

Радиационная биология. Радиоэкология, 2023, T. 63, № 3, стр. 270-284
Ионизирующие излучения и воспалительная реакция. Механизмы формирования и возможные последствия
Д. Б. Пономарев 1, А. В. Степанов 1, А. Б. Селезнёв 1, *, Е. В. Ивченко 2
1 Государственный научно-исследовательский испытательный институт военной медицины
Санкт-Петербург, Россия
2 Военно-медицинская академия им. С.М. Кирова
Санкт-Петербург, Россия
* E-mail: alexseleznov@list.ru
Поступила в редакцию 29.08.2022
После доработки 29.03.2023
Принята к публикации 05.04.2023
- EDN: YAVIJT
- DOI: 10.31857/S0869803123030128
Аннотация
Ионизирующие излучения вызывают комплекс генетических, биохимических, структурных и функциональных изменений в организме. Одним из вариантов проявления системного ответа организма на их воздействие считают развитие постлучевой воспалительной реакции, которая посредством активации иммунитета выполняет как защитную функцию, так и принимает участие в формировании нежелательных ранних, отсроченных и нецелевых эффектов. Молекулярно-клеточные механизмы, составляющие ее основу, обусловлены: повреждением ДНК, изменениями метаболизма свободных радикалов (в первую очередь активных форм кислорода и азота), развитием оксидативного стресса, активацией инфламмасом, высвобождением “сигналов опасности” и секрецией провоспалительных цитокинов. В работе представлены сведения о роли неапоптотических типов гибели клеток (ферроптоза и пироптоза) в генезе постлучевой воспалительной реакции и последующем повреждении тканей, органов, систем. Отмечена способность постлучевой воспалительной реакции, за счет наличия положительной обратной связи между разными звеньями ее патогенеза, приобретать характер устойчивого во времени самоподдерживающегося процесса, увеличивающего степень тяжести радиационно-индуцированных повреждений.
Общеизвестно, что воздействие ионизирующих излучений (ИИ) способно вызывать гибель как отдельных клеток, так и целого организма. Результаты многолетних исследований свидетельствуют, что такой исход наступает вследствие первичных радиационно-химических реакций, обусловленных непосредственным взаимодействием излучений с молекулами-мишенями (в первую очередь, с водой, нуклеиновыми кислотами, липидами, полисахаридами, белками). При ионизации и возбуждении атомов (за счет энергии ИИ) происходит изменение структуры молекул, их свойств и функций, с последующим разобщением механизмов поддержания жизнедеятельности клетки (в том числе, постоянства внутриклеточной среды), нарушением процессов деления, вследствие повреждения ДНК, и ее гибелью. Поскольку “явления молекулярного уровня составляют фундамент, биохимическую основу процессов и проявлений жизнедеятельности” [1], именно радиационно-химические реакции в облученном организме ответственны за последующий каскад ответных реакций на молекулярно-генетическом, клеточном, тканевом и системном уровнях, одним из проявлений которых считают постлучевую воспалительную реакцию (рис. 1) [2–6].
БИОРАДИКАЛЫ И ОКСИДАТИВНЫЙ СТРЕСС
Продолжительность стадии первичных радиационно-химических реакций в организме составляет 10–16–10–3 с, и на этом этапе в результате радиолиза воды в условиях аэробной клеточной среды и значениях pH, соответствующих физиологической норме, происходит образование высоко реагентно-способных кислородсодержащих нестабильных продуктов: ${\text{O}}_{2}^{{\centerdot - }}$, ·OH, Н2О2 и др. [1, 7–10].
Возникшие в результате первичных преобразований радикалы способны взаимодействовать со сложными органическими молекулами (в том числе белками и нуклеиновыми кислотами) с образованием перекисных и гидроперекисных производных, что в условиях наличия кислорода может запускать дальнейший каскад реакций, обусловливающий усиление первичного повреждающего действия ИИ [7, 11].
Хотя ИИ вызывают прямое повреждение ДНК с образованием одно- и (или) двунитевых разрывов, по некоторым данным около 70% повреждений ДНК инициировано действием свободных радикалов как продуктов радиолиза воды [12].
В организме активные формы кислорода (АФК/reactive oxygen species – ROS) служат неотъемлемым элементом внутриклеточных химических реакций, а их уровень динамически контролируется про- и антиоксидантными системами [13, 14]. При радиационном воздействии возникает дисбаланс выработки и инактивации свободных радикалов. Умеренное повышение уровня активных форм кислорода может способствовать канцерогенезу, тогда как высокое содержание АФК станет триггером, запускающим различные типы регулируемой клеточной гибели, в частности, апоптоз, аутофагию и ферроптоз [15].
Вступая в реакции с высокомолекулярными веществами, АФК изменяют их структуру и функции, что чрезвычайно значимо в отношении нуклеиновых кислот (ядерной и митохондриальной ДНК), белков, липидов, повреждение которых может привести к необратимым нарушениям процессов транскрипции и трансляции, поддержания гомеостаза и межклеточных взаимодействий, клеточного роста, деления и гибели [9, 14, 16–18]. Вызванное АФК перекисное окисление липидов приводит не только к нарушению сборки, структуры и функционирования клеточных мембран, но и генерации новых свободных радикалов, способных повреждать ДНК и белки [13, 14, 18]. Следовательно, в результате вызванного воздействием ИИ повышения уровня нестабильных продуктов радиолиза и оксидативного стресса будет происходить дальнейшее увеличение числа клеток с повреждениями ДНК, что способствует формированию устойчивого воспалительного процесса [3, 6, 19, 20]. При воспалении потенцирование повреждения клеток будет связано не только с избытком активных радикалов, но и липопероксидов, металлопротеиназ, простагландинов и лейкотриенов, ферментов лизосом (протеаз, липаз, фосфолипаз, эластаз, коллагеназ и др.), источником которых будут являться клетки поврежденной ткани, находящиеся в ней лейкоциты [21, 22].
Вместе с тем роль АФК, образующихся непосредственно в результате радиолиза воды при облучении, вероятно, второстепенна в отношении дальнейшего усиления радиационно-индуцируемых поражений, поскольку основными источниками АФК в клетках являются митохондриальная дыхательная цепь и ферменты, метаболизирующие кислород (НАДФ-оксидазы, миелопероксидазы, циклооксигеназы и липоксиеназы) [13, 16, 22, 23]. Стоит также отметить, что чрезмерное образование ОН•-радикалов и других АФК в клетках может быть потенцировано ионами металлов переменной валентности (Men+) (а именно, Fe2+, Cu2+, Co2+, Ni2+, Zn2+ , Ti3+, V2+), способствуя формированию очередного каскада свободнорадикальных реакций [13, 14]. Таким образом, наблюдаемые эффекты, вероятно, связаны в первую очередь с развитием метаболических сдвигов и провоспалительных процессов, а не с радиолизом воды [9].
Развитие клеточного оксидативного стресса связано также и с образованием пероксинитрит аниона (ONOO–), который, наряду с АФК, способен повреждать ключевые молекулы-мишени (ДНК, белки, липиды) [8, 9, 13]. Возрастание уровня активных форм кислорода и азота (АФА/reactive nitrogen species – RNOS) на фоне истощения пула “гасителей” свободнорадикальных процессов является важной предпосылкой к развитию воспалительной реакции, поскольку активация процессов перекисного окисления липидов приводит к повреждению плазматических мембран клетки и ее органоидов, нарушению ее барьерной функции и ионного баланса [9]. Более того, установлено, что за счет механизмов межклеточной коммуникации проявления окислительного стресса возникают не только в самих поврежденных клетках, но также в соседних и удаленных (нецелевых) клетках, и даже у их потомков [8, 23]. На основании обобщения экспериментальных данных было сформировано предположение, что существует как минимум два пути передачи стресс-сигнала: через поры щелевых контактов между соседними клетками (коннексоны) или за счет выделения медиаторов в межклеточное пространство [6].
Важную роль в усилении и пролонгировании окислительного стресса, вызванного воздействием ИИ, отводят дисрегуляции функциональной активности митохондрий. Значимость митохондриального окислительно-восстановительного дисбаланса отмечена на всех этапах реализации ответной реакции клетки на облучение: в его инициировании, краткосрочных и долгосрочных перестройках метаболизма, усилении (потенцировании) “первичного” образования свободных радикалов, цитохром-С опосредованных механизмах программированной клеточной гибели [8, 9, 24]. В частности, с продукцией АФК/АФА (ROS/RNOS), обусловленной воспалительной реакцией, а также с возможной “утечкой” из поврежденных митохондрий НАД-оксидаз или индуцибильной синтетазы оксида азота (iNOS) связывают возникновение в течение длительного периода времени повреждений ДНК в клетках легких, обеспечивающее увеличение выраженности первоначальных нарушений [25]. Кроме того, в зависимости от величины поглощенной дозы истощение антирадикальных систем может приобретать затяжной характер. Продолжительное дозозависимое снижение антиоксидантной способности цельной крови в период с двух по 24-е сутки после воздействия выявлено у мышей линии C57BL/6 J, облученных в дозах 0.5–3 Гр, тогда как повышение уровня перекисного окисления липидов отмечали с двух по 6-е сутки наблюдения [26]. В ходе последующих исследований было установлено снижение антиоксидантной способности цельной крови у мышей в течение не менее чем 100 сут после облучения в дозе 3 Гр, а при увеличении дозовой нагрузки до 5 Гр указанные изменения носили хронический характер и были зарегистрированы даже спустя 800 дней после облучения [27].
На основании данных о роли и месте реакций свободнорадикального окисления в генезе постлучевых патологических процессов проводят поиск лекарственных средств, способных оказывать корригирующее действие на метаболический гомеостаз и поддержание баланса про- и антиоксидантных систем. Например, снижение выраженности негативных эффектов постлучевого хронического воспаления и окислительного повреждения тканей достигают терапевтическим применением ROS(RNOS)-скавенджеров [25]. Концепция применения антиоксидантов в качестве средств противорадиационной защиты активно разрабатывается и находит многочисленные экспериментальные подтверждения правильности выбранного направления исследований [28–30].
РАДИАЦИОННО-ИНДУЦИРОВАННАЯ ГИБЕЛЬ КЛЕТОК. МЕХАНИЗМЫ ВОЗНИКНОВЕНИЯ И ЭФФЕКТЫ
Свободные радикалы способны повреждать геном, что может привести к гибели клетки. Вместе с тем выявлен ДНК‑независимый путь гибели клеток, обусловленный индукцией перекисного окисления липидов (в частности, в результате взаимодействия АФК с полиненасыщенными жирными кислотами мембран), получивший название ферроптоз [15, 18, 31–33]. Ферроптоз – неапоптотическая, железозависимая форма регулируемой клеточной гибели, возникающая в результате чрезмерного накопления АФК и активации перекисного окисления липидов [16, 18, 31–33]. К настоящему времени установлена возможность гибели клеток путем ферроптоза при нейродегенеративных заболеваниях (в том числе травмах головного мозга), ишемии/реперфузии почек, печени, сердца, а также гибели опухолевых клеток при облучении [12, 34–36]. Сравнительный анализ реакции различных клеточных линий опухолевых клеток на облучение в широком диапазоне доз (от 2 до 50 Гр) показал их сходство и зависимость от интенсивности воздействия, при этом отмечено дозозависимое увеличение продукции АФК, усиления перекисного окисления липидов и повреждения ДНК [12]. Установлено, что во всех исследованных клеточных линиях (CT26, MCA205, 71-7) имело место усиление перекисного окисления липидов через 24 ч после облучения в дозах 20 и 50 Гр, что приводило к гибели клеток через железозависимые механизмы [12]. Результаты исследований, выполненных на клетках HT‑1080, свидетельствуют, что при облучении в дозах 1, 2 или 4 Гр перекисное окисление липидов и ферроптоз были причинами радиационно-индуцированной гибели клеток данной линии [36]. Роль ферроптоза в генезе постлучевого нарушения кроветворения и гибели организма была продемонстрирована в экспериментах на мышах [37–39]. При облучении мышей в дозах 4, 8 и 10 Гр отмечено повышение уровня перекисного окисления липидов и частоты гибели клеток крови (предшественников гранулацитарно-макрофагальных кроветворных клеток) по механизму ферроптоза [37–39]. Направленное воздействие на механизмы развития ферроптоза, путем введения животным после воздействия ИИ ферростатина‑1 (ferrostatin-1) или LDN193189, способствовало снижению выраженности наблюдаемых биохимических и гематологических изменений и значимому увеличению выживаемости облученных мышей [37, 38]. Радиозащитный эффект выявлен и в случае применения ингибитора липоксигеназы-15, препарата бейкалеин (baicalein/lipoxygenase-15 inhibitor). Однократное введение указанного препарата спустя 24 ч после облучения в дозе 9.25 Гр способствовало увеличению выживаемости животных более чем в 2 раза, а при его использовании в составе схемы лечения из трех препаратов (совместно с ингибиторами некроза и апоптоза) – до 75% [39].
Особо следует отметить, что одной из особенностей ферроптоза является его провоспалительный эффект, обусловленный высвобождением “сигналов опасности” (демпферов), в частности, ядерного белка амфотерина (алармина 1) (HMGB1/high-mobility group box 1 protein/высокомобильный групповой белок 1) [34]. Поскольку HMGB1 через взаимодействие с рецепторами распознавания образов участвует в генезе и потенцировании воспалительной реакции, приведенные данные свидетельствуют о непосредственной взаимосвязи оксидативного стресса с формированием воспалительной реакции после воздействия ИИ.
Несмотря на появление новых данных о возможных механизмах гибели клеток после облучения, традиционно считают данное событие следствием возникновения однонитевых и(или) двунитевых разрывов ДНК, приводящих к апоптозу или формированию некротических изменений [40–42]. Индукция апоптотической гибели клеток может быть обусловлена активацией как внутренних, так и внешних сигнальных путей [43, 44]. Ионизирующие излучения запускают внутренний апоптотический сигнальный путь, в частности, через белки семейства Bcl-2 [45], тогда как внешний инициируется трансмембранными рецептор-опосредованными взаимодействиями с задействованием “рецепторов смерти” суперсемейства фактора некроза опухоли (ФНО) [43, 44, 46]. Гибель клеток через апоптоз сопровождается характерными биохимическими процессами и структурными преобразованиями (округлением клетки, уменьшением ее объема, изменениями и расщеплением ДНК, компактизацией ядерного и цитоплазматического содержимого клетки) [43, 44, 47–49]. Признаки апоптотических изменений клеток выявлены уже в самые ранние сроки после облучения. По данным S.A. Lorimore и соавт. (2001) в селезенке мышей, подвергнутых воздействию ИИ в дозе 4 Гр, апоптотические изменения в клетках выявляют уже спустя 2 ч после облучения, а спустя 6 ч их число достигает наибольших значений [50]. В последующем (через 9 ч после облучения) было отмечено снижение числа гибнущих клеток, а уже спустя 24 ч после воздействия ИИ их число соответствовало исходным значениям, при этом снижение клеточности ткани составило в течение первых 24 ч для линий мышей C57BL/6 и CBA/Ca – 82 и 72% соответственно [50].
При апоптозе сохраняется целостность клеточной мембраны и не происходит выделение из гибнущей клетки в окружающее межклеточное пространство эндогенных провоспалительных факторов, следовательно, не происходит индуцирования воспалительной реакции [43, 49, 51]. Вместе с тем одновременное появление большого количества апоптотических клеток в результате воздействия ИИ может привести к “неспособности” макрофагов их элиминировать за короткий промежуток времени, что, в свою очередь, приведет к запуску процесса, описываемого как “вторичный некроз”, при котором формируется проницаемость мембран клеток для макромолекул [7, 50]. Некротическая же смерть клеток сопровождается увеличением их объема, набуханием органелл и разрывом плазматической мембраны, что приводит к высвобождению внутриклеточного содержимого [33, 48, 52]. Среди принципиальных различий между апоптозом и некрозом следует отметить тот факт, что именно некротическая гибель клеток вызывает развитие выраженной воспалительной реакции [7, 47, 49].
Относительно недавно была идентифицирована провоспалительная форма регулируемой клеточной гибели, которая зависит от ферментативной активности воспалительных протеаз – пироптоз. Установлено два варианта индукции пироптоза: канонический – обусловленный активацией инфламасом и каспазы-1 и, соответственно, неканонический – опосредованный каспазами-4/5/11 [49, 53–56]. Пироптоз является регулируемой формой некроза и отличается от других форм гибели клеток как по механизму возникновения, так и морфологически, поскольку активация каспазы-1 приводит к образованию в плазматической мембране за счет белка Гасдермина Д (Gasdermin D /GSDMD/) пор диаметром 1.1–2.4 Нм, изменению градиента ионов внутри и снаружи клетки, набуханию клетки и ее осмотическому лизису [33, 53–57]. При воздействии ИИ гибель клеток, в частности, макрофагов, дендритных клеток, натуральных киллеров (NK‑клеток), Т- и В-клеток по механизму пироптоза была выявлена в исследованиях, выполненных V.M. Stoecklein и соавт. (2015) [58]. При облучении культуры первичных макрофагов костного мозга также была обнаружена гибель клеток путем пироптоза [59]. В ходе изучения механизмов радиационно-индуцированных повреждений ЦНС в экспериментах in vitro получены данные о развитии пироптоза в клетках микроглии (клеточная линия BV-2) при однократном облучении в дозе 10 Гр [60]. Указанные факты принципиально важны в контексте объяснения механизмов воспалительной реакции при воздействии ИИ, поскольку, в отличие от апоптоза (при котором, как было отмечено, сохраняется целостность мембраны клетки), пироптоз характеризуется “быстрым” разрывом плазматической мембраны и высвобождением провоспалительного внутриклеточного содержимого (в том числе, интерлейкина-1β /ИЛ-1β/ и интерлейкина-18 /ИЛ-18/, HMGB1, белков семейства S100 и др.), что индуцирует развитие воспалительной реакции [46, 53–57, 60].
Несмотря на то что флогогенные факторы могут принципиально различаться по своей природе (биологической, химической и физической), установлено, что механизмы реализации ответной реакции организма на них в значительной степени оказываются схожи. Эволюционный консерватизм и общность механизмов развития воспаления при воздействии самых разнообразных повреждающих агентов обусловлены их направленностью на клетку – как ключевую мишень, и ролью данной системной реакции в ограничении (локализации) повреждений, устранении их первопричин и последующем восстановлении организма [22].
РОЛЬ СИГНАЛЬНЫХ МОЛЕКУЛ В ГЕНЕЗЕ ПОСТЛУЧЕВОЙ ВОСПАЛИТЕЛЬНОЙ РЕАКЦИИ
Уже в 1952 г. результаты обследования лиц, подвергшихся воздействию ИИ, позволили Л. Гемпельману, Г. Лиско и Д. Гофману сделать заключение, что динамика гибели клеток и разрушения тканей после воздействия ИИ имеет сходство с аналогичными явлениями при бактериальных инфекциях, а выраженность системной реакции организма на облучение определяется физиологически активными веществами, выделяемыми как из погибших, так и поврежденных клеток (тканей) [61].
В условиях локального (местного) воздействия ИИ имеет место образование характерных зон поражения, тогда как при общем облучении возникают множественные “очаги” воспаления, затрагивающие все ткани, органы и системы, которые способствуют развитию полиорганной недостаточности и гибели организма. Хотя значимость воспалительной реакции в патогенезе радиационных поражений, нецелевых эффектов и других патологических состояний весьма высока, лишь научные достижения последних двух десятилетий позволили существенно приблизиться к пониманию причин их возникновения и механизмов формирования.
Повреждение, вызванное воздействием ИИ (впрочем, как и другими деструктивными агентами: возбудители инфекции, травма, ожог, обморожение), и последующая гибель клеток (через пирроптоз, ферроптоз и др. формы некротической гибели) приводит к высвобождению “сигналов опасности” [7, 21, 62–65].
Среди сигнальных молекул, свидетельствующих о воздействии на организм потенциально опасных факторов, важная роль принадлежит аларминам (alarmins) или факторам повреждения (damage-associated molecular pattern / DAMP), которые “информируют” соседние клетки о развитии процессов деструкции, активируют иммунную систему и формирование воспалительной реакции [63–70]. Алармины не несут информации о специфичности воздействующего фактора, лишь свидетельствуют о повреждении клеток, поскольку являются эндогенными молекулами. В настоящее время идентифицирован целый ряд молекул, которые соответствуют определенным критериям (роли в воспалении/иммунитете, восстановлении гомеостаза, способе секреции) и могут быть отнесены к аларминам. Среди таковых: ядерный белок HMGB1, белки теплового шока (Heatshockproteins/ HSPs), протеины семейства S100, интерлейкин-1α (ИЛ-1α), гепатомный фактор роста (HDGF), мочевая кислота (микрокристаллы урината натрия/MSU) и др. [21, 64, 69–72]. “Сигналы опасности” воспринимают рецепторы распознавания образов (pattern-recognition receptors/PRRs), локализованные на поверхности клетки (Toll‑подобные рецепторы (TLRs) и лектиновые рецепторы С-типа) и внутри нее (NOD‑ и RIG‑1‑подобные рецепторы /NLRs и RLRs соответственно) [69, 71, 73, 74]. При этом различные алармины вызывают активацию различных типов рецепторов, в частности, ядерный белок HMGB1 и HSPs связываются с рецепторами TLR2 и TLR4, ИЛ-1α через рецепторы ИЛ-1R, некоторые кальцийсвязывающие белки семейства S100 посредством взаимодействия с рецепторами конечных продуктов гликирования белков – RAGE (receptor for advanced glycation end products) [21, 69–71, 73, 75].
Спектр биологически активных факторов, синтезируемых в результате активации TLR, весьма широк и, наряду с провоспалительными цитокинами, включает хемокины, молекулы адгезии, факторы роста, тканедеструктирующие ферменты (в частности, металлопротеиназы), и ферменты, ответственные за генерацию таких медиаторов воспаления, как циклооксигеназа-2 (ЦОГ-2 /COX-2/) и индуцибильная синтетаза оксида азота (iNOS) [70].
Внеклеточные сигнальные молекулы, в частности HMGB1, связываясь с Toll-подобными рецепторами, инициируют формирование каскада реакций, опосредованных белками адаптерами MyD88 (первичный белок миелоидной дифференцировки 88) и TRIF (белок, содержащий TIR-домен, индуцирующий интерферон-β/TIR domain-containing adapter inducing IFN-B/), ядерным фактором‑kB (NF‑kB), транскрипционным фактором АР-1, интерферон регуляторным фактором (IRF) и митоген‑активированными протеинкиназами (mitogen-activated protein kinase/МАРК) [65, 69, 76].
Активация NF-kB отмечена при воздействии различных клеточных стрессоров, в том числе и при повреждении (разрывах) ДНК [77, 78], тем не менее, каноническим и наиболее изученным является механизм активации NF‑kB, индуцированный ФНОα и ИЛ-1 [79, 80]. В большинстве случаев NF‑kB непосредственно связывается с промотором и действует как активатор транскрипции для сотен генов-мишеней [80]. Известно, что NF‑kB, АР‑1 и IRF5 контролируют экспрессию воспалительных цитокинов, тогда как IRF3 и IRF7 определяют активацию интерферона (INF) I типа и IFN-индуцибильных генов [70]. Вместе с тем имеет место взаимная активация между NF‑kB и провоспалительными цитокинами, поскольку данный ядерный фактор регулирует экспрессию ФНО, интерлейкинов (ИЛ-1, ИЛ-2, ИЛ-6, ИЛ-8, ИЛ‑12) [73, 81]. Через регуляцию экспрессии генов, ответственных за апоптоз, пролиферацию клеток, синтез хемокинов, цитокинов, молекул адгезии, белки семейства NF‑kB обеспечивают формирование иммунного ответа и воспалительной реакции [79, 80].
Увеличение уровня провоспалительных цитокинов и экспрессии их генов при воздействии ИИ неоднократно констатировали по результатам исследований in vivo и in vitro [59, 60, 76, 81, 82]. Фактор некроза опухоли и ИЛ-1β обладают широким спектром биологической активности, вызывая аутокринные и паракринные эффекты, приводящие к активации макрофагов и нейтрофилов [68]. При элиминации макрофагами поврежденных клеток, как и в случае уничтожения патогенных организмов, они продуцируют большое количество АФК (свободных радикалов) и провоспалительных факторов. В результате возникает ситуация, когда повышение уровня провоспалительных цитокинов и хемокинов обеспечивает рекрутирование дополнительных иммунных клеток, которые высвобождают все больше свободных радикалов. [23, 68]. Значимое увеличение уровня провоспалительных цитокинов и, вероятно, в первую очередь ФНОα, приводит к гибели клеток вне зависимости от наличия в них повреждений ДНК, обусловленных воздействием ИИ.
При воздействии ИИ в реализации внутриклеточных механизмов передачи сигналов и активации компенсаторных процессов задействованы МАРК трех типов: ERK 1/2 (extracellular signal-regulated kinase 1/2), JNK/SAPK (c‑Jun N‑terminal kinase/stressactivated protein kinase – терминальные киназы клеточных сочленений/стресс-активируемые протеинкиназы) и р38 МАРК [83, 84]. Контроль выживания, роста и дифференцировки клеток обеспечивается через ERK 1/2, тогда как JNK/SAPK и р38 МАРК функционируют как проапоптотические киназы, регулирующие механизмы формирования дисфункции митохондрий и радиационно-индуцированной апоптотической гибели клеток [83, 84].
Митохондрии играют особую роль в формировании воспалительной реакции при повреждении, что обусловлено их участием в поддержании гомеостаза клетки и радиочувствительностью. Облучение даже с крайне низкой мощностью дозы (1 мГр/мин) приводит к изменению структуры и функции митохондрий, что сопровождается увеличением содержания АФК и развитием оксидативного стресса [85]. Следует также отметить, что степень повреждения мтДНК в результате воздействия ИИ существенно выше, чем у защищенной гистонами ДНК ядра клетки (яДНК) [86, 87].
Характеризуя роль митохондрий в генезе воспалительной реакции, следует отметить, что, во-первых, они являются единственными органеллами, которые действуют как DAMP сами по себе, индуцируя секрецию иммунными клетками провоспалительных цитокинов, а во-вторых – при их деструкции выделяется ряд веществ, иммуностимулирующее действие которых реализуется через связывание с PRRs или активацию инфламасом: митохондриальная ДНК (мтДНК), АТФ, митохондриальный фактор транскрипции А (T fam), малые высокореактивные ионы и молекулы (АФК/mROS), формилированные пептиды (formylpeptide) [64, 88–91].
Митохондриальные DAMP являются как внутриклеточными, так и экстраклеточными факторами инициации воспалительной реакции [91, 92]. Радиационно-индуцированная фрагментация мтДНК и выход ее составляющих в цитозоль клеток головного мозга и селезенки выявлены у мышей через 5 ч после облучения в дозе 5 Гр [93]. В результате выхода во внутриклеточную среду фрагменты мтДНК могут вызывать формирование воспалительной реакции как через взаимодействие с TLR9 с передачей сигнала ядерным фактором NF-kB, так и путем взаимодействия с инфламмасомой NLRP3 и активацией каспазы-1, что в обоих случаях приведет к увеличению уровня провоспалительных цитокинов [94–97]. Третий вариант иммуностимулирующего действия мтДНК реализуется через IRF-1 и индукцию синтеза IFNβ и INFλ1 [94–97].
В ходе экспериментальных исследований по оценке влияния рентгеновского излучения на содержание фрагментов мтДНК и яДНК установлено, что уже после облучения в дозе 1 Гр количество фрагментов мтДНК в сыворотке крови мышей-самцов линии BALB/с увеличивается, а соотношение мтДНК/яДНК выше контрольных значений на протяжении не менее 25 сут [86]. Высвобождение мтДНК в кровоток будет способствовать формированию и поддержанию воспалительного процесса на системном уровне. Обобщенные сведения о молекулярных механизмах индукции воспалительной реакции, опосредованных митохондриями, в норме и патологии представлены в ряде обзорных статей [94–97].
ИНФЛАММАСОМЫ И ИХ ЗНАЧЕНИЕ
Одним из путей формирования системных иммунных реакций и воспаления, вызванных повреждением клеток, в том числе и при воздействии ИИ, является активация аларминами внутриклеточных белковых комплексов – “инфламмасом” [74, 88, 98–100]. Инфламмасомы состоят из трех элементов: “сенсорного” (рецепторного) белка, которым являются рецепторы распознавания образов из семейства NOD‑подобных рецепторов (NLRs) или AIM2 (absent in melanoma 2 (AIM2)-like receptors), адапторного белка – апоптоз-ассоциированного спект-подобного белка (apoptosis-associated speck‑like protein /ASC/) и эффекторного белка – киназы‑1 [74, 88, 98–100]. К настоящему времени достигнуты существенные успехи в выяснении структурно-молекулярной организации инфламмасом и путях их активации [98, 99, 101–106]. Установлено, что “классическая” активация инфламмасом является двухкомпонентным процессом и регулируется как на транскрипционном, так и на посттранскрипционном этапах. Первая стадия активации реализуется через путь TLR/NF-kB или TLR4/MyD88. Вторая стадия – через образование инфламмасомы и ее взаимодействие с факторами повреждения [88–90, 99, 105, 106].
В ранних и отсроченных эффектах воздействия ИИ на организм ключевую роль играют инфламмасома NLRP3 и AIM2, индукция которых при облучении может быть обусловлена изменением уровня внутриклеточного пула К+/Са2+, избытком АФК и оксидативным стрессом, высвобождением аларминов из погибших и(или) поврежденных клеток.
Агонистами, индуцирующими образование инфламмасом NLRP3, являются молекулы АТФ, порообразующие токсины, патогены грибковой, бактериальной и(или) вирусной природы, нуклеиновые кислоты, гиалуроновая кислота и кристаллические вещества (диоксид кремния, асбест), кристаллы мочевой кислоты [60, 98, 99, 102]. Мочевая кислота (кристаллы моноурината натрия) является одним из “сигналов опасности”, который вызывает развитие воспалительной реакции через активацию NOD‑подобного рецепторного белка инфламмасомы NLRP3 (стимулируя выработку ИЛ-1β и ИЛ-18) и взаимодействие с Toll‑подобными рецепторами (TLR2 и TLR4) [107]. Повышение содержания мочевой кислоты при воздействии ИИ совпадает с развитием клеточно-опосредованных иммунных реакций, что позволяет считать ее одним из триггеров активации и устойчивого развития воспалительной реакции при облучении [58]. Так, при облучении мышей в дозе 4 Гр уже через 2 ч после воздействия уровень мочевой кислоты становится значимо выше фоновых значений и остается повышенным на протяжении не менее суток [58].
В отличие от инфламмасомы NLRP3, структуры AIM2 ответственны за распознавание двухцепочечных разрывов ДНК, которые появляются в результате воздействия ИИ на генетический аппарат клетки [99, 101, 102]. В результате активации инфламмасом через каспаза-1 зависимый механизм, происходит формирование (из неактивных прекурсоров про-ИЛ-1β и про-ИЛ‑18) и секреция функционально активных форм провоспалительных цитокинов ИЛ-1β и ИЛ-18, что в дальнейшем будет стимулировать высвобождение ФНОα, ИЛ-6 и ИЛ-1α [88, 89]. Другим патогенетически важным итогом активации этих инфламмасом является каспаза-1 зависимая клеточная гибель – пироптоз [58, 59, 108].
Результаты экспериментальных исследований на мышах, облученных в дозе 2 Гр, свидетельствовали об отсутствии активации инфламмасом в ранние сроки после воздействия ИИ (спустя 1–4 ч), тем не менее в период с первых по 7-е сутки она была выраженной, но в последующем (к 14-м суткам) снижалась до уровня фоновых значений. При этом динамика гибели иммунных клеток точно соответствовала кинетике активации инфламмасом [58]. Дозозависимые эффекты воздействия ИИ (при облучении в дозах 5, 10 и 20 Гр) на опосредованную NLRP3 активацию каспазы-1, гибель клеток и продукцию цитокинов: ИЛ-1β, ИЛ-18, ФНОα и IFN-γ подтверждены с использованием в эксперименте культуры первичных макрофагов костного мозга [59].
СТРУКТУРНЫЕ И ФУНКЦИОНАЛЬНЫЕ ИЗМЕНЕНИЯ
Анализ данных по изучению роли цитокинов и инфламмасом в формировании ответной реакции организма на воздействие ИИ свидетельствует об их участии в генезе постлучевых нарушений структуры и функций различных органов и систем.
Предположение о важной роли активации воспалительной реакции в патогенезе радиационно-индуцированного мукозита было сделано в ходе изучения механизмов его развития в модельных экспериментах на мышах [82]. Так, в образцах тканей языка мышей спустя 8 сут после облучения в дозе 15 Гр установлено увеличение более чем в 20 раз экспрессии генов ИЛ‑1β, ИЛ-6, ИЛ-11 и ИЛ-23 [82]. С увеличением дозовой нагрузки до 20 Гр вышеуказанные изменения становились еще более существенными и наряду с этим значимо увеличивался уровень экспрессии генов провоспалительного цитокина – ФНОα, а также инфламмасомы NLRP3 (почти в 60 раз) [82].
Значимость каскада активации инфламмасом как фактора, обусловливающего повреждение костной ткани и селезенки, продемонстрирована при облучении мышей в дозе 9 Гр, помимо этого отмечено, что изменение их весовых показателей коррелирует с уровнем ИЛ‑1β [109].
Получены данные, свидетельствующие о роли активации инфламмасом и воспалительных процессов в радиационно-индуцированном повреждении головного мозга. В частности, выявлено, что воздействие ИИ оказывало стимулирующее влияние на экспрессию генов инфламмасом, особенно NLRP3 в клетках микроглии BV-2, что сопровождалось повышением продукции ИЛ-1β, ИЛ-18 и активацией механизмов каспаза-1 регулируемой клеточной гибели [60].
Установлено наличие взаимосвязи между повышением уровня фенотипических маркеров воспалительной реакции с развитием постлучевого пневмонита и фиброза [110]. Значимость инфламмасомы AIM2 в радиационно-индуцированном поражении легких продемонстрирована в работе Gao J. и соавт. (2019). Выявлено, что облучение грудной клетки мышей в дозе 18 Гр вызывало отек легких, гиперемию и прогрессирующее утолщение стенок альвеол, тогда как ингибирование активации инфламмасомы AIM2 в условиях применения андрографолида (Andrographolide) обеспечивало увеличение выживаемости облученных животных и снижало выраженность острого пневмонита и легочного фиброза [108].
Как известно, прогрессирующие воспалительные реакции являются одной из причин заболеваний сердечно-сосудистой системы, в частности, атеросклеротических процессов [111]. Ионизирующие излучения через активацию инфламмасомы NLRP3 вызывают повреждение эндотелия, ускоряют атеросклеротические поражения и могут приводить к фиброзированию [90].
Обобщенные сведения о значении активации инфламмасомы NLRP3 в формировании лучевых поражений кожи, кишечника, слизистой оболочки полости рта и других систем представлены в обзоре J. Wei и соавт. (2019) [89].
Для организма последствия формирования радиационно-индуцированной воспалительной реакции многогранны и сопряжены не только с повреждением нормальных тканей, но и появлением нецелевых эффектов (Прим. – в литературе при описании феноменов, являющихся косвенными последствиями облучения и не связанными с непосредственным воздействием ИИ на ядерную ДНК клеток-мишеней, используют понятия “эффект свидетеля”) [3].
Наличие взаимосвязи между формированием в постлучевом периоде воспалительной реакции (включающей индукцию активных форм кислорода, провоспалительных цитокинов – ИЛ-1 и ФНОα) и появлением нецелевых эффектов подтверждается рядом исследований [3, 50]. Однако, как отмечено D. Schaue и W.H. McBride (2010), “зачастую может казаться правомочным проведение параллели между процессами воспаления при развитии биологического отклика на воздействие ИИ и радиационно-индуцированными “нецелевыми” (“сторонними”) эффектами”. Однако, по мнению самих авторов, прямых доказательств для такого рода экстраполяции на данный момент нет, по крайней мере, по причине отсутствия однозначности в характеристики этих явлений [23].
Таким образом, в связи с установлением роли АФК/АФА, “сигналов опасности” и инфламмасом в развитии ответа организма на воздействие ИИ стало очевидно, что биохимические и иммунологические процессы, сопровождающие постлучевую воспалительную реакцию, помимо обеспечения защитной функции, могут приводить к геномной нестабильности и способствовать повышению риска канцерогенеза, вызывать преждевременное старение и развитие полиорганной недостаточности, чему в последнее время придают особое значение [21, 25, 112]. рис. 1
Рис. 1.
Схема основных путей формирования радиационно-индуцированной воспалительной реакции.
Fig. 1. Pattern of radiation-induced inflammatory response formation pathways.
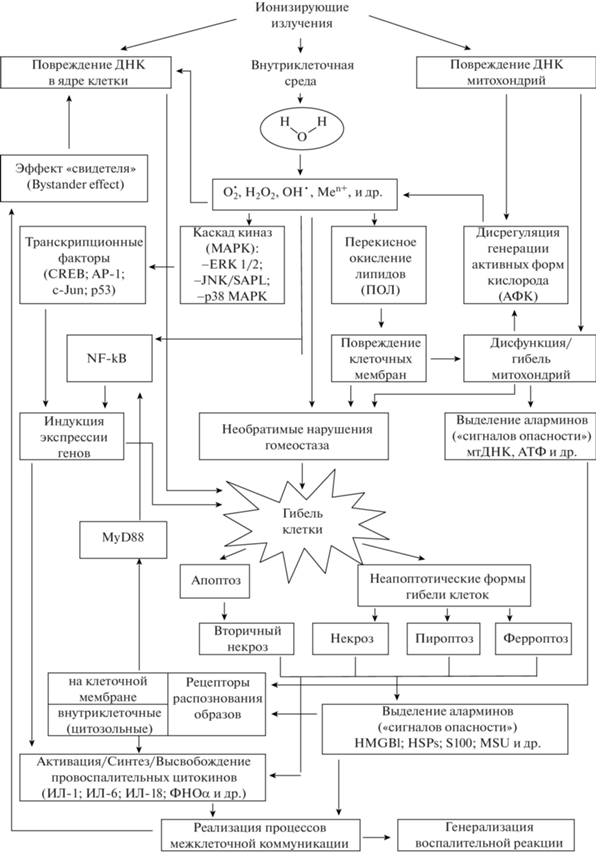
ЗАКЛЮЧЕНИЕ
Представленные материалы свидетельствуют, что воспалительная реакция при облучении является результатом реализации каскада взаимосвязанных радиационно-химических, биохимических и цитогенетических процессов, которые за счет механизмов межклеточной коммуникации приводят к формированию системной реакции. Как и в случае других, обусловленных воздействием ИИ явлениях, первичные (пусковые) факторы (повреждение ДНК и радиолиз воды с образованием АФК/АФА) вызывают нарушение функциональной целостности клеточных элементов (в первую очередь мембран органоидов и самой клетки) и способствуют “вторичному” повреждению генетического материала (ДНК, РНК). Повреждение ДНК и формирование микродефектов клеточных структур, связанные с развитием окислительного стресса, предопределяют гибель клеток путем апоптоза, или через некротические изменения (в том числе, пироптоз, ферроптоз). Ионизирующее излучение при уровнях воздействия более 1 Гр индуцирует гибель клеток и приводит к выделению “сигналов опасности”. Эндогенные “сигналы опасности”, распознаваемые Toll‑подобными рецепторами, через систему “посредников” (MAPs, NF-kB, COX-2) вызывают секрецию воспалительных цитокинов ИЛ-1, ИЛ-6, ИЛ-8, ФНО, ИЛ-33 и IFN-γ, которые за счет механизмов положительной обратной связи предопределяют дальнейшее образование АФК и гибель клеток. Наличие положительной обратной связи между повреждениями генома, нарушением баланса активности про- и антиоксидантных систем, гибелью клеток и выделением провоспалительных факторов (цитокинов, хемокинов, протеаз, простаноидов, а также активных форм кислорода и азота) придает воспалительной реакции характер устойчивого во времени самоподдерживающегося цикла, увеличивающего степень тяжести повреждений тканей, органов и систем. Указанные процессы, вероятно, определяют развитие нецелевых эффектов, радиационно-индуцированной нестабильности генома, канцерогенез и другие отсроченные эффекты [4, 5]. Раскрытие основных составляющих патогенеза радиационно-индуцированной воспалительной реакции и выяснение взаимодействия между различные звеньями патологического процесса позволят в дальнейшем расширить представления о путях и способах коррекции неблагоприятных эффектов воздействия ионизирующих излучений на организм.
Список литературы
Радиационная медицина. Руководство для врачей-исследователей, организаторов здравоохранения и специалистов по радиационной безопасности. Теоретические основы радиационной медицины. Т. 1. М.: Изд. АТ, 2004. 992 с. [Radiacionnaja medicina. Rukovodstvo dlja vrachej-issledovatelej, organizatorov zdravoohranenija i specialistov po radiacionnoj bezopasnosti. Teoreticheskie osnovy radiacionnoj mediciny. V. 1. M.: Izd. AT, 2004. 992 p. (In Russ.)]
Schaue D., Micewicz E.D., Ratikan J.A. et al. Radiation & Inflammation // Seminars in Radiation Oncology. 2015. V. 25. № 1. P. 4–10. https://doi.org/10.1016/j.semradonc.2014.07.007
Mukherjee D., Coates Ph.J., Lorimore S.A. et al. Responses to ionizing radiation mediated by inflammatory mechanisms // J. Pathol. 2014. V. 232. № 3. P. 289–99. https://doi.org/10.1002/path.4299
Multhoff G., Radons J. Radiation, Inflammation, and Immune Responses in Cancer // Front Oncol. 2012. V. 2. P. 58. https://doi.org/10.3389/fonc.2012.00058
Yahyapour R., Amini P., Rezapour S. et al. Radiation-induced inflammation and autoimmune diseases // Milit. Med. Res. 2018. V. 5. P. 9. https://doi.org/10.1186/s40779-018-0156-7
Mavragani I.V., Laskaratou D.A., Frey B. et al. Key mechanisms involved in ionizing radiation-induced systemic effects. A current review // Toxicol. Res. (Camb). 2016. V. 5. № 1. P. 12–33. https://doi.org/10.1039/c5tx00222b
Тимофеев-Ресовский Н.В., Савич А.В., Шальнов М.И. Введение в молекулярную радиобиологию (физико-химические основы). М.: Медицина, 1981. 320 с. [Timofeev-Resovskij N.V., Savich A.V., Shal’nov M.I. Vvedenie v molekuljarnuju radiobiologiju (fiziko-himicheskie osnovy). M.: Medicina, 1981. 320 p. (In Russ.)]
Azzam E.I., Jay-Gerin J.-P., Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury // Cancer Lett. 2012. V. 327. P. 48–60. https://doi.org/10.1016/j.canlet.2011.12.012
Gorbunov N.V., Sharma P. Protracted oxidative altera-tions in the mechanism of hematopoietic acute radiation syndrome // Antioxidants (Basel). 2015 V. 4. № 1. P. 134–52. https://doi.org/10.3390/antiox4010134
Долин П.И. Время жизни промежуточных состояний, возникающих при действии излучений на водные растворы // Роль перекисей и кислорода в начальных стадиях радиобиологического эффекта. М.: Изд-во АН СССР, 1960. С. 20–25. [Dolin P.I. Vremja zhizni promezhutochnyh sostojanij, voznikajushhih pri dejstvii izluchenij na vodnye rastvory // Rol’ perekisej I kisloroda v nachal’nyh stadijah radiobiolo-gicheskogo effekta. M.: Izd-vo AN USSR, 1960. P. 20–25. (InRuss.)]
Кузин А.М. О роли образования перекисей при действии радиации на биологические объекты // Роль перекисей и кислорода в начальных стадиях радиобиологического эффекта. М.: Изд-во АН СССР, 1960. С. 3–8. [Kuzin A.M. O roli obrazovanija perekisej pri dejstvii radiacii na biologicheskie ob’ekty // Rol’ perekiseji kisloroda v nachal’nyh stadijah radiobiologicheskogo effekta. M.: Izd-vo AN USSR, 1960. P. 3–8. (In Russ.)
Adjemian S., Oltean T., Martens S. et al. Ionizing radiation results in a mixture of cellular outcomes including mitotic catastrophe, senescence, methuosis, and iron-dependent cell death // Cell Death Dis. 2020. V. 11. № 11. P. 1003. https://doi.org/10.1038/s41419-020-03209-y
Оксидативный стресс и воспаление: патогенетическое партнерство: Монография / Под ред. О. Г. Хурцилавы, Н. Н. Плужникова, Я. А. Накатиса. СПб.: Изд-во СЗГМУ им. И. И. Мечникова, 2012. 340 с. [Oksidativnyj stress i vospalenie: patogeneticheskoe partnerstvo: Monografija / Pod red. O. G. Hurcilavy, N. N. Pluzhnikova, Ja. A. Nakatisa. SPb.: Izdatel’stvo SZGMU im. I. I. Mechnikova, 2012. 340 p. (In Russ.)]
Мартусевич А.К., Карузин К.А. Оксидативный стресс и его роль в формировании дезадаптации и патологии // Биорадикалы и антиоксиданты. 2015. Т. 2. № 2. С. 5–18. [Martusevich A.K., Karuzin K.A. Oksidativnyj stress i ego rol' v formirovanii dezadaptacii i patologii // Bioradikaly i antioksidanty. 2015. V. 2. № 2. P. 5–18. (In Russ.)]
Chen Y., Li Y., Huang L. et al. Antioxidative stress: inhibiting reactive oxygen species production as a cause of radioresistance and chemoresistance // Oxidat. Med. Cell. Longevity. 2021. V. 2021. P. 6620306. https://doi.org/10.1155/2021/6620306
Kajarabille N., Latunde-Dada G.O. Programmed cell-death by ferroptosis: antioxidants as mitigators // Int. J. Mol. Sci. 2019. V. 20. № 19. P. 4968. https://doi.org/10.3390/ijms20194968
Кузник Б.И., Линькова Н.С., Ивко О.М. Оксидативный стресс, старение и короткие пептиды // Успехи физиол. наук. 2021. Т. 52. № 2. С. 13–20. [Kuznik B.I., Linkova N.S., Ivko O.M. Oxidative stress, aging and short peptides // Progress in Physiological Science. 2021. V. 52. № 2. P. 13–20. (In Russ.)]. https://doi.org/10.31857/S0301179821020041
Gaschlera M.M., Stockwellb B.R. Lipid peroxidation in cell death // Biochem. Biophys. Res. Commun. 2017. V. 482. № 3. P. 419–425. https://doi.org/10.1016/j.bbrc.2016.10.086
Citrin D.E., Mitchell J.B. Mechanisms of normal tissue injury from irradiation // Semin. Radiat. Oncol. 2017. V. 27. № 4. P. 316–324. https://doi.org/10.1016/j.semradonc.2017.04.001
Li P., Chang M. Roles of PRR-mediated signaling pathways in the regulation of oxidative stress and inflammatory diseases // Int. J. Mol. Sci. 2021. V. 19; 22. № 14. P. 7688. https://doi.org/10.3390/ijms22147688
Rock K.L., Kono H. The inflammatory response to cell death // Ann. Rev. Pathol. Mechanisms Diseases. 2008. V. 3. P. 99–126. https://doi.org/10.1146/annurev.pathmechdis.3.121806. 151456
Литвицкий П.Ф. Воспаление // Вопр. совр. педиатрии. 2006. Т. 6. № 3. С. 48–51 [Litvitsky P.F. Inflammation // Curr. Pediatr. 2006. V. 6. № 3. P. 48–51. (In Russ.)].
Schaue D., McBride W.H. Links between innate immunity and normal tissue radiobiology // J. Radiat. Res. 2010. V. 173. № 4. P. 406–417.https://doi.org/10.1667/RR1931.1
Farhood B., Ashrafizadeh M., Khodamoradi E. et al. Targeting of cellular redox metabolism for mitigation of radiation injury // Life Sci. 2020. V. 250. P. 117570. Available at: https://doi.org/ May 24, 2020.https://doi.org/10.1016/j.lfs.2020.117570.Accessed
Hill R.P., Zaidi A., Mahmood J. et al. Investigations into the role of inflammation in normal tissue response to irradiation // Radiother. Oncol. 2011. V. 101. № 1. P. 73–79. https://doi.org/10.1016/j.radonc.2011.06.017
Sun L., Inaba Y., Sato K. et al. Dose-dependent decrease in anti-oxidant capacity of whole blood after irradiation: A novel potential marker for biodosimetry // Sci. Rep. 2018. V. 8. P. 7425. https://doi.org/10.1038/s41598-018-25650-y
Sun L., Inaba Y., Sogo Y. et al. Total body irradiation causes a chronic decrease in antioxidant levels // Sci. Rep. 2021. V. 11. № 1. P. 6716. https://doi.org/10.1038/s41598-021-86187-1
Singh V.K., Beattie L.A., Seed T.M. Vitamin E: toco-pherols and tocotrienols as potential radiation countermeasures // J. Radiat. Res. 2013. V. 54. № 6. P. 973–988. https://doi.org/10.1093/jrr/rrt048
Greenberger J., Kagan V., Bayir H. et al. Antioxidant approaches to management of ionizing irradiation injury // Antioxidants. 2015. V. 4. P. 82–101. https://doi.org/10.3390/antiox4010082
Hofer M., Hoferová Z., Falk M. Pharmacological mo-dulation of radiation damage. Does it exist a chance for other substances than hematopoietic growth factors and cytokines? // Int. J. Mol. Sci. 2017. V. 18. № 7. P. 1385. https://doi.org/10.3390/ijms18071385
Yang W.S., Stockwell B.R. Ferroptosis: death by lipid peroxidation // Trends Cell Biol. 2016. V. 26. № 3. P. 165–176. https://doi.org/10.1016/j.tcb.2015.10.014
Hirschhorn T., Stockwell B.R. The development of the concept of ferroptosis // Free Radical Biol. Med. 2019. V. 133. P. 130–143. https://doi.org/10.1016/j.freeradbiomed.2018.09.043
Galluzzi L., Vitale I., Aaronson S.A. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018 // Cell Death Differentiation. 2018. V. 25. № 3. P. 486–541. https://doi.org/10.1038/s41418-017-0012-4
Xie Y., Hou W., Song X. et al. Ferroptosis: process and function // Cell Death Differentiation. 2016. V. 23. № 3. P. 369–379. https://doi.org/10.1038/cdd.2015.158
Stockwell B.R., Friedmann Angeli J.P., Bayir H. et al. Ferroptosis: a regulated cell death nexus linking meta-bolism, redox biology, and disease // Cell. 2017. V. 171. № 2. P. 273–285. https://doi.org/10.1016/j.cell.2017.09.021
Ye L.F., Chaudhary K.R., Zandkarimi F. et al. Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers // ACS Chem. Biol. 2020. V. 15. № 2. P. 469–484. https://doi.org/10.1021/acschembio.9b00939
Zhang X., Xing X., Liu H. et al. Ionizing radiation indu-ces ferroptosis in granulocyte-macrophage hematopoietic progenitor cells of murine bone marrow // Int. J. Radiat. Biol. 2020. V. 96. № 5. P. 584–595. https://doi.org/10.1080/09553002.2020.1708993
Zhang X., Tian M., Li X. et al. Hematopoietic protection and mechanisms of ferrostatin-1 on hematopoietic acute radiation syndrome of mice // Int. J. Radiat. Biol. 2021. V. 97. № 4. P. 464–473. https://doi.org/10.1080/09553002.2021.1876956
Thermozier S., Hou W., Zhang X. et al. Anti-ferroptosis drug enhances total-body irradiation mitigation by drugs that block apoptosis and necroptosis // J. Radiat. Res. 2020. V. 193. № 5. P. 435–450. https://doi.org/10.1667/RR15486.1
Cohen-Jonathan E., Bernhard E.J., McKenna W.G. How does radiation kill cells? // Curr. Opin. Chem. Biol. 1999. V. 3. № 1. P. 77–83. https://doi.org/10.1016/S1367-5931(99)80014-3
Verheij M., Bartelink H. Radiation-induced apoptosis // Cell Tissue Res. 2000, V. 301. № 1. P. 133–42. https://doi.org/10.1007/s004410000188
Shinomiya N. New concepts in radiation-induced apoptosis: “premitotic apoptosis” and “postmitotic apoptosis” // J. Cell Mol. Med. 2001. V. 5. № 3. P. 240–253. https://doi.org/10.1111/j.1582-4934.2001.tb00158
Elmore S. Apoptosis: A review of programmed cell death // Toxicol. Pathol. 2007. V. 35. № 4. P. 495–516. https://doi.org/10.1080/01926230701320337
Черешнев В.А., Цыган В.Н., Одинак М.М. и др. Фармакологическое регулирование программированной гибели клеток / Под ред. В.А. Черешнева. СПб.: Наука, 2011. 255 с. [Chereshnev V.A., Tsygan V.N., Odinak M.M. et al. Pharmacological regulation of programmed cell death / Ed. V.A. Chereshnev. St. Petersburg: Nauka, 2011. 255 p. (In Russ.)]
Cao X., Wen P., Fu Y. et al. Radiation induces apoptosis primarily through the intrinsic pathway in mammalian cells // Cell Signal. 2019. V. 62. P. 109337. https://doi.org/10.1016/j.cellsig.2019.06.002
Lee K.-H., Kang T.-B. The molecular links between cell death and inflammasome // Cells. 2019. V. 8. № 9. P. 1057. https://doi.org/10.3390/cells8091057
D’Arcy M.S. Cell death: a review of the major forms of apoptosis, necrosis and autophagy // Cell Biol. Int. 2019. V. 43. № 6. P. 582–592. https://doi.org/10.1002/cbin.11137
Kroemer G., Galluzzi L., Vandenabeele P. et al. Classification of cell death recommendations of the Nomenclature Committee on Cell Death 2009 // Cell Death Differentiat. 2009. V. 16. № 1. P. 3–11. https://doi.org/10.1038/cdd.2008.150
Gudipaty S.A., Conner Ch.M., Rosenblatt J. et al. Unconventional ways to live and die: cell death and survi-val in development, homeostasis, and disease // Ann. Rev. Cell Develop. Biol. 2018. V. 34. P. 311–332. https://doi.org/10.1146/annurev-cellbio-100616-060748
Lorimore S.A., Coates Ph.J., Scobie G.E. et al. Inflammatory-type responses after exposure to ionizing radiation in vivo: a mechanism for radiation-induced bystander effects? // Oncogen. 2001. V. 20. P. 7085–7095. https://doi.org/10.1038/sj.onc.1204903
Taylor R.C., Cullen S.P., Martin S.J. Apoptosis: controlled demolition at the cellular level // Nat. Rev. Mol. Cell Biol. 2008. V. 9. № 3. P. 231–241. https://doi.org/10.1038/nrm2312
Silva M.T., do Vale A., dos Santos N.M.N. Secondary necrosis in multicellular animals: an outcome of apoptosis with pathogenic implications // Apoptosis. 2008. V. 13. № 4. P. 463–482. https://doi.org/10.1007/s10495-008-0187-8
Bergsbaken T., Fink S.L., Cookson B.T. Pyroptosis: host cell death and inflammation // Nat. Rev. Microbiol. 2009. V. 7. № 2. P. 99–109. https://doi.org/10.1038/nrmicro2070
Lu F., Lan Zh., Xin Zh. et al. Emerging insights into molecular mechanisms underlying pyroptosis and functions of inflammasomes in diseases // J. Cell Physiol. 2020. V. 235. № 4. P. 3207–3221. https://doi.org/10.1002/jcp.29268
Walle L.V., Lamkanfi M. Pyroptosis // Curr. Biol. 2016. V. 26. № 13. P. R568–R572. https://doi.org/10.1016/j.cub.2016.02.019
Yu P., Zhang X., Liu N. et al. Pyroptosis: mechanisms and diseases // Signal Transduct. Target Ther. 2021. V. 6. № 1. P. 128. https://doi.org/10.1038/s41392-021-00507-5
He W.-T., Wan H., Hu L. et al. Gasdermin D is an exe-cutor of pyroptosis and required for interleukin-1β secretion // Cell Res. 2015. V. 25. № 12. P. 1285–1298. https://doi.org/10.1038/cr.2015.139
Stoecklein V.M., Osuka A., Ishikawa Sh. et al. Radiation exposure induces inflammasome pathway activation in immune cells // J. Immunol. 2015. V. 194. № 3. P. 1178–1189. https://doi.org/10.4049/jimmunol.1303051
Liu Y.-G., Chen J.-K., Zhang Z.-T. et al. NLRP3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages // Cell Death Disease. 2017. V. 8. № 2. P. e2579. https://doi.org/10.1038/cddis.2016.460
Liao H., Wang H., Rong X. et al. Mesenchymal stem cells attenuate radiation-induced brain injury by inhi-biting microglia pyroptosis // Biomed. Res. Inte. 2017. P. 1948985. https://doi.org/10.1155/2017/1948985
Гемпельман Л., Лиско Г., Гофман Д. Острый лучевой синдром: изучение 9 случаев и обзор проблемы / Под ред. А. Бурназяна. М.: Изд-во Иностр. лит-ры, 1954. 290 с. [Gempel’man L., Lisko G., Gofman D. Ostryj luchevoj sindrom: izuchenie 9 sluchaev I obzor problem / Pod red. A. Burnazjana. M.: Izd-vo inostrannoj literatury, 1954. 290 p. (In Russ.)]
McBride W.H., Chiang Chi.-Sh., Olson J.L. et al. A Sense of Danger From Radiation // Radiat. Res. 2004. V. 162. № 1. P. 1–19. https://doi.org/10.1667/rr3196
Богданова И.М. Иммунологические механизмы сепсиса и новые подходы к его терапии // Клин. и эксперим. морфология. 2014. Т. 3. № 11. С. 52–58. [Bogdanova I.M. Immunological mechanisms of sepsis and new approaches to its treatment // Clinical and experimental morphology. 2014. V. 3. № 11. P. 52–58. (In Russ.)]
Schaefer L. Complexity of Danger: The Diverse Nature of Damage-associated Molecular Patterns // J. Biol. Chem. 2014. V. 289. № 51. P. 35237–35245. https://doi.org/10.1074/jbc.R114.619304
Ratikan J.A., Micewicz E.D., Xie M.W. et al. Radiation takes its Toll // Cancer Lett. 2015. V. 368. № 2. P. 238–245. https://doi.org/10.1016/j.canlet.2015.03.031
Черешнев В.А., Гусев Е.Ю. Иммунологические и патофизиологические механизмы системного воспаления // Мед. иммунология. 2012. Т. 14. № 1–2. С. 9–20. [Chereshnev V.A., Gusev E.Ju. Immunolo-gicheskie i patofiziologicheskie mehanizmy sistemnogo vospalenija // Medicinskaja immunologija. 2012. V. 14. № 1–2. P. 9–20. (In Russ.)]
Shi Y., Evans J., Rock K. Molecular identification of a danger signal that alerts the immune system to dying cells // Nature. 2003. V. 425. P. 516–521. https://doi.org/10.1038/nature01991
Netea M.G., Balkwill F., Honchol M. et al. A guiding map for inflammation // Nat. Immunol. 2017. V. 18. № 8. P. 826–831.https://doi.org/10.1038/ni.3790
Тухватулин А.И., Логунов Д.Ю., Щербинин Д.Н. и др. Toll‑подобные рецепторы и их адапторные молекулы. Обзор // Биохимия. 2010. Т. 75. № 9. С. 1224–1243. [Tukhvatulin A.I., Logunov D.Y., Shcherbinin D.N. et al. Toll-like receptors and their adapter molecules // Biochemistry (Moscow). 2010. V. 75. № 9. P. 1098–1114. (In Russ.)]
Piccinini A.M., Midwood K.S. DAMPening inflammation by modulating tlr signaling // Mediators Inflamm. 2010. P. 672395. https://doi.org/10.1155/2010/672395
Bianchi M.E. DAMPs, PAMPs and alarmins: all we need to know about danger // J. Leukocyte Biol. 2007. V. 81. № 1. P. 1–5. https://doi.org/10.1189/jlb.0306164
Бакунина Л.С., Литвиненко И.В., Накатис Я.А. и др. Сепсис: пожар и бунт на тонущем в шторм корабле: монография. В 3 ч. Ч. I. Триггеры воспаления. Рецепция триггеров воспаления и сигнальная трансдукция / Под ред. Н.Н. Плужникова, С.В. Чепура, О.Г. Хурцилава. СПб.: Изд-во СЗГМУ им. И.И. Мечникова, 2018. 232 с. [Bakunina L.S., Litvinenko I.V., Nakatis Y.A. et al. Sepsis: pozhar i bunt na tonuschem v shtorm korable: Recepciya trigerov vos-palenia i signal`naya transdukciya / Pod redakciej N.N. Pluzhnikova, S.V. Chepura, O.G. Xurcilava. Saint Peterburg: Izdatel`stvo SZGMU imeni I.I. Mechnikova, 2018. 232 p. (In Russ.)]
Takeuchi O., Akira Sh. Pattern Recognition Receptors and Inflammation // Cell. 2010. V. 140. № 6. P. 805–820. https://doi.org/10.1016/j.cell.2010.01.022
Ковальчук Л.В., Хорева М.В., Никонова А.С. Распознающие рецепторы врожденного иммунитета (NLR, RLRи CLR) // Журн. микробиологии, эпидемиологии и иммунобиологии. 2011. № 1. С. 93–100. [Koval’chuk L.V., Khoreva M.V., Nikonova A.S. Recognition receptors of innate immunity (NLR, RLR, AND CLR) // J. Microbiol. Epidemiol. Immunobiol. 2011. № 1. 93–100. (In Russ.)]
Успенская Ю.А., Комлева Ю.К., Пожиленкова Е.А. и др. Лиганды RAGE-белков: роль в межклеточной коммуникации и патогенезе воспаления // Вестн. РАМН. 2015. Т. 70. № 6. С. 694–703. [Uspenskaya Yu.A., Komleva Yu.K., Pozhilenkova E.A. et al. Ligands of RAGE-Proteins: Role in Intercellular Communication and Pathogenesis of Inflammation // Vestnik Rossiiskoi Akademii Meditsinskikh Nauk (Annals of the Russian Academy of Medical Sciences). 2015. V. 70. № 6. P. 694–703 (In Russ.)]. https://doi.org/10.15690/vramn566
Schaue D., Kachikwu E.L., McBride W.H. Cytokines in Radiobiological Responses: A Review // Radiat. Res. 2012. V. 178. № 6: 505–523. https://doi.org/10.1667/RR3031.1
Janssens S., Tschopp J. Signals from within: the DNA-damage-induced NF-kappaB response // Cell Death Different. 2006. V. 13. № 5. P. 773–84. https://doi.org/10.1038/sj.cdd.4401843
Janssens S., Tinel A., Lippens S. et al. PIDD mediates NF-κB activation in response to DNA damage // Cell. 2005. V. 123. № 6. P. 1079–1092.
Lin Y., Bai L, Chen W. et al. The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy // Exp. Opin. Therap. Targets. 2010. V. 14. № 1. P. 45–55. https://doi.org/10.1517/14728220903431069
Zhang Q., Lenardo M.J., Baltimore D. 30 Years of NF-κB: A blossoming of relevance to human pathobiology // Cell. 2017. V. 168. № 1–2. P. 37–57. https://doi.org/10.1016/j.cell.2016.12.012
Di Maggio F.M., Minafra L., Forte G.I. et al. Portrait of inflammatory response to ionizing radiation treatment // J. Inflamm. (Lond). 2015. V. 12. P. 14. https://doi.org/10.1186/s12950-015-0058-3
Pulsipher A., Savage J.R., Kennedy T.P. et al. GM-1111 reduces radiation-induced oral mucositis in mice by targeting pattern recognition receptor-mediated inflammatory signaling // PLoS One. 2021. V. 16. № 3. P. e0249343. https://doi.org/10.1371/journal.pone.0249343
Dent P., Yacoub A., Fisher P.B. et al. MAPK pathways in radiation responses // Oncogene. 2003. V. 22. № 37. P. 5885–96. https://doi.org/10.1038/sj.onc.1206701
Munshi A., Ramesh R. Mitogen-activated protein kina-ses and their role in radiation response // Genes Cancer. 2013. V. 4. № 9–10. P. 401–408. https://doi.org/10.1177/1947601913485414
Meng Q., Karamfilova Zaharieva E., Sasatani M. et al. Possible relationship between mitochondrial changes and oxidative stress under low dose-rate irradiation // Redox Rep. 2021. V. 26. № 1. P. 160–169. https://doi.org/10.1080/13510002.2021.1971363
Евдокимовский Э.В., Губина Н.Е., Ушакова Т.Е. и др. Изменение соотношения мтДНК/яДНК в сыворотке крови после рентгеновского облучения мышей в различных дозах // Радиац. биология. Радиоэкология. 2012. Т. 52. № 6. С. 565–571. [Evdokimovsky E.V., Gubina N.E., Ushakova T.E. et al. Changes of Mitochondrial DNA/Nuclear DNA Ratio in the Blood Serum Following X-Ray Irradiation of Mice at Various Doses // Radiation biology. Radioecology. 2012. V. 52. № 6. P. 565–571. (InRuss.)]
Евдокимовский Э.В., Губина Н.Е., Абдуллаев С.А., и др. Изменения в уровне метилирования ДНК, а также экспрессии генов в митохондриях разных отделов головного мозга крыс, облученных протонами 150 МэВ // Мат. междунар. конф. “Современные вопросы радиационной генетики”. Дубна, 2019. С. 53–54. [Evdokimovskij Je.V., Gubina N.E., Abdullaev S.A. et al. Izmenenija v urovne metilirovanija DNK, a takzhej ekspressii genov v mitohondrijah raznyh otdelov golovnogo mozga krys, obluchennyh protonami 150 Mjev. Sovremennye voprosy radiacionnoj genetiki // Materialy rossijskoj konferencii s mezhdu-narodnym uchastiem. Dubna, 2019. P. 53–54. (In Russ.)]
Yang L., Hu M., Lu Y. et al. Inflammasomes and the maintenance of hematopoietic homeostasis: new perspectives and opportunities // Molecules. 2021. V. 26. № 2. P. 309. https://doi.org/10.3390/molecules26020309
Wei J., Wang H., Wang H. et al. The role of NLRP3 inflammasome activation in radiation damage // Biomed. Pharmacotherap. 2019. V. 118. P. 109217. https://doi.org/10.1016/j.biopha.2019.109217
Huang Sh., Che J., Chu Q. et al. The Role of NLRP3 inflammasome in radiation-induced cardiovascular injury // Front. Cell Develop. Biol. 2020. V. 8. P. 140. https://doi.org/10.3389/fcell.2020.00140
Колмычкова К.И., Желанкин А.В., Карагодин В.П. и др. Митохондрии и воспаление // Патол. физиология и эксперим. терапия. 2016. Т. 60. № 4. С. 114–121 [Kolmychkova K.I., Zhelankin A.V., Karagodin V.P. et al. Mitochondria and inflammation // Patologicheskaya Fiziologiya i Eksperimental’naya Terapiya (Pathological physiology and experimental therapy). 2016. V. 60. № 4. P. 114–121. (In Russ.)]
Zhang Q., Raoof M., Chen Y. et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury // Nature. 2010. V. 464. № 7285. P. 104–107. https://doi.org/10.1038/nature08780
Патрушев М.В., Патрушева В.Е., Касымов В.А. и др. Элиминация мтДНК из митохондрий и активация ее репликации в клетках тканей облученных мышей // Цитология. 2006. Т. 48. № 8. С. 684–691. [Patrushev M.V., Patrusheva V.E., Kasymov V.А. et al. Release of mtDNA from mitochondria and activation of its replication in tissues of irradiated mice // Tsitologiya. 2006. V. 48. № 8. P. 684–691. (In Russ.)]
Picca A., Calvani R., Coelho-Junior H.J. et al. Cell death and inflammation: the role of mitochondria in health and disease // Cells. 2021. V. 10. № 3. P. 537. https://doi.org/10.3390/cells10030537
De Gaetano A., Solodka K., Zanini G. et al. Molecular mechanisms of mtDNA-mediated inflammation // Cells. 2021. V. 10. № 11. P. 2898. https://doi.org/10.3390/cells10112898
Kong C., Song W., Fu T. Systemic inflammatory response syndrome is triggered by mitochondrial damage // Mol. Med. Rep. 2022. V. 25. № 4. P. 147. https://doi.org/10.3892/mmr.2022.12663
Riley J.S., Tait S.W.G. Mitochondrial DNA in inflammation and immunity // EMBO Rep. 2020. V. 21. № 4. P. e49799. https://doi.org/10.15252/embr.201949799
Guo H., Callaway J.B., Ting J. P.-Y. Inflammasomes: mechanism of action, role in disease, and therapeutics // Nat. Med. 2015. V. 21. № 7. P. 677–687.
Sharma D., Kanneganti Th.-D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation // J. Cell Biol. 2016. V. 213. № 6. P. 617–629. https://doi.org/10.1083/jcb.201602089
He Y., Hara H., Núñez G. Mechanism and regulation of NLRP3 inflammasome activation // Trends Biochem. Sci. 2016. V. 41. № 12. P. 1012–1021. https://doi.org/10.1016/j.tibs.2016.09.002
Vanaja S., Rathinam V.K., Fitzgerald K.A. Mechanisms of inflammasome activation: recent advances and no-vel insights // Trends Cell Biol. 2015. V. 25. № 5. P. 308–315. https://doi.org/10.1016/j.tcb.2014.12.009
Man S.M., Kanneganti Th.-D. Regulation of inflammasome activation // Immunol. Rev. 2015. V. 265. № 1. P. 6–21. https://doi.org/10.1111/imr.12296
Гариб Ф.Ю., Ризопулу А.П., Кучмий А.А., и др. Инактивация инфламмасом патогенами регулирует воспаление (обзор) // Биохимия. 2016. Т. 81. № 11. С. 1578–1592. [Garib F.Yu., Rizopulu A.P., Kuchmiy A.A. et al. Inactivation of inflammasomes by pathogens regulates inflammation // Biochemistry (Moscow). 2016. V. 81. № 11. P. 1326–1339. (In Russ.)]
Гариб Ф.Ю., Ризопулу А.П. Инфламмасомы и воспаление // Рос. иммунол. журн. 2017. Т. 11 (20). № 4. С. 620–626. [Garib F.Yu., Rizopulu A.P. Inflammasomes and inflammation // Russian Journal of Immunology (Rossiyskiy Immunologicheskiy Zhurnal). 2017. V. 11 (20). № 4. P. 620–626. (In Russ.)]
Богданова И.М. Инфламмасомы: внутриклеточные регуляторы противоинфекционного и воспалительного ответа в системе врожденного иммунитета // Клин. и эксперим. морфология. 2016. Т. 1. № 17. С. 63–69. [Bogdanova I.M. Inflammasomes: intracellular regulators of anti-infectious and inflammatory responses in the innate immune system // Clin. Exp. Morphology. 2016. V. 1. № 17. P. 63–69. (In Russ.)]
Кувачева Н.В., Моргун А.В., Хилажева Е.Д. и др. Формирование инфламмасом: новые механизмы регуляции межклеточных взаимодействий и секреторной активности клеток // Сиб. мед. обозрение. 2013. № 5 (83). С. 3–10. [Kuvacheva N.V., Morgun A.V., Hilazheva E.D. Inflammasomes forming: new mechanisms of intercellular interactions regulation and secretory activity of the cells // Siberian Me-dical Review. 2013. № 5 (83). P. 3–10. (In Russ.)]
Ghaemi-Oskouie F., Shi Y. The role of uric acid as an endogenous danger signal in immunity and inflammation // Curr. Rheumatol. Rep. 2011. V. 3. № 2. P. 160–166. https://doi.org/10.1007/s11926-011-0162-1
Gao J., Peng Sh., Shan X. et al. Inhibition of AIM2 inflammasome-mediated pyroptosis by Andrographolide contributes to amelioration of radiation-induced lung inflammation and fibrosis // Cell Death Diseases. 2019. V. 10. № 12. P. 957. https://doi.org/10.1038/s41419-019-2195-8
Xiao J., Wang Ch., Yao J.-Ch. et al. Radiation causes tissue damage by dysregulating inflammasome-gasdermin D signaling in both host and transplanted cells // PLoS Biol. 2020. V. 18. № 8. P. e3000807. https://doi.org/10.1371/journal.pbio.3000807
Sohn S.-H., Lee J.M., Park S. et al. The inflammasome accelerates radiation-induced lung inflammation and fibrosis in mice // Environ. Toxicol. Pharmacol. 2015. V. 39. № 2. P. 917–926. https://doi.org/10.1016/j.etap.2015.02.019
Цыган В.Н., Бубнов В.А., Цыган Н.В. и др. Врожденный иммунитет и активация атерогенеза // Воен.-мед. журн. 2016. Т. 337. № 3. С. 47–54. [Tsygan V.N., Bubnov V.A., Tsygan N.V. et al. The innate immunity and activation of the atherogenesis // Voenno-med. zhurn. (J. Mil. Med). 2016. V. 337. № 3. P. 47–54. (In Russ.)]
Zhao W., Robbins M.E.C. Inflammation and chronic oxidative stress in radiation-induced late normal tissue injury: therapeutic implications // Curr. Med. Chem. 2009. V. 16. № 2. P. 130–143. https://doi.org/10.2174/092986709787002790
Дополнительные материалы отсутствуют.
Инструменты
Радиационная биология. Радиоэкология