

Журнал аналитической химии, 2019, T. 74, № 3, стр. 201-210
Жидкостная хроматография со свободной неподвижной фазой в элементном анализе: от нефти до особо чистых веществ
Т. А. Марютина 1, *, П. С. Федотов 1
1 Институт геохимии и аналитической химии им. В.И. Вернадского Российской академии наук
119991 Москва, ул. Косыгина, 19, Россия
* E-mail: t_maryutina@mail.ru
Поступила в редакцию 26.04.2017
После доработки 15.09.2018
Принята к публикации 15.09.2018
Аннотация
Показаны возможности использования жидкостной хроматографии со свободной неподвижной фазой (ЖХСНФ) в элементном анализе различных по химическому и фазовому составу образцов. Сочетание характерных особенностей экстракционной хроматографии и многоступенчатой экстракции с проведением процессов разделения и концентрирования в закрытой системе (тефлоновой спиральной колонке) открывает перспективы применения метода для анализа широкого круга объектов: от особо чистых веществ до нефти и горных пород. Использование ЖХСНФ на стадии пробоподготовки позволяет снизить пределы обнаружения примесных элементов в высокочистых материалах до уровня пг/г, т.е. на 2–3 порядка по сравнению с прямым определением масс-спектрометрией с индуктивно связанной плазмой. При анализе нефтяных образцов ЖХСНФ позволяет определять их полный микроэлементный состав (включая платиновые, редкие и редкоземельные элементы) за счет концентрирования микроэлементов в небольшой объем неподвижной водной фазы, удерживаемый в колонке.
С начала 70-х годов прошлого века жидкостную хроматографию со свободной неподвижной фазой успешно применяют для разделения органических и биоорганических веществ [1]. Метод основан на удерживании одной из фаз двухфазной жидкостной системы (неподвижной фазы) во вращающейся спиральной колонке (ВСК) под действием центробежных сил, возникающих при вращении колонки вокруг своей оси и ее одновременном обращении вокруг центральной оси прибора (так называемой “планетарной центрифуги”). Другую (подвижную) фазу при этом непрерывно пpокачивают через колонку. Вращающаяся спиральная колонка обычно представляет собой тефлоновый или стальной капилляр c внутренним диаметром от 0.4 до 8.0 мм, спирально намотанный на жесткий цилиндрический сердечник. В англоязычной литературе ЖХСНФ известна как противоточная хроматография (countercurrent chromatography); это название дал методу его основоположник Ито [1].
В нашей стране ЖХСНФ стала развиваться с 1986 г. в Институте геохимии и аналитической химии им. В.И. Вернадского РАН в лаборатории концентрирования. Впервые показано, что ВСК можно успешно использовать для разделения не только органических, но и неорганических веществ с близкими свойствами с применением двухфазных жидкостных систем сложного состава (экстрагент в органическом растворителе–водный раствор соли, кислоты или комплексообразующего реагента) [2, 3]. Такие системы также впервые в мировой практике использованы и для решения радиохимических задач [4].
В отличие от других хроматографических методов, неподвижная фаза в ЖНСНФ удерживается без использования твердого носителя и, по сути, является условно неподвижной, поскольку обе фазы перемешиваются внутри колонки и находятся в постоянном движении. При этом неподвижная жидкая фаза из колонки не вымывается. Данные отличительные черты, а также возможность рассматривать процесс разделения веществ в колонках с использованием двухфазных жидкостных систем как многоступенчатую экстракцию обусловливают своеобразие теории ЖХСНФ [3, 5–9] и особенности ее практического применения. Удерживание неподвижной жидкой фазы в колонке зависит от конструктивных параметров планетарной центрифуги, скорости вращения колонки, скорости прокачивания подвижной фазы и физико-химических свойств двухфазных жидкостных систем (разности плотностей и вязкостей фаз, значения межфазного натяжения).
По эффективности разделения неорганических веществ ЖХСНФ значительно уступает ВЭЖХ, однако метод имеет ряд неоспоримых достоинств, в частности, необычайно широкий спектр исследуемых образцов – от таких сложных и вязких матриц, как сырая нефть, до особо чистых веществ. Кроме того, проведение процесса многоступенчатой экстракции в закрытой системе (тефлоновой колонке) при использовании дополнительно очищенных реагентов дает возможность выделять ультраследовые количества примесных элементов при анализе особо чистых веществ.
Цель настоящей работы − показать возможности и перспективы использования ЖХСНФ при элементном анализе различных по химическому и фазовому составу материалов: от особо чистых веществ до нефти и горных пород.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Вещества разделяли при помощи планетарных центрифуг, оснащенных вертикально расположенной однослойной ВСК емкостью ~20 мл [2, 3]. Одна из таких центрифуг, изготовленных в Институте аналитического приборостроения РАН, представлена на рис. 1. Внутренний диаметр колонки равен 1.5 мм, если это не оговорено дополнительно. Скорость вращения колонки равна ее скорости обращения и составляет 400−800 об/мин. Скорость потока подвижной фазы 1 мл/мин.

Двухфазную жидкостную систему уравновешивали для взаимного насыщения фаз, после чего водную фазу использовали в качестве подвижной, а органическую − в качестве неподвижной фазы (вариант обращенно-фазовой хроматографии). Исключением является выделение элементов из нефти и нефтепродуктов. В данном случае водные растворы неорганических кислот использовали как неподвижную, а образец нефти – как подвижную фазу.
Перед началом хроматографического эксперимента колонку заполняли неподвижной фазой, затем во время вращения на ее вход подавали подвижную фазу. После установления гидродинамического равновесия (удерживание в колонке постоянного объема неподвижной фазы при непрерывном прокачивании подвижной) содержимое колонки извлекали, измеряли объемы обеих фаз и определяли фактор удерживания Sf как отношение объема неподвижной фазы Vs в колонке к общему объему колонки Vc. При концентрировании элементов после установления гидродинамического равновесия вводили пробу, для чего через колонку прокачивали раствор образца (10−250 мл). Затем меняли состав подвижной фазы для элюирования элементов из неподвижной фазы. Объем отбираемых фракций подвижной фазы, прошедшей через колонку, составлял в зависимости от решаемой задачи и вида детектирования 5−20 мл. При концентрировании элементов из нефти и нефтепродуктов после окончания эксперимента неподвижную водную фазу извлекали из колонки для дальнейшего элементного анализа.
Фракции водной фазы анализировали методами атомно-эмиссионной спектрометрии и масс-спектрометрии с индуктивно связанной плазмой (АЭС-ИСП и МС-ИСП).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Анализ особо чистых веществ. Анализ особо чистого хлорида кальция. Задачу группового выделения микропримесей редкоземельных элементов для их определения в высокочистом хлориде кальция решали по предложению фирмы “Мерк”, производящей соли кальция особой чистоты. Применение твердофазной экстракции с последующим анализом концентрата методом МС-ИСП позволяет определять ряд переходных элементов, но не редкоземельные элементы (РЗЭ) и иттрий [10]. Использование ВСК дало возможность предложить альтернативный способ концентрирования большой группы переходных и послепереходных металлов, а также выбрать системы и условия выделения и концентрирования РЗЭ из растворов хлорида кальция.
Для извлечения суммы РЗЭ из растворов хлорида кальция изучены системы, содержащие ди-2-этилгексилфосфорную кислоту (Д2ЭГФК) и дифенил(дибутилкарбамоилметил)фосфиноксид (КМФО) − высокоэффективный экстракционный реагент для РЗЭ и актинидов, предложенный в лаборатории радиохимии ГЕОХИ РАН. Показано, что экстракционные системы на основе 0.5 М раствора Д2ЭГФК в декане не обеспечивают количественного извлечения группы РЗЭ из растворов CaCl2. Групповое концентрирование РЗЭ из растворов хлорида кальция также изучено в системе КМФО в хлороформе−HNO3. В этой системе хлорид кальция не только не мешает экстракции, но и ведет себя как высаливающий агент, поэтому извлечение РЗЭ увеличивается с ростом концентрации растворов соли (от 1 до 5%). Добавление небольших количеств хлорной кислоты к азотнокислому раствору образца позволяет полностью экстрагировать РЗЭ из более концентрированных растворов хлорида кальция и уменьшить объем реэкстракта [11]. На рис. 2 приведены значения степени извлечения РЗЭ из 5%-ного раствора хлорида кальция в смеси хлорной и азотной кислот. В качестве неподвижной фазы использовали 0.12 и 0.5 М растворы КМФО в хлороформе. Из рис. 2 видно, что экстракционная система 0.5 М раствор КМФО в хлороформе–3.0 М HNO3 + + 0.1 М HClO4 позволяет количественно выделить все РЗЭ.
Рис. 2.
Результаты выделения РЗЭ из 30 (1) и 150 мл (2) 5%-ного раствора CaCl2 в смеси 3 М HNO3 и 0.1 М HClO4 во вращающейся спиральной колонке с неподвижной фазой: 1 – 0.12 М КМФО в хлороформе (Vs = 8.7 мл, Sf = 0.59), 2 – 0.5 М КМФО в хлороформе (Vs = 13.0 мл, Sf = 0.87). Подвижная фаза на стадии элюирования – 3 мл воды.
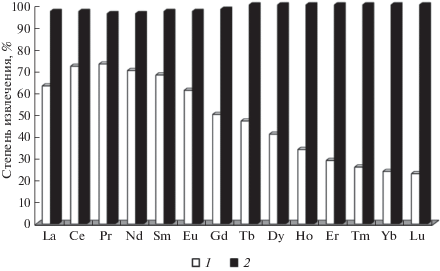
На основании полученных результатов предложена методика группового выделения микропримесей РЗЭ методом ЖХСНФ с их последующим МС-ИСП-определением в высокочистом хлориде кальция. Правильность предложенной методики проверяли методом введено–найдено. Полученные значения степеней извлечения всех РЗЭ находятся в пределах 95–100%. Значения стандартного отклоненя не превышают 0.20, что является удовлетворительным для ультраследовых концентраций элементов. В табл. 1 приведены результаты определения содержания РЗЭ в реальном образце хлорида кальция фирмы “Мерк”, а также пределы обнаружения РЗЭ, которые находятся на уровне долей пг/г, что на два–три порядка ниже возможностей прямого инструментального определения методом МС-ИСП [11].
Таблица 1.
Результаты определения методом масс-спектрометрии с индуктивно связанной плазмой микропримесей РЗЭ в высокочистом CaCl2 ⋅ 2H2O (Мерк, Германия) после их выделения с помощью жидкостной хроматографии со свободной неподвижной фазой (n = 5, P = 0.95)
| Элемент | сmin, нг/г | Найдено РЗЭ, нг/г |
|---|---|---|
| Y | 0.016 | 0.03 ± 0.01 |
| La | 0.014 | 0.20 ± 0.07 |
| Ce | 0.015 | 0.4 ± 0.1 |
| Pr | 0.001 | 0.03 ± 0.01 |
| Nd | 0.003 | 0.21 ± 0.07 |
| Sm | 0.011 | 0.07 ± 0.02 |
| Eu | 0.016 | 0.02 ± 0.01 |
| Gd | 0.007 | 0.05 ± 0.02 |
| Tb | 0.004 | 0.021 ± 0.009 |
| Dy | 0.003 | 0.04 ± 0.02 |
| Ho | 0.001 | 0.015 ± 0.006 |
| Er | 0.005 | 0.021 ± 0.006 |
| Tm | 0.004 | 0.008 ± 0.003 |
| Yb | 0.002 | <0.002 |
| Lu | 0.002 | <0.002 |
Проведено также выделение и концентрирование группы переходных металлов и некоторых непереходных элементов из растворов хлорида кальция. Для решения данной задачи наиболее приемлемыми оказались растворы дигексилдитиофосфата тетрабутиламмония (ДГДТФТБА) в хлороформе. Оптимальна система 0.05 М ДГДТФТБА в хлороформе–3 М HNO3. В этих условиях удается количественно извлекать Zn, Cd, Cu, Pb, Ni, In, Tl и другие элементы из 5%-ного раствора хлорида кальция.
Анализ особо чистого свинца. Применение особо чистого свинца необходимо при решении одной из важнейших задач современной ядерной физики – изучении свойств нейтрино. Информация о массе и взаимопревращениях нейтрино позволяет понять, как субатомные частицы повлияли на развитие вселенной. Для проведения экспериментов нужны детекторы, находящиеся глубоко под землей и изготовленные из высокочистых материалов. В частности, защитный кожух должен быть изготовлен из сверхчистого свинца, содержание урана и тория в котором не превышало бы 1 пг/г (10–10 мас. %) [12, 13]. Однако свинец, произведенный в наши дни, имеет фон от радиоактивного 210Pb с Т1/2 = 22.2 года, а также его дочерних изотопов 210Bi и 210Po с Т1/2 5.012 и 138.376 дня соответственно, входящих в состав естественного радиоактивного ряда 238U–226Ra, которые в достаточно больших количествах имеются в любой Pb-содержащей руде. Для уменьшения радиоактивного фона от 210Pb в 1000 раз необходимо выдерживать свинец в течение более 10 периодов полураспада, т.е. более 222 лет. Одним из перспективных источников такого выдержанного свинца является античный свинец, полученный на Римских рудниках в Испании и Англии более 2000 лет назад и найденный на местах кораблекрушений.
Для оценки чистоты материала необходимо применение комбинированных методов, так как ни один инструментальный вид анализа не позволяет достичь столь низких пределов обнаружения. Показано, что при прямом определении урана и тория методом МС-ИСП их пределы обнаружения находятся на уровне нг/г и составляют 5 × 10–7 и 2 × × 10–8 мас. % соответственно.
Для снижения предела обнаружения применяют сочетание МС-ИСП с такими методами концентрирования, как ионный обмен, экстракция, флотация, сорбция [14]. Однако при концентрировании микрокомпонентов необходим тщательный контроль окружающей среды, чистоты используемых реактивов, лабораторной посуды и т.п., чтобы получить достоверные результаты. Также заметно возрастает трудоемкость и продолжительность анализа. Наиболее приемлемым методом концентрирования и выделения урана и тория при анализе высокочистого свинца можно считать экстракционную хроматографию, однако предложенная методика многостадийна, трудоемка и требует проведения всего эксперимента в условиях “чистой комнаты” [13]. Альтернативой экстракционной хроматографии может быть ЖХСНФ, в которой разделение и концентрирование проводят в закрытой системе в колонке из тефлона – чистого инертного материала, использование которого практически исключает дополнительное загрязнение анализируемого раствора.
Первоначально для концентрирования и выделения урана и тория была выбрана экстракционная система Д2ЭГФК в н-гексане–водный раствор HCl. Изучали влияние концентрации экстракционного реагента на разделение урана, тория и других элементов. Показано, что при уменьшении концентрации экстрагента от 0.1 до 0.01 М Co, Ni, Cu, Zn, Ge, Ag, Cd, Te, Cs, Ba, Re, Tl, Pb не экстрагируются и вымываются из колонки со свободным объемом подвижной фазы, а уран, торий и РЗЭ экстрагируются количественно. Разделение проводили при ступенчатом увеличении концентрации HCl в элюенте (подвижной фазе) от 1 до 5 М. Количественной реэкстракции урана, тория и РЗЭ в этих условиях достичь не удалось. При ступенчатом элюировании азотной кислотой реэкстракция суммы РЗЭ составила 90–100%, урана – 89%, а тория – 9%. При изучении извлечения элементов из азотнокислой среды с последующим элюированием растворами азотной кислоты различных концентраций показано, что степени экстракции и реэкстакции значительно снижаются для всех элементов, кроме иттрия и РЗЭ, для которых степень извлечения остается на уровне 85–100%. Уран и торий реэкстрагируются неколичественно. Для количественного извлечения урана и тория необходима другая экстракционная система: тетрафенилметилендифосфин диоксид (ДФО) в хлороформе–водный раствор HNO3. Предварительно в условиях статической экстракции изучено влияние времени контакта фаз, кислотности среды и концентрации реагента на коэффициенты распределения урана и тория. Оптимальны концентрация реагента 0.01 М и концентрация азотной кислоты 1 М. В динамических условиях изучено влияние сопутствующих элементов на извлечение урана и тория. Установлено, что Pb, Mg, Al, Ti, V, Cr, Mn, Co, Ni, Cu, Zn, Ga, Ge, Rb, Sr, Ag, Mo, Cd, Sn, Sb, Te, Cs, Ba, Re, W, Tl, Zr, Nb, Hf, Ta в этих условиях не экстрагируются. Реэкстракцию урана и тория проводили при элюировании 0.01 М раствором этидроновой кислоты. Степень извлечения урана и тория составила 100%.
Разработанный комбинированный метод применен для анализа образца античного свинца. Полученные пределы обнаружения составили для урана 3 пг/г (3 × 10–10 мас. %), а для тория – 1 пг/г (1 × 10–10 мас. %). Содержание урана и тория в образце античного свинца не превышало пределов обнаружения [15].
Таким образом, применение методического подхода, основанного на выделении урана и тория методом ЖХСНФ и их последующем МС-ИСП-определении, позволило на 2–3 порядка снизить пределы обнаружения (по сравнению с прямым инструментальным определением) и достичь уровня пг/г. Возможность использования практически любых экстракционных систем в ЖХСНФ открывает широкие перспективы применения предложенного подхода при анализе различных высокочистых материалов.
Анализ твердых геологических образцов. Благодаря высокой препаративной емкости вращающихся колонок (объем удерживаемой неподвижной жидкой фазы может составлять до 80% от общего объема колонки), ЖХСНФ может быть использована как для выделения следов элементов из сложных многокомпонентных матриц, так и для извлечения матричных элементов [3]. Применение жидкостных систем на основе экстракционных реагентов различных классов позволило предложить ряд методик группового концентрирования и выделения редких, редкоземельных и других элементов из растворенных геологических образцов перед последующим определением элементов методом АЭС-ИСП. При групповом выделении РЗЭ из образцов руд, базальта, гранита и т.д. использовали системы, содержащие Д2ЭГФК, триоктилфосфиноксид, КМФО [3]. Разработанные методики применяли для анализа геологических образцов в ГЕОХИ РАН.
Экстракционная система на основе 0.1 М раствора тетраоктилэтилендиамина в хлороформе позволяет проводить концентрирование Zr, Hf, Nb, и Ta и их последующее выделение в небольшой (7 мл) объем элюата при анализе сложных геологических объектов [3]. Сброс матричных элементов достигается на стадии концентрирования редких элементов (элюенты 0.1 М HCl + 0.01 М H2C2O4 и 0.1 М HCl + 5%-ная аскорбиновая кислота). После выделения редких элементов (элюент 2.0 М HCl) колонку промывали 1.0 М HNO3 для регенерации. Выделение редких элементов осуществляют в ходе одного хроматографического эксперимента.
Нужно отметить, что в связи с быстрым развитием инструментальной базы АЭС-ИСП и МС-ИСП не все методики концентрирования и выделения элементов для анализа твердых геологических образцов полностью сохранили свою актуальность. Тем не менее, предложенные методы и подходы являются перспективными и могут найти применение при решении ряда насущных задач аналитической химии и геохимии.
Анализ нефти и нефтепродуктов. Нефтяное сырье может содержать более 60 микроэлементов, большая часть которых представлена металлами (Be, Li, Mo, Zr, Se, Ge, Sb, Ni, V, Co, Mn, Cr, Nb, Re, U, Y, РЗЭ). При этом определение полного элементного состава нефти крайне затруднено из-за низких концентраций ряда элементов (платиновых, редких и редкоземельных). Благодаря развитию современного приборостроения, на рынок аналитического оборудования поступают новые приборы с улучшенными характеристиками. Высокая чувствительность к определяемым элементам в различных матрицах (на фоне содержания солей до 10–12 мас. %) отчасти позволяет решить указанную выше проблему. Тем не менее, инструментальное определение элементов в нефти невозможно без использования стадии пробоподготовки.
Для пробоподготовки нефти перед определением микроэлементов наиболее часто используют разбавление растворителями, эмульгирование и кислотное разложение [16]. Кислотное разложение – наиболее доступный и распространенный способ пробоподготовки, применяемый для определения РЗЭ и других элементов в нефтях [17], который, однако, является длительным и не лишен риска потерь летучих соединений элементов в процессе озоления образца. Использование традиционных способов пробоподготовки часто не позволяет обнаружить низкие содержания ряда микроэлементов.
Эффективность выделения микроэлементов из нефтяного сырья во многом определяется формами их нахождения в объекте анализа. Это – соли металлов, элементоорганические соединения, внутримолекулярные комплексы, полилигандные комплексы или π-комплексы с ароматическими ядрами. Известно, что ряд металлов, содержащихся в нефти, концентрируется преимущественно в ее асфальтово-смолистых фракциях. Так, ванадий и никель в основном находятся в нефти в виде порфириновых и непорфириновых соединений, прочно связанных с компонентами асфальтеновых структур нефти по типу “сэндвич-комплексов”, что значительно затрудняет их полное экстракционное извлечение. Изменяя полярность растворителей при реализации экстракционного процесса, можно извлекать разные формы элементов из нефти без изменения структуры нефтяного образца. Так, полярные органические растворители (спирты, кетоны, диметилкарбонат, водные и спиртовые растворы диметилформамида, тетрагидрофурана, пиридина, уксусной кислоты и ее производных и т.д.) можно использовать для выделения порфириновых и непорфириновых соединений ванадия и никеля из нефтяного сырья. Для данной цели применяют также растворы неорганических кислот (H2SO4, HCl, HBr), солей (хлориды олова, титана), комплексообразующих реагентов.
Тяжелые нефти, битумы и тяжелые нефтяные остатки (ТНО) отличаются от обычных нефтей повышенным содержанием металлов (V, Ni, Fe, Mo, Cu, Na), S, N и асфальтенов. Например, содержание V, Ni, Ag в смолах нефти Русского месторождения Томской области находится на уровне 35.2, 35.0, 34.2 мкг/г соответственно.
Показано, что ЖХСНФ позволяет проводить концентрирование микропримесей при элементном анализе нефтяных образцов (включая высоковязкие нефти и тяжелые нефтяные фракции). Нефть при этом прокачивают через колонку как подвижную фазу, а в качестве неподвижной используют водный раствор HNO3 [18, 19]. Предварительное концентрирование микроэлементов из нефти с помощью ВСК позволяет определять РЗЭ в концентрациях на уровне пг/г. Данный способ пробоподготовки довольно прост в исполнении, не требует большого расхода реагентов, время экстрагирования зависит от исходной концентрации элементов в нефти и пределов определения метода детектирования (чем ниже концентрация РЗЭ в нефти, тем больший объем нефти используют для извлечения элементов).
Описаны зависимости удерживания систем нефть/нефтепродукт–водная фаза от конструкционных и рабочих параметров ВСК, а также от физико-химических свойств самих образцов [20]. Определены пограничные условия применения ВСК в зависимости от вязкости и плотности фаз. Показано, что в ВСК достигается значение Sf > > 0.9 при Δρ > 0.210 г/см3 и Δη < 0.470 мм2/с. Необходимые параметры разности плотностей и вязкостей фаз в ВСК достигаются путем разбавления нефтяных образцов (подвижная фаза) растворителями. При необходимости использования растворителя для разбавления ТНО до требуемых параметров разностей плотностей и вязкостей фаз наилучшие результаты определения элементов были достигнуты при добавлении толуола.
В табл. 2 приведены результаты извлечения ряда элементов из раствора ТНО Черниговской нефти в толуоле и о-ксилоле. Добавки определяемых элементов вводили в растворы ТНО, используя стандартный раствор Коностан S-21. Раствор ТНО до и после прокачивания через ВСК анализировали методом АЭС-ИСП. Полученные данные позволяют судить о полноте перехода элементов в неподвижную водную фазу. Из табл. 2 видно, что при использовании ВСК возможно выделение практически всех металлов, за исключением Ni и V. Для выделения данных элементов должна быть использована неподвижная фаза другого состава.
Таблица 2.
Результаты (мкг/г) анализа методом атомно-эмиссионной спектрометрии с индуктивно связанной плазмой раствора тяжелого нефтяного остатка Черниговской нефти в органическом растворителе (11 мг/мл) до и после извлечения элементов методом жидкостной хроматографии со свободной неподвижной фазой (n = 3, P = 0.95)
| Элемент | Исходный раствор | Раствор после ЖХСНФ | ||
|---|---|---|---|---|
| толуол | о-ксилол | толуол | о-ксилол | |
| Mg | 0.47 ± 0.03 | 0.41 ± 0.04 | – | – |
| Al | 3.1 ± 0.6 | 2.9 ± 0.3 | – | – |
| Ca | 2.7 ± 0.1 | 2.8 ± 0.1 | 0.21 ± 0.03 | 0.15 ± 0.03 |
| V | 29 ± 2 | 25.7 ± 0.9 | 29 ± 2 | 26 ± 1 |
| Mn | 0.17 ± 0.05 | 0.12 ± 0.02 | – | – |
| Fe | 15 ± 3 | 9.8 ± 0.1 | – | – |
| Ni | 33 ± 5 | 26 ± 3 | 33 ± 5 | 26 ± 3 |
| Zn | 1.8 ± 0.2 | 1.5 ± 0.2 | – | – |
| Ba | 0.5 ± 0.1 | 0.31 ± 0.08 | – | – |
В табл. 3 приведены результаты МС-ИСП-определения элементов в пробах нефтей различных месторождений, полученные после экстракционного концентрирования в ВСК. Видно, что использование экстракционного концентрирования в ВСК в качестве пробоподготовки дает возможность определять в исследуемых образцах нефтей большинство элементов, в том числе РЗЭ.
Таблица 3.
Результаты (нг/г) определения методом масс-спектрометрии с индуктивно связанной плазмой микроэлементов в сырых нефтях после экстракционного концентрирования во вращающейся спиральной колонке (n = 3, P = 0.95)
| Элемент/образец | Нефть Ромашкинского месторождения | Нефть Арланского месторождения | Нефть Приобского месторождения | Нефть Лабаганского месторождения |
|---|---|---|---|---|
| Nb | 11 ± 1 | 2.6 ± 0.4 | 3.9 ± 0.6 | 0.37 ± 0.02 |
| Ru | 11 ± 1 | 0.09 ± 0.01 | 0.14 ± 0.02 | 0.082 ± 0.008 |
| Rh | 41 ± 5 | 0.13 ± 0.02 | 0.19 ± 0.03 | 0.28 ± 0.03 |
| Ag | 7.5 ± 0.8 | 1.2 ± 0.2 | 1.8 ± 0.2 | 0.56 ± 0.07 |
| Sb | 45 ± 7 | 4.6 ± 0.3 | 10 ± 2 | 0.67 ± 0.08 |
| Te | 3.7 ± 0.6 | 0.52 ± 0.03 | 1.6 ± 0.5 | 0.070 ± 0.009 |
| Hf | 0.56 ± 0.07 | 0.39 ± 0.07 | 0.58 ± 0.08 | 0.061 ± 0.007 |
| Ir | 1.1 ± 0.2 | 0.071 ± 0.008 | 0.10 ± 0.03 | 0.012 ± 0.003 |
| Pt | 0.56 ± 0.06 | 0.13 ± 0.03 | 0.19 ± 0.01 | 0.020 ± 0.002 |
| Tl | 1.9 ± 0.5 | 19 ± 2 | 6.3 ± 0.4 | 0.091 ± 0.001 |
| La | 3.5 ± 0.4 | 8.5 ± 0.6 | 1.6 ± 0.1 | 0.34 ± 0.02 |
| Ce | 8.6 ± 0.7 | 20 ± 3 | 4.5 ± 0.6 | 0.28 ± 0.02 |
| Pr | 1.1 ± 0.2 | 2.4 ± 0.5 | 1.3 ± 0.1 | 0.082 ± 0.009 |
| Nd | 5.2 ± 0.6 | 10 ± 2 | 0.39 ± 0.07 | 0.35 ± 0.02 |
| Sm | 0.95 ± 0.08 | 2.5 ± 0.5 | 0.47 ± 0.05 | 0.11 ± 0.01 |
| Eu | 0.28 ± 0.06 | 0.39 ± 0.07 | 0.06 ± 0.01 | 0.042 ± 0.006 |
| Tb | 0.22 ± 0.02 | 0.38 ± 0.07 | 0.10 ± 0.03 | 0.020 ± 0.003 |
| Gd | 0.37 ± 0.04 | 1.8 ± 0.3 | 0.18 ± 0.02 | 0.13 ± 0.02 |
| Dy | 0.19 ± 0.03 | 2.0 ± 0.1 | 0.19 ± 0.02 | 0.093 ± 0.006 |
| Ho | 0.30 ± 0.05 | 0.47 ± 0.03 | 0.023 ± 0.004 | 0.031 ± 0.002 |
| Er | 2.2 ± 0.2 | 1.6 ± 0.1 | 2.3 ± 0.1 | 0.17 ±0.01 |
| Tm | 0.19 ± 0.03 | 0.16 ± 0.03 | 0.060 ± 0.004 | 0.011 ± 0.002 |
| Yb | 1.7 ± 0.3 | 1.0 ± 0.2 | 0.39 ± 0.04 | 0.040 ± 0.004 |
| Lu | 0.74 ± 0.07 | 0.52 ± 0.01 | 0.78 ± 0.05 | 0.072 ± 0.003 |
| Σрзэ | 25.26 | 51.72 | 12.34 | 1.76 |
| Σрзэлегк | 19.35 | 43.79 | 8.32 | 1.2 |
| Σрзэтяж | 5.91 | 7.93 | 4.02 | 0.56 |
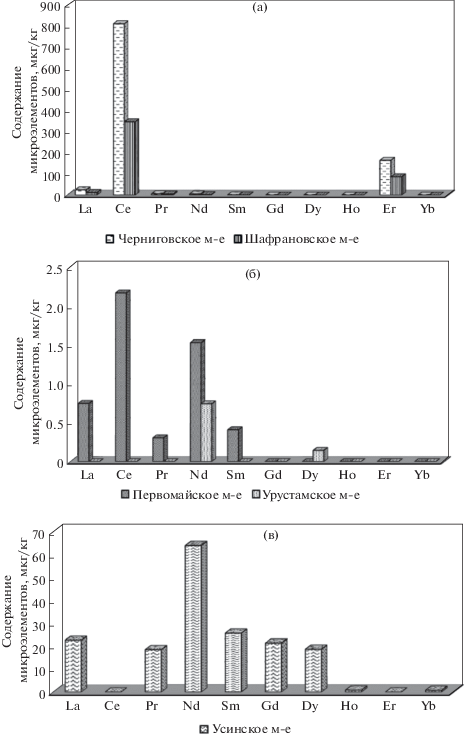
Использование предложенного подхода позволяет изучить характер распределения РЗЭ в нефтях различных месторождений (рис. 3). В нефтях Черниговского и Шафрановского месторождений (рис. 3а) выявлены аналогичные закономерности распределения РЗЭ. В обеих нефтях обнаружены La, Pr, Nd, Sm в незначительных количествах, в максимальных количествах содержатся Ce и Er. В пробе Первомайского месторождения (рис. 3б) найдены La, Ce, Pr, Nd, Sm с преобладанием Ce (2.16 мкг/кг). В нефти Урус-Тамакского месторождения из всего набора РЗЭ обнаружены только Nd и Dy. По имеющимся сведениям нефть Урус-Тамакского месторождения отличается наименьшими значениями плотности и вязкости среди всех исследуемых образцов. При этом в нефти Усинского месторождения, отличающейся самыми высокими значениями плотности и вязкости, найдено наибольшее число РЗЭ и самые высокие концентрации Pr, Nd, Sm, Gd, Dy, Ho и Yb среди всех исследуемых образцов нефтей.
Рис. 3.
Распределение РЗЭ в нефтях различных месторождений: (а) – Черниговское, Шафрановское месторождения, (б) – Первомайское, Урус-Тамакское месторождения, (в) – Усинское месторождение.
Таким образом, показано, что применение ЖХ-СНФ на стадии пробоподготовки позволяет определять содержание РЗЭ в нефти на уровне нг/г, что невозможно при использовании других способов пробоподготовки (например, при автоклавном разложении). Подробная информация о микроэлементном составе нефтей может быть полезна для геохимических исследований, в частности, для изучения генезиса нефтей.
В целом метод ЖХСНФ имеет свою особую нишу в элементом анализе. Среди широкого спектра объектов, при пробоподготовке которых его можно использовать, наиболее интересны крайние случаи – “самые чистые” и “самые грязные” образцы. Проведение разделения и концентрирования микропримесей в закрытой системе, изготовленной из инертного материала (тефлона), в ходе одного хроматографического эксперимента практически исключает дополнительное загрязнения образца (при использовании растворителей и реагентов соответствующей квалификации). Это имеет большое значение при пробоподготовке особо чистых веществ. С другой стороны, реализация процесса многоступенчатой экстракции в лабораторных условиях дает уникальную возможность выделять микроэлементы из таких вязких многокомпонентных образцов, как нефть, когда многие другие методы бессильны. Достоинства ЖХСНФ, ее отличительные черты и уже полученные результаты обусловливают возможность дальнейшего применения метода в элементом анализе.
Список литературы
Mandava N.B., Ito Y. Countercurrent Chromatography. Theory and Practice. New York: Marcel Dekker Inc., 1988. 595 p.
Zolotov Yu.A., Spivakov B.Ya., Maryutina T.A., Bashlov V.L., Pavlenko I.V. Partition countercurrent chromatography in inorganic analysis // Fresenius Z. Anal. Chem. 1989. V. 335. № 8. P. 938.
Maryutina T.A., Fedotov P.S., Spivakov B.Ya. Application of countercurrent chromatography in inorganic analysis / Countercurrent chromatography. Chromatographic Science Series / Eds. Menet J.-M., Thiebaut D. New York: Marcel Dekker Inc., 1999. V. 82. Ch. 6. P. 171.
Чмутова М.К., Марютина Т.А., Спиваков Б.Я., Мясоедов Б.Ф. Разделение америция(III) и европия(III) в системах с нейтральными бидентатными фосфороорганическими экстрагентами методом жидкостной хроматографии со свободной неподвижной фазой // Радиохимия. 1992. № 6. С. 56.
Maryutina T.A., Spivakov B.Ya. CCC Two-phase solvent systems / Encyclopedia of Chromatography / Ed. Cazes J. New York: Marcel Dekker Inc., 2001. P. 137.
Fedotov P.S., Spivakov B.Ya. Hydrodynamic equilibrium in CCC / Encyclopedia of Chromatography / Ed. Cazes J. New York: Marcel Dekker Inc., 2001. P. 414.
Марютина Т.А., Федотов П.С., Спиваков Б.Я. Использование метода жидкостной хроматографии со свободной неподвижной фазой для концентрирования и разделения неорганических веществ. Двухфазные жидкостные системы // Журн. аналит. химии. 1997. Т. 52. № 12. С. 1263.
Марютина Т.А., Игнатова С.Н., Федотов П.С., Спиваков Б.Я. Влияние физико-химических свойств двухфазных жидкостных систем на удерживание органической фазы во вращающейся спиральной колонке // Журн. аналит. химии. 1999. Т. 54. № 8. С. 825.
Fedotov P.S., Kronrod V.A., Maryutina T.A., Spivakov B.Ya. On the mechanism of stationary phase retention in rotating coil column // J. Liq. Chromatogr. Related Technol. 1996. V. 19. № 20. P. 3237.
Keil O., Dahmen J., Volmer D.A. Automated matrix separation and preconcentration for the trace level determination of metal impurities in ultrapure inorganic salts by high-resolution ICP-MS // Fresenius J. Anal. Chem. 1999. V. 364. № 8. P. 694.
Ignatova S.N., Maryutina T.A., Spivakov B.Ya., Karandashev V.K. Group separation of trace rare earth elements by countercurrent chromatography for their determination in high-purity calcium chloride // Fresenius J. Anal. Chem. 2001. V. 370. № 8. P. 1109.
Leonard D.S., Grinberg P., Weber P. et al. Systematic study of trace radioactive impurities in candidateconstruction materials for EXO-200 // Nucl. Instrum. Methods Phys. Res. A. 2008. V. 591. P. 490.
Grinberg P., Willie S., Sturgeon R.E. Determination of thorium and uranium in ultrapure lead by inductively coupled plasma mass spectrometry // Anal. Chem. 2005. V. 77. P. 2432.
Rao T.P., Metilda P., Gladis J.M. Preconcentration techniques for uranium(VI) and thorium(IV) prior toanalytical determination // Talanta. 2006. V. 68. P. 1047.
Федюнина Н.Н., Федотов П.С., Философов Д.В., Якушев Е.А. МС-ИСП определение ультранизких содержаний урана и тория в античном свинце после их выделения методом жидкостной хроматографии со свободной неподвижной фазой // Заводск. лаборатория. Диагностика материалов. 2018. Т. 84. № 4. С. 12.
Sanchez R., Todoli J.L., Lienemann C.P., Mermet J.M. Determination of trace elements in petroleum products by inductively coupled plasma techniques: a critical review // Spectrochim. Acta B. 2013. V. 88. P. 104.
Akinlua A., Sigedle A., Buthelezi T., Fadipe O.A. Trace element geochemistry of crude oils and condensates from South African Basins // Marine Petrol. Geol. 2015. V. 59. P. 286.
Maryutina T.A., Soin A.V. Novel approach to the elemental analysis of crude and diesel oil // Anal. Chem. 2009. V. 81. № 14. P. 5896.
Soin A., Maryutina T., Musina N., Soin An. New possibility for REE determination in oil // Int. J. Spectrosc. 2012. URL: http://dx.org/10.1155/2012/174697 (13.09.2018).
Соин А.В. Применение вращающихся спиральных колонок при определении микроэлементов в нефтях и нефтепродуктах. Дис. … канд. хим. наук. М.: ГЕОХИ РАН, 2010. 125 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии