

Вестник Военного инновационного технополиса «ЭРА», 2022, T. 3, № 2, стр. 116-123
ДНК ИЗ ОКРУЖАЮЩЕЙ СРЕДЫ (environmental DNA): МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЙ ПОДХОД К ИЗУЧЕНИЮ СООБЩЕСТВ ОРГАНИЗМОВ
М. В. Гладышева-Азгари 1, *, С. М. Расторгуев 1
1 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
* E-mail: marglader@gmail.com
Поступила в редакцию 21.03.2022
После доработки 28.03.2022
Принята к публикации 07.04.2022
Аннотация
За последние два десятилетия произошли значительные изменения в изучении биологического разнообразия за счет развития методов молекулярной биологии и генетики и их имплементации в экологические науки. Одним из подходов к изучению сообществ является анализ ДНК из окружающей среды (environmental DNA, eDNA), ранее использовавшийся для исследования водных и почвенных сред, но в настоящее время перенятый для исследований наземно-воздушных сообществ. Анализ eDNA – это технология, которая находит широкий спектр применения при проведении экологических исследований, дополняя картину биоразнообразия сообществ и взаимосвязей видов в них. Методология анализа eDNA включает в себя сбор образцов, выделение ДНК, амплификацию целевых регионов, секвенирование полученных ампликонов и последующий анализ данных. Каждый этап имеет особенности в зависимости от поставленных целей и задач исследования. Анализ eDNA является быстрым и малоинвазивным методом, но из-за вариативности его методологии желательно предварительное понимание биологии и экологии изучаемых объектов.
ОГЛАВЛЕНИЕ
Введение
1. Применение анализа ДНК из окружающей среды
2. Методология работы с ДНК из окружающей среды
2.1. Субстраты и сбор образцов
2.2. Фильтровальные системы и выделение ДНК
2.3. Амплификация целевых последовательностей из образца ДНК из окружающей среды
2.4. Баркоды и праймеры
2.5. Секвенирование и анализ данных
Заключение
ВВЕДЕНИЕ
Оценка биологического разнообразия и идентификация таксонов в экосистемах до некоторого времени были задачами экологии и смежных наук. Для их решения использовались данные, в основном полученные обсервационными методами – учетом особей в природе с помощью биноклей и ловушек, анализом микробных сообществ с помощью микроскопа и других приборов. Однако эти традиционные методы мониторинга биоразнообразия сильно зависят от таксономических и диагностических знаний [1]. Целые таксоны внутри изучаемого сообщества могут быть неправильно идентифицированы или не обнаружены вследствие невозможности проведения достаточных наблюдений, что может быть связано с образом жизни объектов исследования, их небольшим размером, испугом от присутствия исследователя и с прочими причинами. Традиционные обсервационные методы часто являются предвзятыми, инвазивными и деструктивными; они также трудозатратны, требуют присутствия специалистов-морфологов и занимают много времени [2].
С развитием технологий молекулярной биологии и диагностики стало возможным применять методы молекулярной биологии в экологических исследованиях. На данный момент в фондах, предоставляющих информацию о биоразнообразии, помимо морфологических, анатомических и экологических особенностей вида и его вариантов предоставляется информация о нуклеотидных последовательностях. Например, Глобальный фонд информации о биоразнообразии имеет 420 миллионов записей и 1.45 миллиона названий видов (www.gbif.org). Записи попадают в базы данных не только основными путями (сбор образцов из музеев и гербариев, краудсорсинговые наблюдения, изображения и звуки, полученные с помощью методов дистанционного наблюдения при помощи камер или диктофонов), но и благодаря отбору образцов ДНК [3].
Одним из наиболее популярных молекулярно-генетических подходов к изучению биоразнообразия является изучение ДНК из окружающей среды (environmental DNA, eDNA). В настоящее время eDNA определяют как генетический материал, полученный непосредственно из образцов окружающей среды (почвы, донных отложений, воды и т.д.). ДНК может сохраняться в окружающей среде в течение нескольких часов в проточных водоемах или тысяч лет в вечной мерзлоте, что позволяет проводить прямую изоляцию без каких-либо других явных признаков присутствия организма. При этом подходе к получению генетического материала может быть применен набор молекулярных методологий и инструментов анализа для ответа на вопросы различных биологических наук, связанных с изучением окружающей среды [4].
На рубеже XX–XXI веков концепция eDNA относилась в основном к изучению бактериальных геномов и была ассоциирована с метагеномикой микробных сообществ и исследованиями их разнообразия [5–8]. Позднее этот термин стал использоваться в работах, посвященных биоразнообразию эукариот [9–12] и детекции эукариотических видов в окружающей среде [13]. Увеличение количества исследований с использованием ДНК из окружающей среды связывают со стадиями прогресса молекулярной биологии, последовавшими за изобретением ПЦР-амплификации, а затем за развитием методов высокопроизводительного секвенирования [14].
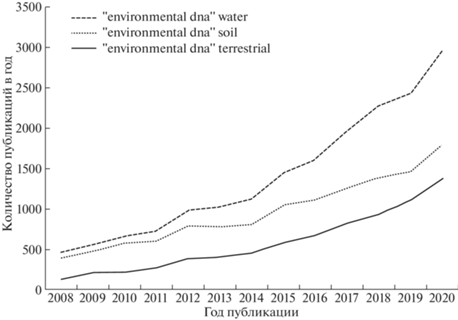
До недавнего времени анализ eDNA ограничивался исследованиями водных или почвенных сред, что приводило к преобладанию изученности водных и почвенных сообществ и входящих в них видов – рыб, амфибий, морских млекопитающих и прочих [15–17]. Для наземных организмов отсутствие субстрата, сопоставимого с водой с точки зрения простоты отбора проб и репрезентативности содержания eDNA, может препятствовать эффективности исследований на основе анализа eDNA в наземных местообитаниях. На данный момент появились работы, в которых наземные виды исследуются путем отбора водных проб, например у водопоя наземных млекопитающих [18–20]. Также в последнее время получают распространение работы, в которых пробы для исследований наземных сообществ отбираются не из воды, а из иных субстратов, поэтому общее количество публикаций, посвященных изучению различных экологических аспектов наземных экосистем посредством eDNA, растет с каждым годом (рис. 1).
Рис. 1.
Результаты поисковых запросов в базе данных научных публикаций Google Scholar. Поисковые запросы включали в себя ключевые слова, связанные с ДНК из окружающей среды (environmental DNA) и изученной средой/местом обитания организмов – водой, почвой и наземным местообитанием (water, soil, terrestrial).
1. ПРИМЕНЕНИЕ АНАЛИЗА ДНК ИЗ ОКРУЖАЮЩЕЙ СРЕДЫ
Изучение eDNA имеет широкий спектр применения за счет малой инвазивности методов, их относительной дешевизны, быстроты применения, возможности обнаружения ДНК редких видов и оценки биоразнообразия для широкого таксономического диапазона [2]. Результаты работ показывают, что изучение eDNA представляет собой огромный потенциал для решения вопросов фундаментальных исследований в области экологии как при изучении водных и почвенных, так и наземных экосистем. ДНК из окружающей среды используется для улучшения оценки представленности таксонов в сообществе, когда трудно получить полную картину биоразнообразия с помощью традиционных методов [21–24], а также может применяться для изучения причин снижения биоразнообразия [25, 26], влияния загрязнений [27] и биомониторинга [28]. Изучение e-DNA в некоторых случаях позволяет обнаружить присутствие видов, считавшихся исчезнувшими [29] или находящихся под угрозой исчезновения и, как следствие, представленных в сообществе малым числом особей, практически неотслеживаемых с помощью обсервационных методов [30–32]. Посредством анализа ДНК из окружающей среды можно обнаружить виды, населяющие экстремальные условия [33]. Подобным образом возможно оценить представленность видов и их распределение в сообществе как временное, так и пространственное [34–38], а также возникновение инвазий и их влияние на нативные популяции [39]. С помощью eDNA становится возможным оценить трофические связи между видами в наземном сообществе, определить пищевую нишу вида и его образ питания [40–42]. Особенно интересным в наземно-воздушной среде представляется биомониторинг патогенов и аллергенов посредством секвенирования eDNA, необходимый для изучения и смягчения их воздействия на здоровье человека [43, 44].
2. МЕТОДОЛОГИЯ РАБОТЫ С ДНК ИЗ ОКРУЖАЮЩЕЙ СРЕДЫ
2.1. Субстраты и сбор образцов
Методики сбора образцов и дальнейшей экстракции eDNA сильно варьируются в зависимости от исследуемых биотопа и объектов. Как было отмечено выше, в наибольшем количестве работ изучается eDNA в образцах воды и почвы [45–47], однако в последнее время исследователи обращают особое внимание на иные интересные источники генетического материала в окружающей среде для изучения наземных видов, например воздух [48–50], фекалии хищников [51], слюна хищников [52], мед [53].
В случае наземных экосистем субстрат и методика сбора образца сильно влияют на количество таксонов, обнаруженных в дальнейшем при помощи анализа eDNA [54], поэтому результаты исследования сильно зависят от изначального понимания экологических взаимодействий ключевых исследуемых таксонов. К примеру, для изучения распространения конкретного вида или его обнаружения в среде проводят генетический анализ экскретов или крови его паразитов [55], и наоборот, сообщества наземных беспозвоночных исследуют путем анализа экскретов млекопитающих [56]. В некоторых случаях возможно оценить представленность наземных таксонов путем совмещения анализа eDNA из водных проб и результатов традиционных обсервационных методов [57, 58]; механизмы, с помощью которых ДНК наземных животных переносится в водоток, сложны и, вероятно, зависят от поведения животного – к появлению его ДНК в воде могут приводить плавание, выделение слюны во время питья и отложение мочи или фекалий в воду.
2.2. Фильтровальные системы и выделение ДНК
Для отбора водных или почвенных проб методики фильтрации и выделения eDNA отработаны, но постоянно модифицируются в зависимости от целей исследования. В большинстве случаев методика включает в себя перегонку образца через фильтр с микропорами при помощи вакуумного насоса, лизис либо фильтрата, либо оставшихся на фильтре частиц и дальнейшие очистку и растворение преципитата или элюирование с мембраны колонки.
Для разных водных субстратов используются разные фильтровальные системы и фильтры из различных материалов и с разным диаметром микропор. Это связано с присутствием в образцах возможных ингибиторов ПЦР, затрудняющих амплификацию и дальнейший анализ [59]. Выбор фильтра, способы предварительной фильтрации и хранения фильтра влияют на качество дальнейшей экстракции eDNA и анализа состава сообщества с помощью ДНК-баркодов [60]. Кроме того, из воды и почвы берутся разные слои с разными временными промежутками в зависимости от поставленной исследователями цели [61, 62], к примеру для изучения разных категорий размерности видов (макро-, мейо- и микрофауны) используются фильтры с разными размерами пор, или образец не фильтруется вовсе [63, 64].
Из-за разнообразия наземных субстратов способы лизиса и выделения из них сильно различаются. Выделение eDNA подразумевает лизис с использованием либо заготавливаемых буферов для выделения [65], либо коммерческих буферов из наборов для экстракции ДНК (например, наборы линейки “QIAGEN”) [66]. Способы выделения ДНК также влияют на итог: во многих случаях предварительный лизис позволяет получить более полный результат [67], видимая представленность таксонов различается при выделении коммерческими наборами и с использованием фенол-хлороформа [68, 69].
2.3. Амплификация целевых последовательностей из образца ДНК из окружающей среды
ПЦР-методы. Выделенный образец eDNA обычно анализируют путем амплификации с использованием полимеразной цепной реакции (polymerasechainreaction, PCR) и последующего секвенирования ДНК. Амплификация выполняется либо с помощью одновидового подхода с использованием специфических праймеров, либо с помощью многовидового (множественного таксона) подхода с использованием общих праймеров для данной целевой группы организмов. В случаях, когда необходимо быстрое выявление присутствия ДНК организмов в образце (к примеру, ДНК патогенного организма в водоеме), используется петлевая изотермическая амплификация (loop-mediatedisothermalamplification, LAMP), при которой визуализация амплификата возможна без использования лабораторного оборудования [70, 71].
Для амплификации последовательностей из образцов с eDNA применяется несколько модификаций ПЦР-метода из-за присутствия большого числа ингибиторов в отобранных пробах, деградации ДНК в среде и, как следствие, сильной фрагментированности ее молекул. Помимо традиционно используемой стандартной ПЦР в работах встречаются такие модификации, как ПЦР в режиме реального времени (real-time PCR, RT-PCR) [72–74], капельная цифровая ПЦР (droplet digital PCR, ddPCR) [75, 76], вложенная ПЦР (nested PCR) [77, 78] и иные вариации [79].
Баркоды и праймеры. Помимо баз данных по биоразнообразию, основанных на традиционных методах наблюдения и детекции, существуют базы, направленные на сбор информации о генетических маркерах видов, называемых ДНК-баркодами. Наиболее крупной базой данных такого плана является International Barcode of Life (IBoL, http://ibol.org/). Последовательности ДНК-баркодов помещаются в базу данных, которая включает в себя справочную библиотеку ДНК-баркодов и может использоваться для присвоения идентичности последовательностям неизвестного происхождения. База данных предоставляет механизм идентификации, основанный на текущей библиотеке баркодов.
Одним из наиболее важных этапов в исследованиях метабаркодирования eDNA является дизайн праймера для ПЦР. Идентификация таксонов в образце eDNA происходит посредством метабаркодирования ДНК – массового секвенирования ДНК для одновременной молекулярной идентификации нескольких таксонов в сложном образце. Метабаркодирование eDNA основано на том, что короткие стандартизованные участки ДНК – обычно гены митохондриальной, хлоропластной или рибосомной РНК (мтДНК, хпДНК или рРНК) – могут быть амплифицированы с помощью ПЦР, секвенированы и впоследствии использованы в качестве баркодов для идентификации и различения таксонов.
Различные праймеры и локусы различаются по охвату, разрешению и систематической ошибке для разных таксонов. Различные участки митохондриального гена COI для животных, пластидных генов rbcL и matK для растений и внутренний транскрибируемый спейсер (ITS) для грибов являются стандартными и наиболее распространенными баркодами; эти участки в основном были выбраны из-за их высокого разрешения на уровне видов и высокой копийности внутри клетки [4]. Однако подобные предлагаемые стандартизованные баркоды ДНК имеют размер >500 пар нуклеотидов (п. н.), тогда как образец eDNA обычно включает в себя сильно фрагментированные последовательности [80]. К примеру, для некоторых исследований COI может оказаться слишком вариабельным, что приводит к трудностям в дизайне праймеров, а ген 28S рРНК – слишком консервативным, что не позволяет различать виды [81], поэтому используются участки генов 12S и 16S рРНК, более подходящие для разрешения на межвидовом уровне [82, 83].
В некоторых случаях гораздо более короткая последовательность баркода может оказаться более информативной на уровне видов, что позволяет метабаркодировать eDNA с высоким таксономическим разрешением. Таким образом, сложность метабаркодирования eDNA также заключается в нахождении коротких амплифицируемых маркеров (~100–300 п. н.), по которым будет возможно различать таксоны, при этом амплифицируемые последовательности должны быть в одной справочной базе данных [84]. Локусы, используемые для метабаркодирования e-DNA, должны быть достаточно короткими для амплификации деградированных образцов, идентичными внутри вида, но различающимися между видами и фланкированными высококонсервативными областями для амплификации множества таксонов без ущерба для специфичности целевой группы [85, 86].
Выбор определенных праймеров для амплификации одного региона может привести к смещению результатов за счет предпочтительной амплификации одних целевых последовательностей в большей степени, чем других [87]. Одним из возможных решений этой проблемы является использование нескольких наборов праймеров, совпадающих со стандартизованными баркодами для целевых таксономических групп [88]. Подобные смещения также исследуются посредством введения искусственных сообществ (mock communities), имитирующих природные сообщества, в качестве контроля для специфичности праймеров и амплификации регионов разных видов [89, 90]. Для исключения амплификации регионов нецелевых таксономических групп и при высокой вероятности контаминации образцов могут использоваться блокирующие праймеры [81, 91] – особенно это важно при изучении наземных сообществ из экскретов, где необходимо исключить амплификацию последовательностей организма-хозяина.
2.4. Секвенирование и анализ данных
На этапе секвенирования доступен ряд платформ, но секвенирование по технологии Illumina в настоящее время является наиболее популярным среди исследователей и превосходит другие платформы NGS с точки зрения глубины и точности [92]. С появлением технологии Oxford Nanopore, позволяющей генерировать длинные прочтения на портативных приборах, стало возможным получать длинные прочтения последовательностей как в лаборатории, так и в полевых условиях [93, 94]. Данные, генерируемые платформами секвенирования во время этих исследований, обрабатываются последовательным программным обеспечением для преобразования необработанных последовательностей. Полученные прочтения возможно сравнить с имеющимися в базах данных последовательностями. Однако при анализе eDNA исследователи часто сталкиваются с последовательностями, не представленными в базах данных. Поэтому чаще всего при анализе результатов секвенирования полученные прочтения не соотносятся с конкретными таксонами, а группируются в молекулярные операционные таксономические единицы (molecular operational taxonomic units, MOTU) на основе сходства последовательностей [57]. Прочтения переводят в статистически пригодную для использования условную матрицу, которая содержит MOTU в виде строк и образцы в виде столбцов. Как было упомянуто выше, для наземных сообществ субстрат, методика сбора и метод выделения могут сильно повлиять на количество различных обнаруженных последовательностей. Это может быть проанализировано по количеству вариантов последовательностей ампликонов (amplicon sequence variants), которые не группируются, как традиционные MOTU, но анализируются во многом таким же образом [54].
Обработку результатов можно условно разделить на несколько этапов. Первым является демультиплексирование образцов, т.е. разделение их прочтений по отдельным файлам в соответствии с заранее включенными для каждого конкретного образца метками в праймерах перед ПЦР-амплификацией. Второй этап подразумевает объединение прочтений в полноразмерные контиги в случае парных прочтений. Третьим этапом проводится фильтрация по качеству, имеющая решающее значение для удаления технических шумов данных – проводится попытка исключить ошибки, связанные с ПЦР, химерными последовательностями или ошибками секвенирования [95, 96]. Затем проводится кластеризация MOTU – этот этап наиболее вариативен и до сих пор является активной областью биоинформатических исследований. Этот шаг имеет решающее значение для получения максимального количества биологически значимой информации и оказывает сильное влияние на измерения разнообразия и последующий анализ. Последний этап включает в себя таксономическое присвоение каждой MOTU, что возможно благодаря базам данных последовательностей.
Различные инструменты, такие как MOTHUR, USEARCH, QIIME, OBITools или VSEARCH, были разработаны специально для анализа данных метабаркодирования eDNA и широко используются в настоящее время [97]. Появляются и новые вариации способов обработки данных – как отдельные программные скрипты [98], так и инструменты, даже не требующие знания командной строки [97].
ЗАКЛЮЧЕНИЕ
Анализ ДНК из окружающей среды является быстрым и малоинвазивным методом для экологических исследований, имеющим широкий спектр применения при изучении как водных, так и наземных сообществ. Мета-анализ работ, включающих в себя сравнение традиционных методов и анализа eDNA, показал, что при прямом сравнении анализ ДНК из окружающей среды является более точным и эффективным, однако в большинстве публикаций прямого сравнения не проводится [99]. Тем не менее при изучении e-DNA желательно знание особенностей биологии и экологии объектов исследования и сред их обитания, так как использование этого анализа обладает рядом особенностей методологии и применения. Таким образом, изучение ДНК из окружающей среды вкупе с использованием традиционных обсервационных методов является интенсивно развивающимся и перспективным способом проведения экологических исследований.
Список литературы
Paknia O., Rajaei Sh H., Koch A. // Org. Divers Evol. 2015. V. 15. № 3. P. 619. https://doi.org/10.1007/s13127-015-0202-1
Beng K.C., Corlett R.T. // Biodivers. Conserv. 2020. V. 29. № 7. https://doi.org/10.1007/s10531-020-01980-0
Pimm S.L., Alibhai S., Bergl R. et al. // Trends Ecol. Evol. 2015. V. 30. № 11. https://doi.org/10.1016/j.tree.2015.08.008
Thomsen P.F., Willerslev E. // Biol. Conserv. 2015. V. 183. https://doi.org/10.1016/j.biocon.2014.11.019
Giovannoni S.J., Britschgi T.B., Moyer C.L. et al. // Nature. 1990. V. 345. № 6270. https://doi.org/10.1038/345060a0
Picard C., Ponsonnet C., Paget E. et al. // Appl. Environ. Microbiol. 1992. V. 58. № 9. https://doi.org/10.1128/aem.58.9.2717-2722.1992
Sebat J.L., Colwell F.S., Crawford R.L. // Appl. Environ. Microbiol. 2003. V. 69. № 8. https://doi.org/10.1128/AEM.69.8.4927-4934.2003
Rappé M.S., Giovannoni S.J. // Annu. Rev. Microbiol. 2003. V. 57. № 1. https://doi.org/10.1146/annurev.micro.57.030502.090759
Holzmann M., Habura A., Giles H. et al. // J. Eukaryot. Microbiol. 2003. V. 50. № 2. P. 135. https://doi.org/10.1111/j.1550-7408.2003.tb00248.x
Bass D., Cavalier-Smith T. // Int. J. Syst. Evol. Microbiol. 2004. V. 54. № 6. https://doi.org/10.1099/ijs.0.63229-0
Berney C., Fahrni J., Pawlowski J. // BMC Biol. 2004. V. 2. № 1. https://doi.org/10.1186/1741-7007-2-13
Mincer T.J., Fenical W., Jensen P.R. // Appl. Environ. Microbiol. 2005. V. 71. № 11. https://doi.org/10.1128/AEM.71.11.7019-7028.2005
Ficetola G.F., Miaud C., Pompanon F. et al. // Biol. Lett. 2008. V. 4. № 4. https://doi.org/10.1098/rsbl.2008.0118
Pawlowski J., Apothéloz-Perret-Gentil L., Altermatt F. // Mol. Ecol. 2020. V. 29. № 22. https://doi.org/10.1111/mec.15643
Thomsen P.F., Kielgast J., Iversen L.L. et al. // PLoS One. 2012. V. 7. № 8. https://doi.org/10.1371/journal.pone.0041732
Takahara T., Minamoto T., Yamanaka H. et al. // PLoS One. 2012. V. 7. № 4. https://doi.org/10.1371/journal.pone.0035868
Pont D., Rocle M., Valentini A. et al. // Sci. Rep. 2018. V. 8. № 1. P. 10361. https://doi.org/10.1038/s41598-018-28424-8
Seeber P.A., McEwen G.K., Löber U. et al. // Mol. Ecol. Resour. 2019. V. 19. № 6. https://doi.org/10.1111/1755-0998.13069
Williams K.E., Huyvaert K.P., Vercauteren K.C. et al. // Ecol. Evol. 2018. V. 8. № 1. P. 688. https://doi.org/10.1002/ece3.3698
Rodgers T.W., Mock K.E. // Conserv. Genet. Resour. 2015. V. 7. № 3. https://doi.org/10.1007/s12686-015-0478-7
Cilleros K., Valentini A., Allard L. et al. // Mol. Ecol. Resour. 2019. V. 19. № 1. https://doi.org/10.1111/1755-0998.12900
Boussarie G., Bakker J., Wangensteen O.S. et al. // Sci. Adv. 2018. V. 4. № 5. https://doi.org/10.1126/sciadv.aap9661
Ahn H., Kume M., Terashima Y. et al. // PLoS One. 2020. V. 15. № 10. https://doi.org/10.1371/journal.pone.0231127
de Menezes G.C.A., Câmara P.E.A.S., Pinto O.H.B. et al. // Extremophiles. 2021. V. 25. № 2. https://doi.org/10.1007/s00792-021-01221-4
Clark D.E., Pilditch C.A., Pearman J.K. et al. // Environ. Pollut. 2020. V. 267. https://doi.org/10.1016/j.envpol.2020.115472
DiBattista J.D., Reimer J.D., Stat M. et al. // Sci. Rep. 2020. V. 10. № 1. https://doi.org/10.1038/s41598-020-64858-9
Kavehei A., Hose G.C., Chariton A.A. et al. // J. Hazard. Mater. 2021. V. 416. https://doi.org/10.1016/j.jhazmat.2021.125794
Veilleux H.D., Misutka M.D., Glover C.N. // Sci. Total Environ. 2021. V. 782. https://doi.org/10.1016/j.scitotenv.2021.146891
Lopes C.M., Baêta D., Valentini A. et al. // Mol. Ecol. 2021. V. 30. P. 3289. https://doi.org/10.1111/mec.15594
Bonfil R., Palacios-Barreto P., Vargas O.U.M. et al. // Mar. Biol. 2021. V. 168. № 5. https://doi.org/10.1007/s00227-021-03862-7
Plough L.V., Bunch A.J., Lee B.B. et al. // Environ. DNA. 2021. V. 3. № 4. P. 800. https://doi.org/10.1002/edn3.186
Schmidt B.C., Spear S.F., Tomi A. et al. // Freshw. Sci. 2021. V. 40. № 2. https://doi.org/10.1086/714411
Howell L., LaRue M., Flanagan S.P. // New Zeal. J. Zool. 2021. V. 48. № 3–4. P. 245. https://doi.org/10.1080/03014223.2021.1900299
Bálint M., Pfenninger M., Grossart H.-P. et al. // Trends Ecol. Evol. 2018. V. 33. № 12. https://doi.org/10.1016/j.tree.2018.09.003
Zilius M., Samuiloviene A., Stanislauskienė R. et al. // Microbiol. Ecol. 2021. V. 81. № 1. https://doi.org/10.1007/s00248-020-01562-1
Carraro L., Mächler E., Wüthrich R. et al. // Nat. Commun. 2020. V. 11. № 1. https://doi.org/10.1038/s41467-020-17337-8
Di Capua I., Piredda R., Mazzocchi M.G. et al. // ICES J. Mar. Sci. Published online April 16, 2021. https://doi.org/10.1093/icesjms/fsab059
Mariani S., Fernandez C., Baillie C. et al. // Conserv. Sci. Pract. 2021. V. 3. № 6. https://doi.org/10.1111/csp2.407
Troth C.R., Burian A., Mauvisseau Q. et al. // Sci. Total Environ. 2020. V. 748. https://doi.org/10.1016/j.scitotenv.2020.141394
Rowney F.M., Brennan G.L., Skjøth C.A. et al. // Curr. Biol. 2021. V. 31. № 9. https://doi.org/10.1016/j.cub.2021.02.019
Clyde D. // Nat. Rev. Genet. 2021. V. 22. № 5. https://doi.org/10.1038/s41576-021-00353-9
Lopes C.M., De Barba M., Boyer F. et al. // Mol. Ecol. 2020. V. 29. № 16. https://doi.org/10.1111/mec.15549
D’Alessandro S., Mariani S. // Fish Fish. 2021. V. 22. № 4. https://doi.org/10.1111/faf.12553
Schmack J.M., Lear G., Astudillo-Garcia C. et al. // J. Appl. Ecol. 2021. V. 58. № 6. https://doi.org/10.1111/1365-2664.13856
Rees H.C., Maddison B.C., Middleditch D.J. et al. // J. Appl. Ecol. 2014. V. 51. № 5. https://doi.org/10.1111/1365-2664.12306
Parsons K.M., Everett M., Dahlheim M. et al. // R. Soc. Open. Sci. 2018. V. 5. № 8. https://doi.org/10.1098/rsos.180537
Buxton A.S., Groombridge J.J., Griffiths R.A. // PLoS One. 2018. V. 13. № 1. https://doi.org/10.1371/journal.pone.0191737
Johnson M.D., Cox R.D., Barnes M.A. // Environ. DNA. 2019. V. 1. № 2. https://doi.org/10.1002/edn3.19
Clare E.L., Economou C.K., Faulkes C.G. et al. // PeerJ. 2021. V. 9. https://doi.org/10.7717/peerj.11030
Banchi E., Ametrano C.G., Tordoni E. et al. // Sci. Total Environ. 2020. V. 738. https://doi.org/10.1016/j.scitotenv.2020.140249
Nørgaard L., Olesen C.R., Trøjelsgaard K. et al. // Sci. Rep. 2021. V. 11. № 1. https://doi.org/10.1038/s41598-021-85488-9
Hopken M.W., Orning E.K., Young J.K. et al. // BMC Res. Notes. 2016. V. 9. № 1. https://doi.org/10.1186/s13104-015-1797-1
Ribani A., Utzeri V.J., Taurisano V. et al. // Vet. Sci. 2020. V. 7. № 3. https://doi.org/10.3390/vetsci7030113
Heyde M., Bunce M., Wardell-Johnson G. et al. // Mol. Ecol. Resour. 2020. V. 20. № 3. https://doi.org/10.1111/1755-0998.13148
Nguyen T.V., Tilker A., Nguyen A. et al. // Environ. DNA. 2021. V. 3. № 4. P. 780–791. https://doi.org/10.1002/edn3.182
Sigsgaard E.E., Olsen K., Hansen M.D.D. et al. // Mol. Ecol. 2021. V. 30. P. 3374. https://doi.org/10.1111/mec.15734
Sales N.G., Kaizer M. da C., Coscia I. et al. // Mamm. Rev. 2020. V. 50. № 3. https://doi.org/10.1111/mam.12183
Lyet A., Pellissier L., Valentini A. et al. // Sci. Rep. 2021. V. 11. № 1. https://doi.org/10.1038/s41598-021-90598-5
Takasaki K., Aihara H., Imanaka T. et al. // PLoS One. 2021. V. 16. № 5. https://doi.org/10.1371/journal.pone.0250162
Majaneva M., Diserud O.H., Eagle S.H.C. et al. // Sci. Rep. 2018. V. 8. № 1. https://doi.org/10.1038/s41598-018-23052-8
Koziol A., Stat M., Simpson T. et al. // Mol. Ecol. Resour. 2019. V. 19. № 2. https://doi.org/10.1111/1755-0998.12971
Brandt M.I., Pradillon F., Trouche B. et al. // Sci. Rep. 2021. V. 11. № 1. https://doi.org/10.1038/s41598-021-86396-8
Castro L.R., Meyer R.S., Shapiro B. et al. // Hydrobiologia. 2021. V. 848. № 15. P. 3407. https://doi.org/10.1007/s10750-021-04576-z
Stoeck T., Frühe L., Forster D. et al. // Mar. Pollut. Bull. 2018. V. 127. https://doi.org/10.1016/j.marpolbul.2017.11.065
Shahraki A.H., Chaganti S.R., Heath D. // J. Water Health. 2019. V. 17. № 1. https://doi.org/10.2166/wh.2018.108
Tsuji S., Takahara T., Doi H. et al. // Environ. DNA. 2019. V. 1. № 2. https://doi.org/10.1002/edn3.21
Klunder L., Duineveld G.C.A., Lavaleye M.S.S. et al. // J. Sea Res. 2019. V. 152. https://doi.org/10.1016/j.seares.2019.101764
Hinlo R., Gleeson D., Lintermans M. et al. // PLoS One. 2017. V. 12. № 6. https://doi.org/10.1371/journal.pone.0179251
Djurhuus A., Port J., Closek C.J. et al. // Front. Mar. Sci. 2017. V. 4. https://doi.org/10.3389/fmars.2017.00314
Williams M.R., Stedtfeld R.D., Engle C. et al. // PLoS One. 2017. V. 12. № 10. https://doi.org/10.1371/journal.pone.0186462
Alzaylaee H., Collins R.A., Shechonge A. et al. // Parasit. Vectors. 2020. V. 13. № 1. https://doi.org/10.1186/s13071-020-3941-6
Minamoto T., Hayami K., Sakata M.K. et al. // Ecol. Res. 2019. V. 34. № 1. https://doi.org/10.1111/1440-1703.1018
Wang X., Lu G., Zhao L. et al. // PLoS One. 2020. V. 15. № 12. https://doi.org/10.1371/journal.pone.0244495
Doi H., Minamoto T., Takahara T. et al. // Ecol. Res. 2021. V. 36. № 3. P. 379. https://doi.org/10.1111/1440-1703.12217
Schweiss K.E., Lehman R.N., Drymon J.M. et al. // E-nviron. DNA. 2020. V. 2. № 1. https://doi.org/10.1002/edn3.39
Mauvisseau Q., Davy-Bowker J., Bulling M. et al. // Sci. Rep. 2019. V. 9. № 1. https://doi.org/10.1038/s41598-019-50571-9
Stoeckle M.Y., Das Mishu M., Charlop-Powers Z. // PLoS One. 2018. V. 13. № 12. https://doi.org/10.1371/journal.pone.0198717
Fernandez S., Sandin M.M., Beaulieu P.G. et al. // PeerJ. 2018. V. 6. https://doi.org/10.7717/peerj.4486
Tsuji S., Iguchi Y., Shibata N. et al. // Sci. Rep. 2018. V. 8. № 1. https://doi.org/10.1038/s41598-018-27434-w
Dejean T., Valentini A., Duparc A. et al. // PLoS One. 2011. V. 6. № 8. https://doi.org/10.1371/journal.pone.0023398
Klymus K.E., Marshall N.T., Stepien C.A. // PLoS One. 2017. V. 12. № 5. https://doi.org/10.1371/journal.pone.0177643
Milan D.T., Mendes I.S., Damasceno J.S. et al. // Sci. Rep. 2020. V. 10. № 1. https://doi.org/10.1038/s41598-020-74902-3
Miya M., Sato Y., Fukunaga T. et al. // R. Soc. Open. Sci. 2015. V. 2. № 7. https://doi.org/10.1098/rsos.150088
Collins R.A., Bakker J., Wangensteen O.S. et al. // Methods Ecol. Evol. 2019. V. 10. № 11. https://doi.org/10.1111/2041-210X.13276
Wilcox T.M., McKelvey K.S., Young M.K. et al. // PLoS One. 2013. V. 8. № 3. https://doi.org/10.1371/journal.pone.0059520
Epp L.S., Boessenkool S., Bellemain E.P. et al. // Mol Ecol. 2012. V. 21. № 8. P. 1821. https://doi.org/10.1111/j.1365-294X.2012.05537.x
Cristescu M.E. // Trends Ecol. Evol. 2014. V. 29. № 10. https://doi.org/10.1016/j.tree.2014.08.001
Drummond A.J., Newcomb R.D., Buckley T.R. et al. // Gigascience. 2015. V. 4. № 1. https://doi.org/10.1186/s13742-015-0086-1
Coghlan S.A., Shafer A.B.A., Freeland J.R. // Environ. DNA. 2021. V. 3. № 2. https://doi.org/10.1002/edn3.120
Deiner K., Renshaw M.A., Li Y. et al. // Methods Ecol. Evol. 2017. V. 8. № 12. https://doi.org/10.1111/2041-210X.12836
Juhel J., Marques V., Polanco Fernández A. et al. // Ecol. Evol. 2021. V. 11. № 7. https://doi.org/10.1002/ece3.7057
Ruppert K.M., Kline R.J., Rahman M.S. // Glob. Ecol. Conserv. 2019. V. 17. https://doi.org/10.1016/j.gecco.2019.e00547
Davidov K., Iankelevich-Kounio E., Yakovenko I. et al. // Sci. Rep. 2020. V. 10. № 1. https://doi.org/10.1038/s41598-020-74180-z
Ames C.L., Ohdera A.H., Colston S.M. et al. // Front. Mar. Sci. 2021. V. 8. https://doi.org/10.3389/fmars.2021.640527
Edgar R.C., Haas B.J., Clemente J.C. et al. // Bioinformatics. 2011. V. 27. № 16. https://doi.org/10.1093/bioinformatics/btr381
Callahan B.J., McMurdie P.J., Rosen M.J. et al. // Nat. Methods. 2016. V. 13. № 7. https://doi.org/10.1038/nmeth.3869
Dufresne Y., Lejzerowicz F., Perret-Gentil L.A. et al. // BMC Bioinformatics. 2019. V. 20. № 1. https://doi.org/10.1186/s12859-019-2663-2
Mousavi-Derazmahalleh M., Stott A., Lines R. et al. // Mol. Ecol. Resour. 2021. V. 21. № 5. https://doi.org/10.1111/1755-0998.13356
Fediajevaite J., Priestley V., Arnold R. et al. // Ecol. Evol. 2021. V. 11. № 9. https://doi.org/10.1002/ece3.7382
Дополнительные материалы отсутствуют.
Инструменты
Вестник Военного инновационного технополиса «ЭРА»