable and efficient first principles–based method for predicting pKa values. 4. Organic bases // J. Comput. Chem. 2012. V. 33. № 31. P. 2469.
Ульяновский Н.В., Косяков Д.С., Покрышкин С.А., Боголицын К.Г. Определение продуктов трансформации 1,1-диметилгидразина методом тандемной газовой хроматомасс-спектрометрии // Масс-спектрометрия. Т. 11. № 3. С. 155. (Ul’yanovskii N.V., Kosyakov D.S., Pokryshkin S.A., Bogolitsyn K.G. Determination of transformation products of 1,1-dimethylhydrazine by gas chromatography–tandem mass spectrometry // J. Analyt. Chem. 2015. V. 70. № 13. P. 1553.)
ГОСТ Р 52985-2008. Экологическая безопасность ракетно-космической техники. Общие технические требования. М.: Стандартинформ, 2008. 27 с.
Жубатов Ж., Кенесов Б.Н., Товасаров А.Д., Козловский В.А., Батырбекова С.Е. Система экологического нормирования ракетно-космической деятельности космодрома Байконур / Под ред. Наурызбаева М.К. Алматы, 2017. 146 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии