

Журнал аналитической химии, 2020, T. 75, № 4, стр. 348-359
Вольтамперометрическое определение кверцетина и рутина при совместном присутствии на электроде, модифицированном политимолфталеином
Е. В. Гусс a, Г. К. Зиятдинова a, *, А. С. Жупанова a, Г. К. Будников a
a Химический институт им. А.М. Бутлерова, Казанский федеральный университет
420008 Казань, ул. Кремлевская, 18, Россия
* E-mail: Ziyatdinovag@mail.ru
Поступила в редакцию 18.01.2019
После доработки 25.02.2019
Принята к публикации 30.10.2019
Аннотация
Разработан вольтамперометрический способ определения кверцетина и рутина при совместном присутствии на стеклоуглеродном электроде, модифицированном углеродными нановолокнами и политимолфталеином. Найдены условия потенциодинамического электролиза тимолфталеина, обеспечивающие максимальную разность потенциалов окисления кверцетина и рутина, равную 136 мВ. Электрополимеризацию тимолфталеина следует проводить из 100 мкМ раствора 10-кратным циклированием потенциала от 0 до 1.7 В при скорости развертки 75 мВ/с на фоне фосфатного буферного раствора с рН 7.0. Электрод, модифицированный политимолфталеином, охарактеризован методами сканирующей электронной микроскопии, циклической вольтамперометрии и электрохимического импеданса. Установлено, что окисление кверцетина протекает необратимо, а рутина – квазиобратимо с участием двух электронов и двух протонов и контролируется адсорбцией аналитов на поверхности электрода. Диапазон определяемых содержаний составляет 0.025–1.00 мкМ для обоих аналитов, а пределы обнаружения – 7.3 нМ для кверцетина и 4.7 нМ для рутина. Показана селективность отклика электрода на кверцетин и рутин в присутствии неорганических ионов, сахаридов, аскорбиновой и п-кумаровой кислот. Разработанный способ апробирован на реальных объектах (липы Tilia L. цветках и луке репчатом).
Определение изомеров и структурно родственных биологически активных органических соединений при совместном присутствии является актуальной задачей функционального анализа, для решения которой традиционно используют хроматографические методы. Однако исследования последних лет в области вольтамперометрии с химически модифицированными электродами показали, что подбор подходящего модификатора может обеспечить разрешение аналитических сигналов структурно близких соединений [1–3].
Среди большого разнообразия биологически активных соединений можно выделить группу фенольных антиоксидантов, в частности, флавоноиды, одними из наиболее распространенных представителей которых являются кверцетин и рутин [4]. Присутствие в структуре фенольных фрагментов обусловливает их способность окисляться на различных электродах, в том числе химически модифицированных. Для индивидуального вольтамперометрического определения кверцетина и рутина предложен широкий круг электродов на основе различных сочетаний модификаторов, среди которых необходимо отметить полимерные покрытия поли(L-лизина) [5], поли(галловой кислоты) [6], поли(винилпирролидона) [7], поли(сульфаниловой кислоты) [8, 9], поли(акридинового оранжевого) [10], а также полимеры с молекулярными отпечатками на основе пиррола [11, 12] и метакриловой кислоты и диметакрилата этиленгликоля [13] (табл. 1). Описаны лишь единичные примеры определения кверцетина и рутина при совместном присутствии (стеклоуглеродный электрод, СУЭ, модифицированный функционализированными п-аминотиофенолом многостенными углеродными нанотрубками и наночастицами золота [29], угольно-пастовый электрод, модифицированный многостенными углеродными нанотрубками в условиях реверсивной дифференциально-импульсной вольтамперометрии [30] и СУЭ, модифицированный композитом из восстановленного оксида графена, каликс [4]амидокрауна-5 и наночастиц рутения [31]). Однако практически все эти электроды апробированы на таблетках рутина и биологических жидкостях с искусственно внесенными аналитами. Кроме того, для большинства электродов не рассматривается селективность отклика в присутствии структурно родственных соединений. Таким образом, дальнейшие исследования в этой области представляют актуальную задачу, для решения которой предлагается СУЭ, модифицированный углеродными нановолокнами и политимолфталеином (ПТФ).
Таблица 1.
Аналитические характеристики известных методов вольтамперометрического определения кверцетина и рутина на химически модифицированных электродах
| Электрод | Метод | ПрО, мкМ | Диапазон определяемых содержаний, мкМ | Литература |
|---|---|---|---|---|
| Кверцетин | ||||
| Нанолисты графена/СУЭ | АдАДИВ | 0.0011 | 0.08–1.0, 1.0–50 | [14] |
| Co3O4 НЧ/СУЭ | ВА с полу- дифференцированием | 0.1 | 0.5–330 | [15] |
| Микроволокна Al/УПЭ | ДИВ | 0.01 | 0.025–1.5 | [16] |
| 1-Метил-3-бутилимидазолий бромид–NiO–УНТ/УПЭ | КВВ | 0.03 | 0.08–400 | [17] |
| Гексадецилтриметиламмоний бромид/Fe- МУНТ/УПЭ | ДИВ | 0.0012 | 0.06–3000 | [18] |
| Pt НЧ-полидопамин@SiO2/СУЭ | КВВ | 0.016 | 0.05–0.383 | [19] |
| МУНТ-полиакриловая кислота/СУЭ | АдКВВ | 0.0075 | 0.1–5 | [20] |
| Поли(L-лизин)/полисилоксан–поли(пропилен) оксид/СУЭ | КВВ | 0.00276 | 0.01–1 | [5] |
| Поли(галловая кислота)/МУНТ/СУЭ | ДИВ | 0.054 | 0.075–25, 25–100 | [6] |
| Поли(винилпирролидон)/УПЭ | КВВ | 0.17 | 0.5–5.5 | [7] |
| Поли(сульфаниловая кислота)/СУЭ | ДИВ | 20.3 | 70–900 | [9] |
| ПМО на основе полипиррола–оксид графена/СУЭ | ДИВ | 0.048 | 0.60–15 | [11] |
| ПМО на основе метакриловой кислоты и диметакрилата этиленгликоля/In-Ti оксидный электрод | КВВ | 0.05 | 0.05–100 | [13] |
| Рутин | ||||
| Графенвос/УПЭ с ионной жидкостью | ДИВ | 0.024 | 0.07–100.0 | [21] |
| Графен/Au НЧ/Игла для аккупунктуры | ДИВ | 0.025 | 0.08–10, 20–200 | [22] |
| 1-Додецил-3-метилимидазолиий гексафтор-фосфат–МУНТ пастовый электрод | КВВ | 0.01 | 0.03–1.5 | [23] |
| ОУНТ/УПЭ с ионной жидкостью | ДИВ | 0.07 | 0.1–800.0 | [24] |
| Au НЧ/Этилендиамин/МУНТ/СУЭ | АдИДИВ | 0.032 | 0.48–0.96 | [25] |
| NiCo2O4/Оксид графенавос/СУЭ | ДИВ | 0.01 | 0.1–8, 8–150 | [26] |
| Поли(сульфаниловая кислота)/СУЭ | ДИВ | 0.1 | 0.25–10.0 | [8] |
| Поли(акридиновый оранжевый)/Графен/УПЭ с ионной жидкостью | ДИВ | 0.0083 | 0.03–800.0 | [27] |
| Фосфорен–поли(3,4-этилендиокситиофен)–поли(стиролсульфонат)/СУЭ | ДИВ | 0.007 | 0.02–15, 15–80 | [28] |
Обозначения: ПрО – предел обнаружения; СУЭ – стеклоуглеродный электрод; НЧ – наночастицы; АдАДИВ – адсорбционная анодная дифференциально-импульсная вольтамперометрия; ВА – вольтамперометрия; УПЭ – угольно-пастовый электрод; ДИВ – дифференциально-импульсная вольтамперометрия; УНТ – углеродные нанотрубки; КВВ – квадратно-волновая вольтамперометрия; МУНТ – многостенные углеродные нанотрубки; АдКВВ – адсорбционная квадратно-волновая вольтамперометрия; ПМО – полимер с молекулярными отпечатками; ОУНТ – одностенные углеродные нанотрубки; АдИДИВ – адсорбционная инверсионная дифференциально-импульсная вольтамперометрия.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты и растворы. Использовали тимолфталеин, кверцетин (95%, Sigma, Германия) и рутина тригидрат (97%, Alfa Aesar, Великобритания), стандартные 10.0 мМ растворы которых готовили по точной навеске растворением в 5.0 мл метанола (х. ч.). Для оценки мешающего влияния использовали аскорбиновую (99%), галловую (99%), кофейную (98%), п-кумаровую кислоты (98%) и арабиногалактан (Sigma, Германия), хлорогеновую кислоту (95%, Aldrich, Германия). Их стандартные 1.0 мМ растворы готовили по точной навеске растворением в метаноле и в дистиллированной воде в случае аскорбиновой кислоты и арабиногалактана. Остальные реактивы были марки х. ч.
Модифицирование электрода. Использовали углеродные нановолокна (d × l 100 нм × 20–200 мкм и средний диаметр 130 нм) (Aldrich, Германия), гомогенную 1.0 мг/мл суспензию которых получали ультразвуковым диспергированием в петролейном эфире в течение 15 мин. На поверхность СУЭ наносили 2 мкл суспензии методом капельного испарения. Перед модификацией рабочую поверхность СУЭ обновляли механически, полируя оксидом алюминия с размером частиц 0.05 мкм. Затем электрод ополаскивали ацетоном и дистиллированной водой.
Пленку ПТФ получали потенциодинамическим электролизом после пятикратного сканирования фонового электролита. Условия электрополимеризации (природа фонового электролита, концентрация мономера, диапазон поляризации, скорость сканирования потенциала, число циклов сканирования потенциала) варьировались.
Электрохимические измерения проводили на потенциостате/гальваностате Autolab PGSTAT 302N с модулем FRA32M (Eco Chemie B.V., Нидерланды) с программным обеспечением NOVA 1.10.1.9 в электрохимической ячейке объемом 10.0 мл. Использовали рабочий СУЭ (площадь поверхности 7.07 мм2), СУЭ, модифицированный углеродными нановолокнами (УНВ/СУЭ) или СУЭ, модифицированный углеродными нановолокнами и политимолфталеином (ПТФ/УНВ/СУЭ), насыщенный хлоридсеребряный электрод сравнения и вспомогательный платиновый электрод. Для расчетов в вольтамперометрии применяли коррекцию базовой линии по программе NOVA 1.10.1.9., что позволило точнее измерять параметры пиков. В электрохимическую ячейку вносили фоновый электролит и 5.0–50 мкл растворов кверцетина и рутина (общий объем раствора в ячейке 5.0 мл) и регистрировали циклические или дифференциально-импульсные вольтамперограммы в диапазоне от –0.2 до 0.6 В или oт –0.05 дo 0.4 В. Параметры импульса варьировали.
Для определения рН фонового электролита использовали рН-метр Эксперт-001 (ООО “Эконикс-Эксперт”, Россия).
Электрохимический импеданс измеряли в присутствии 1.0 мМ K4[Fe(CN)6]/K3[Fe(CN)6] на фоне 0.1 М фосфатного буферного раствора с рН 7.0 в диапазоне частот 10 кГц–0.04 Гц с амплитудой 5 мВ при потенциале поляризации 0.213 В. Потенциал рассчитывали как полусумму пиков окисления и восстановления редокс пары [Fe(CN)6]3–/4–.
Сканирующая электронная микроскопия (СЭМ). Поверхность электродов характеризовали с помощью полевого эмиссионного электронного микроскопа высокого разрешения Merlin™ (Carl Zeiss, Германия) при ускоряющем напряжении 5 кВ и токе эмиссии 300 пА.
Пробоподготовка образцов. Точную навеску очищенного лука репчатого (10.0 ± 0.5 г) тщательно измельчали и отжимали сок, фиксируя его объем. Водный настой липы (Tilia L.) цветков готовили по стандартной фармакопейной методике [32]. Точную навеску лекарственного растительного сырья (5.0000 ± 0.0005 г) помещали в эмалированную посуду, заливали 100 мл кипящей дистиллированной воды и настаивали в течение 15 мин, охлаждали при комнатной температуре, отфильтровывали и доводили водой до первоначального объема.
Для вольтамперометрического определения кверцетина и рутина в электрохимическую ячейку вносили 25 или 10 мкл образца и регистрировали дифференциально-импульсные вольтамперограммы в диапазоне от –0.1 до 0.4 В при амплитуде импульса 25 мВ и времени импульса 25 мс и скорости изменения потенциала 10 мВ/с.
Статистическую обработку результатов проводили для 5 измерений при доверительной вероятности 0.95. Результаты представляли как X ± ΔX, где X – среднее значение и ΔX – доверительный интервал. Случайную погрешность определения оценивали по величине относительного стандартного отклонения (sr). Регрессионный анализ проводили в программе OriginPro 8.0 (OriginLab, США).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
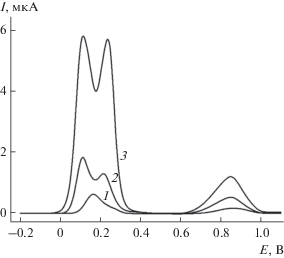
Изучено вольтамперометрическое поведение кверцетина и рутина на СУЭ и УНВ/СУЭ на фоне фосфатного буферного раствора с рН 7.0 в условиях дифференциально-импульсной вольтамперометрии. Кверцетин и рутин окисляются на СУЭ по двум ступеням. Разность потенциалов окисления не позволяет проводить их определение при совместном присутствии (рис. 1, кривая 1). На УНВ/СУЭ наблюдается смещение потенциалов окисления в область меньших значений, а также увеличение токов окисления по сравнению с СУЭ (в 5.3 раза для кверцетина и 4.7 раза для рутина). Разность потенциалов окисления кверцетина и рутина составляет 124 мВ, но при их совместном присутствии наблюдается сближение пиков (рис. 1, кривая 2). Кроме того, токи окисления рутина существенно ниже, чем кверцетина. Для решения этой задачи предложен электрод, модифицированный ПТФ. Для этого предварительно найдены условия полимеризации, обеспечивающие наилучший отклик смеси кверцетина и рутина.
Рис. 1.
Дифференциально-импульсные вольтамперограммы с коррекцией базовой линии 50 мкМ смеси кверцетина и рутина на СУЭ (1), УНВ/СУЭ (2) и ПТФ/УНВ/СУЭ (3) на фоне фосфатного буферного раствора с рН 7.0. Амплитуда импульса 50 мВ, время импульса 50 мс, скорость изменения потенциала 10 мВ/с.
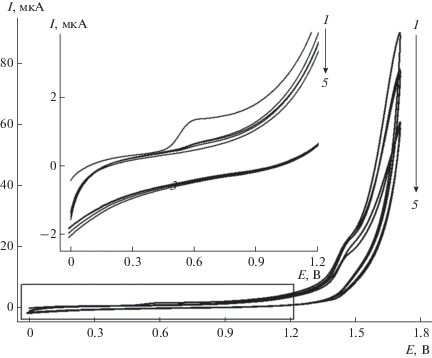
Тимолфталеин является кислотно-основным индикатором (pKa = 9.9), содержащим в своей структуре электрохимически активные тимольные фрагменты. Установлено, что он необратимо окисляется при 0.570 В на УНВ/СУЭ на фоне фосфатного буферного раствора с рН 7.0 (рис. 2, кривая 1). В окислении участвуют тимольные фрагменты, отдающие по одному протону и электрону, что приводит к образованию феноксильных радикальных фрагментов, которые затем могут участвовать в реакциях димеризации и полимеризации. Это согласуется с данными для электроокисления фенолов [33] и бромтимолового синего [34]. На последующих циклах сканирования потенциала наблюдается постепенное уменьшение токов окисления тимолфталеина (рис. 2, кривые 2–4), подтверждающее формирование непроводящего полимерного покрытия. Такое поведение типично для электрополимеризации фенольных соединений [35]. Пик окисления практически исчезает к десятому циклу.
Рис. 2.
Циклические вольтамперограммы 100 мкМ тимолфталеина на фоне фосфатного буферного раствора с рН 7.0 (5) на УНВ/СУЭ: 1 – скан 1, 2 – скан 3, 3 – скан 5, 4 – скан 10. Скорость изменения потенциала 75 мВ/с.
Поскольку отклик целевых аналитов зависит от условий получения полимерного покрытия, оценено влияние числа циклов сканирования потенциала, фонового электролита, концентрации мономера и параметров электролиза на отклик смеси 50 мкМ кверцетина и рутина. В качестве контролируемых параметров использовали разность потенциалов и токи их окисления.
Варьирование числа циклов от 5 до 15 показало постепенное увеличение разности потенциалов и токов окисления кверцетина и рутина с максимальными значениями при 10 сканах. Дальнейшее увеличение числа циклов приводит к блокированию поверхности электрода вследствие увеличения толщины полимерной пленки.
Так как в щелочной среде в присутствии кислорода воздуха тимольные фрагменты достаточно легко окисляются, а в кислой среде индикатор существует в протонированной форме, что затрудняет электроокисление, электрополимеризацию проводили на фоне фосфатного буферного раствора с рН 7.0.
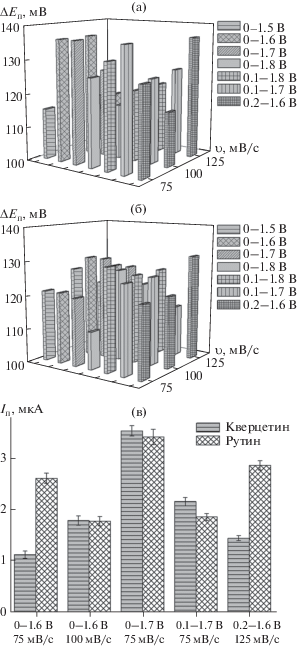
Концентрацию тимолфталеина варьировали в диапазоне 75–200 мкМ. Установлено, что для 75 мкМ тимолфталеина наблюдается слабо выраженная ступень окисления, что не позволяет получить полимерное покрытие достаточной толщины при 10-кратном циклировании потенциала. Поэтому использовали 100 и 200 мкМ растворы тимолфталеина, варьируя для этих концентраций скорость и окно поляризации электрода (рис. 3а, 3б). Максимальная разность потенциалов окисления кверцетина и рутина наблюдается для электродов, полученных при 100 мкМ концентрации тимолфталеина. При этом на дифференциально-импульсных вольтамперограммах смеси наблюдаются четко выраженные пики окисления аналитов, разность потенциалов которых достигает 136 мВ. Сопоставление токов окисления кверцетина и рутина при этой разности потенциалов для различных диапазонов и скорости поляризации электрода (рис. 3в) показало, что наибольшие токи окисления регистрируются для окна поляризации 0–1.7 В при скорости сканирования потенциала 75 мВ/с (3.5 ± 0.2 и 3.4 ± 0.1 мкА для кверцетина и рутина соответственно). Таким образом, электрополимеризацию тимолфталеина следует проводит из 100 мкМ раствора 10-кратным циклированием потенциала от 0 до 1.7 В при скорости развертки 75 мВ/с на фоне фосфатного буферного раствора с рН 7.0.
Рис. 3.
Влияние условий электрополимеризации 100 (а) и 200 (б) мкМ тимолфталеина на разность потенциалов окисления 50 мкМ смеси кверцетина и рутина на фоне фосфатного буферного раствора с рН 7.0 в условиях дифференциально-импульсной вольтамперометрии. (в) – Токи окисления 50 мкМ рутина и кверцетина в смеси для различных условий полимеризации 100 мкМ тимолфталеина, обеспечивающих разность потенциалов окисления аналитов 136 мВ.
На дифференциально-импульсных вольтамперограммах смеси кверцетина и рутина на ПТФ/УНВ/СУЭ на фоне фосфатного буферного раствора с рН 7.0 наблюдаются четко выраженные пики окисления при 0.107 и 0.243 В соответственно (рис. 1, кривая 3), токи которых статистически достоверно увеличиваются по сравнению с УНВ/СУЭ (рис. 1, кривая 2) и СУЭ (рис. 1, кривая 1). Это объясняется увеличением эффективной площади поверхности электрода с полимерным покрытием, а также накоплением аналитов на электродной поверхности за счет гидрофобных взаимодействий ароматических фрагментов в структуре кверцетина и рутина и полимерной пленки.
Для характеристики электродов использовали СЭМ, циклическую вольтамперометрию, хроноамперометрию и спектроскопию электрохимического импеданса. Данные СЭМ (рис. 4) подтверждают однородность покрытия электродной поверхности модификаторами и значительное увеличение шероховатости поверхности модифицированных электродов по сравнению с СУЭ. Политимолфталеин представляет собой равномерно распределенные шарообразные гранулы диаметром 30–50 нм (рис. 4в). Для оценки эффективной площади поверхности рабочих электродов использовали окисление [Fe(CN)6]4–-ионов на фоне 0.1 М KCl в условиях циклической вольтамперометрии (для ПТФ/УНВ/СУЭ) и хроноамперометрии (для СУЭ и УНВ/СУЭ). Рассчитанная по уравнениям Рэндлса−Шевчика и Коттрелла эффективная площадь поверхности составляет 8.2 ± 0.3 мм2 для СУЭ, 14.1 ± 0.4 мм2 для УНВ/СУЭ и 19 ± 1 мм2 для ПТФ/УНВ/СУЭ, что объясняет увеличение токов окисления кверцетина и рутина на модифицированных электродах по сравнению с СУЭ. Кроме того, на ПТФ/УНВ/СУЭ может происходить накопление аналитов в порах полимерной пленки.

Параметры электронного переноса оценивали методом спектроскопии электрохимического импеданса, используя для количественной характеристики эквивалентные ячейки Рэндлса (табл. 2). Для модифицированных электродов наблюдается значительное уменьшение сопротивления переносу заряда, что свидетельствует об увеличении скорости переноса электрона. Величина элемента постоянной фазы для ПТФ-модифицированного электрода статистически достоверно выше, что, вероятно, обусловлено пористой структурой электродной поверхности. Это косвенно подтверждается значением фактора гетерогенности n.
Таблица 2.
Параметры электрохимического импеданса электродов (n = 5, P = 0.95)
| Электрод | Rs, Ом | Rct, кОм | Q, мкОм–1 | n | W, мкОм–1 |
|---|---|---|---|---|---|
| СУЭ | 245 ± 5 | 72 ± 3 | 3.7 ± 0.2 | 0.789 | – |
| УНВ/СУЭ | 318 ± 8 | 30 ± 1 | 0.56 ± 0.03 | 0.859 | 8.0 ± 0.5 |
| ПТФ/УНВ/СУЭ | 326 ± 6 | 23 ± 1 | 4.4 ± 0.2 | 0.862 | 83 ± 4 |
Изучено электроокисление кверцетина и рутина на ПТФ/УНВ/СУЭ. Варьирование рН фонового электролита от 4.8 до 8.0 показало, что потенциалы окисления кверцетина и рутина смещаются в область меньших значений по мере увеличения рН фонового электролита, что свидетельствует об участии протонов в электродной реакции. Потенциалы окисления кверцетина и рутина линейно зависят от рН фонового электролита (уравнения (1) и (2) соответственно):
(1)
$\begin{gathered} E\left( В \right) = (0.{\text{7}}0{\text{6}} \pm 0.00{\text{5}})--(0.0{\text{618}} \pm 0.000{\text{7}}){\text{pH}}, \\ {{R}^{{\text{2}}}} = 0.{\text{9988}}; \\ \end{gathered} $(2)
$\begin{gathered} E\left( В \right) = (0.{\text{583}} \pm 0.00{\text{3}})--(0.0{\text{64}}0 \pm 0.000{\text{5}}){\text{pH}}, \\ {{R}^{{\text{2}}}} = 0.{\text{9994}}. \\ \end{gathered} $Полученные значения тангенсов углов наклона свидетельствуют о равном числе протонов и электронов, участвующих в реакциях. Максимальные токи окисления кверцетина и рутина наблюдаются при рН 6.5 и 7.5 соответственно. Дальнейшие исследования проводили при рН 7.5, поскольку в этих условиях достигается наибольшая разность потенциалов окисления аналитов, составляющая 141 мВ.
Для установления природы электрохимического процесса оценено влияние скорости изменения потенциала на токи окисления кверцетина и рутина. Показано, что окисление кверцетина протекает необратимо, а рутина – квазиобратимо и контролируется адсорбцией аналитов, что подтверждается линейной зависимостью токов окисления от υ (уравнения (3) и (4) соответственно) и значениями тангенсов угла наклона зависимости lnIп от lnυ (уравнения (5) и (6) соответственно):
(3)
$\begin{gathered} {{I}_{{\text{п}}}}\left( {{\text{мкА}}} \right) = \\ = \left( {--0.0{\text{3}} \pm 0.0{\text{3}}} \right) + \left( {0.0{\text{126}} \pm 0.000{\text{3}}} \right)\upsilon {\text{ }}({\text{мВ/с}}),~ \\ {{R}^{{\text{2}}}}{\text{ }} = {\text{ }}0.{\text{9961}};~ \\ \end{gathered} $(4)
$\begin{gathered} {{I}_{{\text{п}}}}\left( {{\text{мкА}}} \right) = \\ = \left( {0.{\text{13}} \pm 0.0{\text{9}}} \right) + \left( {0.0{\text{4}}0{\text{3}} \pm 0.000{\text{7}}} \right)\upsilon {\text{ }}({\text{мВ/с}}), \\ {{R}^{{\text{2}}}} = 0.{\text{9974}}; \\ \end{gathered} $(5)
$\begin{gathered} {\text{ln}}{{I}_{п}}{\text{ }}({\text{мкA}}) = \\ = \left( {{\text{2}}.{\text{39}} \pm 0.0{\text{8}}} \right) + \left( {0.{\text{95}} \pm 0.0{\text{3}}} \right){\text{ln}}\upsilon {\text{ }}({\text{В/с}}), \\ {{R}^{{\text{2}}}} = 0.{\text{9936}}; \\ \end{gathered} $(6)
$\begin{gathered} {\text{ln}}{{I}_{{\text{п}}}}{\text{ }}({\text{мкA}}) = \\ = \left( {{\text{3}}.{\text{5}}0 \pm 0.0{\text{5}}} \right) + \left( {0.{\text{89}} \pm 0.0{\text{1}}} \right){\text{ln}}\upsilon {\text{ }}({\text{В/с}}),{\text{ }} \\ {{R}^{{\text{2}}}} = 0.{\text{9975}}. \\ \end{gathered} $В этом случае число участвующих в реакции электронов рассчитывается согласно формуле I = nFQ$\upsilon $/4RT, где I – ток окисления кверцетина или рутина (А), n – число электронов, участвующих в реакции, F – константа Фарадея (96485 Кл/моль), Q – количество электричества (Кл), $\upsilon $ – скорость изменения потенциала (В/с), R – универсальная газовая постоянная (8.314 Дж/моль К), T – температура (298 К) [36]. Расчеты показали, что электроокисление кверцетина и рутина протекает с участием двух электронов.
На основе полученных результатов и данных [37, 38] можно сделать вывод, что в реакции окисления кверцетина и рутина участвуют гидроксильные группы кольца В с образованием соответствующих о-хинонов.
Количественное определение кверцетина и рутина при совместном присутствии проводили методом дифференциально-импульсной вольтамперометрии. Варьирование амплитуды и времени импульса показало, что наилучшее разрешение пиков и максимальные токи окисления регистрируются при амплитуде импульса 25 мВ и времени импульса 25 мс. При этом на вольтамперограммах смесей кверцетина и рутина наблюдаются четко выраженные пики окисления при 0.110 и 0.230 В соответственно (рис. 5а), токи которых линейно увеличиваются с ростом концентрации аналитов в диапазоне от 0.025 до 1.0 мкМ согласно уравнениям (7) и (8):
(7)
$\begin{gathered} {{I}_{{\text{п}}}}\left( {{\text{мкА}}} \right) = \left( {0.00{\text{43}} \pm 0.000{\text{8}}} \right) + \\ + \,\,\left( {{\text{32}}.{\text{7 }}0.{\text{2}}} \right) \times {\text{1}}{{0}^{{\text{4}}}}{{с}_{{{\text{кверцетина}}}}}{\text{ }}({\text{M}}),{\text{ }}{{R}^{{\text{2}}}} = 0.{\text{9998}}; \\ \end{gathered} $(8)
$\begin{gathered} {{I}_{{\text{п}}}}\left( {{\text{мкА}}} \right) = \left( {--0.000{\text{1}} \pm 0.000{\text{6}}} \right) + \\ + \,\,\left( {{\text{37}}.{\text{9}} \pm 0.{\text{1}}} \right) \times {\text{1}}{{0}^{{\text{4}}}}{{с}_{{{\text{рутина}}}}}\,\,({\text{M}}),{\text{ }}{{R}^{{\text{2}}}}{\text{ = }}0.{\text{9999}}. \\ \end{gathered} $Рис. 5.
(а): Дифференциально-импульсные вольтамперограммы с коррекцией базовой линии эквимолярных смесей кверцетина и рутина (1 – 0.025, 2 – 0.050, 3 – 0.075, 4 – 0.10, 5 – 0.25, 6 – 0.50, 7 – 0.75, 8 – 1.0 мкМ) на ПТФ/УНВ/СУЭ на фоне фосфатного буферного раствора с рН 7.5. (б): cкверцетина = 0.050 (1), 0.10 (2), 0.25 (3), 0.50 (4) и 1.0 (5) мкМ, срутина = 1.0 мкМ. (в): cкверцетина = 1.0 мкМ, срутина = 0.05 (1), 0.10 (2), 0.25 (3), 0.50 (4) и 1.0 (5) мкМ. Амплитуда импульса 25 мВ, время импульса 25 мс, скорость изменения потенциала 10 мВ/с.
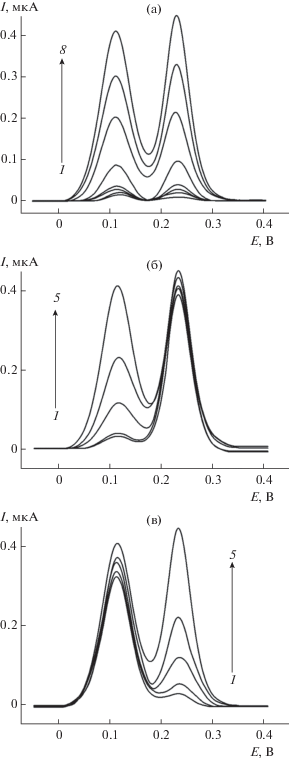
Пределы обнаружения по 3σ-критерию составляют 7.3 и 4.7 нМ для кверцетина и рутина соответственно. Полученные аналитические характеристики совместного определения кверцетина и рутина превосходят описанные для других химически модифицированных электродов (табл. 1), в том числе для угольно-пастового электрода, модифицированного многостенными углеродными нанотрубками [30]. Преимуществом предложенного электрода является также простота изготовления и применение доступных реагентов для получения полимерного покрытия.
Варьирование концентрации одного из компонентов при фиксированном значении концентрации второго не приводит к наложению аналитических сигналов (рис. 5б, 5в). Ток окисления компонента, взятого в избытке, остается постоянным, а токи окисления второго компонента линейно увеличиваются, причем значения токов окисления обоих компонентов совпадают с таковыми для эквимолярных смесей. Это подтверждает, что окисление кверцетина и рутина протекает независимо и для их определения можно использовать градуировочные зависимости для эквимолярных смесей.
Проведено определение кверцетина и рутина при совместном присутствии в модельных системах (табл. 3). Величина относительного стандартного отклонения не превышает 5%. Полученные значения меры правильности свидетельствуют о высокой точности разработанного подхода.
Таблица 3.
Результаты определения кверцетина и рутина в модельных растворах при совместном присутствии на ПТФ/УНВ/СУЭ на фоне фосфатного буферного раствора с рН 7.5 (n = 5, P = 0.95)
| Введено, мкг | Найдено, мкг | sr | R, % |
|---|---|---|---|
| Кверцетин | |||
| 0.038 | 0.040 ± 0.002 | 0.05 | 105 |
| 0.11 | 0.112 ± 0.003 | 0.02 | 102 |
| 0.15 | 0.152 ± 0.009 | 0.05 | 101 |
| 0.76 | 0.761 ± 0.009 | 0.009 | 100 |
| 1.5 | 1.50 ± 0.06 | 0.03 | 100 |
| Рутин | |||
| 0.076 | 0.073 ± 0.004 | 0.04 | 96.1 |
| 0.23 | 0.223 ± 0.004 | 0.01 | 97.0 |
| 0.31 | 0.308 ± 0.003 | 0.008 | 99.4 |
| 1.5 | 1.52 ± 0.02 | 0.01 | 101 |
| 3.1 | 3.08 ± 0.06 | 0.02 | 99.4 |
Показана селективность определения 0.5 мкМ кверцетина и рутина в присутствии 1000-кратных избытков неорганических ионов (K+, Na+, Mg2+, Ca2+, ${\text{NO}}_{3}^{ - },$ Clˉ и ${\text{SO}}_{4}^{{2 - }}$), 100-кратных избытков глюкозы, рамнозы и сахарозы, арабиногалактана, а также аскорбиновой и п-кумаровой кислот. Галловая, кофейная и хлорогеновая кислоты окисляются ступенчато в области 0.070–0.250 В, что приводит к перекрыванию пиков их окисления и аналитов.
Разработанный подход применен для определения кверцетина и рутина в липы (Tilia L.) цветках и в луке репчатом. Как известно [39–41], их основными флавоноидами являются кверцетин и рутин. Кроме того, настой липы цветков содержит большое количество полисахаридов [40], но, как показано выше, они не мешают определению кверцетина и рутина на ПТФ/УНВ/СУЭ.
На вольтамперограммах водного настоя липы цветков и сока лука репчатого наблюдаются четко выраженные пики окисления кверцетина и рутина при 0.12 и 0.23 В (рис.6, кривая 1), что подтверждено методом стандартных добавок. При добавлении стандартных растворов кверцетина и рутина в электрохимическую ячейку происходит пропорциональное увеличение токов окисления при сохранении потенциалов пиков (рис. 6, кривые 2 и 3). Следует отметить, что для лука желтого токи окисления рутина очень низкие, что не позволяет провести количественное определение, однако можно сделать вывод о его присутствии. Степень открытия составляет 97.1–103% (табл. 4), что свидетельствует о высокой точности определения и об отсутствии матричных эффектов.
Рис. 6.
Дифференциально-импульсные вольтамперограммы с коррекцией базовой линии настоя липы цветков (а), сока репчатого лука красного (б) и желтого (в) на ПТФ/УНВ/СУЭ на фоне фосфатного буферного раствора с рН 7.5. (а) – 1 – 10 мкл настоя липы цветков; 2 – 10 мкл настоя + 0.24 мкМ кверцетина + 0.054 мкМ рутина; 3 – 10 мкл настоя + 0.49 мкМ кверцетина + 0.11 мкМ рутина. (б): 1 – 25 мкл сока лука красного; 2 – 25 мкл сока + 0.046 мкМ кверцетина + 0.026 мкМ рутина; 3 – 25 мкл сока + 0.092 мкМ кверцетина + 0.052 мкМ рутина. (в): 1 – 25 мкл сока лука желтого; 2 – 25 мкл сока + 0.050 мкМ кверцетина + 0.007 мкМ рутина; 3 – 25 мкл сока + 0.10 мкМ кверцетина + + 0.014 мкМ рутина. Амплитуда импульса 25 мВ, время импульса 25 мс, скорость изменения потенциала 10 мВ/с.
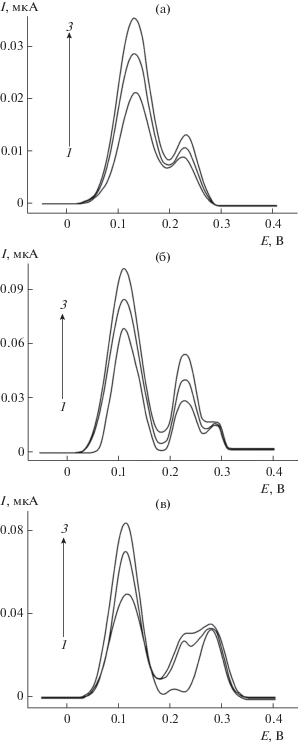
Таблица 4.
Степень открытия кверцетина и рутина в реальных объектах по данным вольтамперометрии на ПТФ/УНВ/СУЭ на фоне фосфатного буферного раствора с рН 7.5 (n = 5, P = 0.95)
| Образец | Аналит | Введено, мкг | Расчетное значение, мкг | Найдено, мкг | R, % |
|---|---|---|---|---|---|
| Настой липы цветков | Кверцетин | 0 | 0.73 ± 0.01 | ||
| 0.37 | 1.10 | 1.10 ± 0.09 | 100 | ||
| 0.74 | 1.47 | 1.48 ± 0.02 | 101 | ||
| Рутин | 0 | 0.34 ± 0.01 | |||
| 0.16 | 0.50 | 0.50 ± 0.01 | 100 | ||
| 0.33 | 0.67 | 0.67 ± 0.02 | 100 | ||
| Лук репчатый красный | Кверцетин | 0 | 0.30 ± 0.01 | ||
| 0.070 | 0.37 | 0.37 ± 0.02 | 100 | ||
| 0.14 | 0.44 | 0.44 ± 0.02 | 100 | ||
| Рутин | 0 | 0.17 ± 0.01 | |||
| 0.079 | 0.249 | 0.25 ± 0.02 | 100 | ||
| 0.19 | 0.33 | 0.34 ± 0.02 | 103 | ||
| Лук репчатый желтый | Кверцетин | 0.20 ± 0.01 | |||
| 0.076 | 0.276 | 0.28 ± 0.01 | 101 | ||
| 0.15 | 0.35 | 0.34 ± 0.02 | 97.1 |
Результаты определения кверцетина и рутина в липы (Tilia L.) цветках и в луке репчатом представлены в табл. 5. Полученное содержание кверцетина в луке репчатом хорошо согласуются с данными [42]. Предложенный метод характеризуется высокими чувствительностью и селективностью, а также простотой и доступностью, что позволяет применять его в анализе растительных материалов.
Таблица 5.
Результаты вольтамперометрического определения кверцетина и рутина в реальных объектах (n = 5, P = 0.95)
| Объект анализа | Аналит | Содержание, мг/г | sr | |
|---|---|---|---|---|
| Липы цветки: | образец 1 | Кверцетин | 1.44 ± 0.03 | 0.02 |
| Рутин | 0.73 ± 0.06 | 0.07 | ||
| образец 2 | Кверцетин | 3.43 ± 0.09 | 0.03 | |
| Рутин | 1.68 ± 0.04 | 0.02 | ||
| Лук репчатый красный | Кверцетин | 0.0049 ± 0.0002 | 0.02 | |
| Рутин | 0.0027 ± 0.0001 | 0.02 | ||
| Лук репчатый желтый | Кверцетин | 0.0023 ± 0.0001 | 0.03 | |
Работа выполнена при финансовой поддержке РФФИ (проект 18-33-00220-мол_а).
Список литературы
Будников Г.К., Евтюгин Г.А., Майстренко В.Н. Модифицированные электроды для вольтамперометрии в химии, биологии и медицине. М.: Бином – Лаборатория знаний, 2010. 416 с.
Зиятдинова Г.К., Козлова Е.В., Зиганшина Э.Р., Будников Г.К. Применение электрода, модифицированного углеродными нановолокнами и катионным ПАВ, для вольтамперометрического определения сиреневого альдегида и ванилина при совместном присутствии // Бутлеровские сообщения. 2015. Т. 42. № 6. С. 132.
Figueiredo-Filho L.C., Silva T.A., Vicentini F.C., Fatibello-Filho O. Simultaneous voltammetric determination of dopamine and epinephrine in human body fluid samples using a glassy carbon electrode modified with nickel oxide nanoparticles and carbon nanotubes within a dihexadecylphosphate film // Analyst. 2014. V. 139. № 11. P. 2842.
Зиятдинова Г.К., Будников Г.К. Природные фенольные антиоксиданты в биоаналитической химии: состояние проблемы и перспективы развития // Успехи химии. 2015. Т. 84. № 2. С. 194. (Ziyatdinova G.K., Budnikov H.C. Natural phenolic antioxidants in bioanalytical chemistry: state of the art and prospects of development // Russ. Chem. Rev. 2015. V. 84. № 2. P. 194.)
Pereira E.R.D.V., Bessegato G.G., Yamanaka H., Zanoni M.V.B. Determination of quercetin by a siloxane-polyester/poly-L-lysine nanocomposite modified glassy carbon electrode // Anal. Lett. 2016. V. 49. № 9. P. 1398.
Ziyatdinova G., Kozlova E., Budnikov H. Poly(gallic acid)/MWNT-modified electrode for the selective and sensitive voltammetric determination of quercetin in medicinal herbs // J. Electroanal. Chem. 2018. V. 821. P. 73.
Franzoi A.C., Spinelli A., Vieira L.C. Rutin determination in pharmaceutical formulations using a carbon paste electrode modified with poly(vinylpyrrolidone) // J. Pharm. Biomed. Anal. 2008. V. 47. № 4-5. P. 973.
Chen X., Wang Z., Zhang F., Zhu L., Li Y., Xia Y. Determination of rutin on the poly(p-aminobenzene sulfonic acid) modified glassy carbon electrode // Chem. Pharm. Bull. 2010. V. 58. № 4. P. 475.
Selvi B., Sadikoglu M., Soylu U.I., Yılmaz S., Onal A., Eser F. Sensitive determination of quercetin in onion peel by voltammetry using a poly(4-aminobenzene sulfonic acid) modified glassy carbon electrode // Anal. Bioanal. Electrochem. 2017. № 5. V. 9. P. 574.
Sun W., Wang Y., Gong S., Cheng Y., Shi F., Sun Z. Application of poly(acridine orange) and graphene modified carbon/ionic liquid paste electrode for the sensitive electrochemical detection of rutin // Electrochim. Acta. 2013. V. 109. P. 298.
Sun S., Zhang M., Li Y., He X. A molecularly imprinted polymer with incorporated graphene oxide for electrochemical determination of quercetin // Sensors. 2013. V. 13. № 5. P. 5493.
Lu B., Xia J., Wang Z., Zhang F., Yang M., Li Y., Xia Y. Molecularly imprinted electrochemical sensor based on an electrode modified with an imprinted pyrrole film immobilized on a β-cyclodextrin/gold nanoparticles/graphene layer // RSC Adv. 2015. V. 5. № 101. P. 82930.
Salmi Z., Benmehdi H., Lamouri A., Decorse P., Jouini M., Yagci Y., Chehimi M.M. Preparation of MIP grafts for quercetin by tandem aryl diazonium surface chemistry and photopolymerization // Microchim. Acta. 2013. V. 180. № 15-16. P. 1411.
Arvand M., Anvari M. Graphene nanosheets as a sensing platform for amplified electrochemical measurement of quercetin and uric acid in biological fluids // Can. J. Chem. 2014. V. 92. № 11. P. 1074.
Wang M., Zhang D., Tong Z., Xu X., Yang X. Voltammetric behavior and the determination of quercetin at a flowerlike Co3O4 nanoparticles modified glassy carbon electrode // J. Appl. Electrochem. 2011. V. 41. № 2. P. 189.
Li Y., Huang W. Electrode modified with porous alumina microfibers as a highly sensitive electrochemical sensor for quercetin // Anal. Methods. 2015. V. 7. № 6. P. 2537.
Gupta V.K., Golestani F., Ahmadzadeh S., Karimi-Maleh H., Fazli G., Khosravi S. NiO/CNTs nanocomposite modified ionic liquid carbon paste electrode as a voltammetric sensor for determination of quercetin // Int. J. Electrochem. Sci. 2015. V. 10. № 4. P. 3657.
Erady V., Mascarenhas R.J., Satpati A.K., Detriche S., Mekhalif Z., Delhalle J., Dhason A. A novel and sensitive hexadecyltrimethylammoniumbromide functionalized Fe decorated MWCNTs modified carbon paste electrode for the selective determination of quercetin // Mater. Sci. Eng. C. 2017. V. 76. P. 114.
Manokaran J., Muruganantham R., Muthukrishnaraj A., Balasubramanian N. Platinum-polydopamine@SiO2 nanocomposite modified electrode for the electrochemical determination of quercetin // Electrochim. Acta. 2015. V. 168. P. 16.
Gutierrez F., Ortega G., Cabrera J.L., Rubianes M.D., Rivas G.A. Quantification of quercetin using glassy carbon electrodes modified with multiwalled carbon nanotubes dispersed in polyethylenimine and polyacrylic acid // Electroanalysis. 2010. V. 22. № 22. P. 2650.
Gao F., Qi X., Cai X., Wang Q., Gao F., Sun W. Electrochemically reduced graphene modified carbon ionic liquid electrode for the sensitive sensing of rutin // Thin Solid Films. 2012. V. 520. № 15. P. 5064.
Niu X., Wen Z., Li X., Zhao W., Li X., Huang Y., Li Q., Li G., Sun W. Fabrication of graphene and gold nanoparticle modified acupuncture needle electrode and its application in rutin analysis // Sens. Actuators B. 2018. V. 255. № 1. P. 471.
Wang X., Cheng C., Dong R., Hao J. Sensitive voltammetric determination of rutin at a carbon nanotubes–ionic liquid composite electrode // J. Solid State Electrochem. 2012. V. 16. № 8. P. 2815.
Zhu Z., Sun X., Zhuang X., Zeng Y., Sun W., Huang X. Single-walled carbon nanotubes modified carbon ionic liquid electrode for sensitive electrochemical detection of rutin // Thin Solid Films. 2010. V. 519. № 2. P. 928.
Yang S., Qu L., Li G., Yang R., Liu C. Gold nanoparticles/ethylenediamine/ carbon nanotube modified glassy carbon electrode as the voltammetric sensor for selective determination of rutin in the presence of ascorbic acid // J. Electroanal. Chem. 2010. V. 645. № 2. P. 115.
Cui S., Li L., Ding Y., Zhang J., Yang H., Wang Y. Mesoporous NiCo2O4-decorated reduced graphene oxide as a novel platform for electrochemical determination of rutin // Talanta. 2017. V. 164. P. 291.
Sun W., Wang Y., Gong S., Cheng Y., Shi F., Sun Z. Application of poly(acridine orange) and graphene modified carbon/ionic liquid paste electrode for the sensitive electrochemical detection of rutin // Electrochim. Acta. 2013. V. 109. P. 298.
Niu X., Weng W., Yin C., Niu Y., Li G., Dong R., Men Y., Sun W. Black phosphorene modified glassy carbon electrode for the sensitive voltammetric detection of rutin // J. Electroanal. Chem. 2018. V. 811. P. 78.
Yola M.L., Atar N. A novel voltammetric sensor based on gold nanoparticles involved in p-aminothiophenol functionalized multi-walled carbon nanotubes: application to the simultaneous determination of quercetin and rutin // Electrochim. Acta. 2014. V. 118. P. 24.
Lin X.-Q., He J.-B., Zha Z.-G. Simultaneous determination of quercetin and rutin at a multi-wall carbon-nanotube paste electrodes by reversing differential pulse voltammetry // Sens. Actuators B. 2006. V. 119. № 2. P. 608.
Elçin S., Yola M.L., Eren T., Girgin B., Atar N. Highly selective and sensitive voltammetric sensor based on ruthenium nanoparticle anchored calix[4]amidocrown-5 functionalized reduced graphene oxide: simultaneous determination of quercetin, morin and rutin in grape wine // Electroanalysis. 2016. V. 28. № 3. P. 611.
Japanese Pharmacopoeia XVI. Tokyo: Pharmaceutical and medical device regulatory science society of Japan, 2011. 2326 p.
Murphy M.A., Wilcox G.D., Dahm R.H., Marken F. Adsorption and redox processes at carbon nanofiber electrodes grown onto a ceramic fiber backbone // Electrochem. Commun. 2003. V. 5. № 1. P. 51.
Chandrashekar B.N., Swamy B.E.K., Mahesh K.R.V., Chandra U., Sherigara B.S. Electrochemical studies of bromothymol blue at surfactant modified carbon paste electrode by using cyclic voltammetry // Int. J. Electrochem. Sci. 2009. V. 4. № 3. P. 471.
Electropolymerization / Ed. Schab-Balcerzak E. In Tech, 2011. 214 p.
Bard A.J., Faulkner L.R. Electrochemical Methods, Fundamentals and Applications. 2nd ed. New York: John Wiley& Sons Inc., 2001. 850 p.
Ziyatdinova G., Aytuganova I., Nizamova A., Morozov M., Budnikov H. Cyclic voltammetry of natural flavonoids on MWNT-modified electrode and their determination in pharmaceuticals // Collect. Czech. Chem. Commun. 2011. V. 76. № 12. P. 1619.
Wang M.Y., Zhang D., Tong Z., Xu X., Yang X. Voltammetric behavior and the determination of quercetin at a flowerlike Co3O4 nanoparticles modified glassy carbon electrode // J. Appl. Electrochem. 2011. V. 41. № 2. P. 189.
Kurzawa M. Determination of quercetin and rutin in selected herbs and pharmaceutical preparations // Anal. Lett. 2010. V. 43. № 6L. P. 993.
Augšpole I., Dūma M., Ozola B. Bioactive compounds in herbal infusions // Agron. Res. 2018. V. 16. № S2. P. 1322.
Lu T.-M., Chiu H.-F., Shen Y.-C., Chung C.-C., Venkatakrishnan K., Wang C.-K. Hypocholesterolemic efficacy of quercetin rich onion juice in healthy mild hypercholesterolemic adults: a pilot study // Plant Foods Hum. Nutr. 2015. V. 70. № 4. P. 395.
Gomez F.J.V., Espino M., de los Angeles Fernandez M., Raba J., Silva M.F. Enhanced electrochemical detection of quercetin by natural deep eutectic solvents // Anal. Chim. Acta. 2016. V. 936. P. 91.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии