

Прикладная биохимия и микробиология, 2022, T. 58, № 6, стр. 537-550
Твердофазная биодеградация полилактида (обзор)
В. В. Миронов 1, *, Е. С. Трофимчук 3, Н. А. Загустина 2, О. А. Иванова 3, А. В. Вантеева 1, Е. А. Бочкова 1, В. В. Острикова 1, Ш. Чжан 4
1 Институт микробиологии им. С.Н. Виноградского, Федеральный исследовательский центр
“Фундаментальные основы биотехнологии” Российской академии наук
119071 Москва, Россия
2 Институт биохимии им. А.Н. Баха, Федеральный исследовательский центр
“Фундаментальные основы биотехнологии” Российской академии наук
119071 Москва, Россия
3 Химический факультет МГУ имени М.В. Ломоносова
119991 Москва, Россия
4 Институт городской среды Китайской Академии наук
361021 Сямынь, Китай
* E-mail: 7390530@gmail.com
Поступила в редакцию 02.06.2022
После доработки 26.06.2022
Принята к публикации 04.07.2022
- EDN: BKBDKS
- DOI: 10.31857/S0555109922060101
Аннотация
Основной проблемой, связанной с использованием полилактида (ПЛ) в различных областях, является требование контролируемой деградации его в составе отходов. Однако в настоящее время возможности биодеструкции ПЛ не изучены в достаточной степени. Предполагается, что микроорганизмы могут разлагать полимер только после его предварительного химического гидролиза и уменьшения его молекулярной массы до 10000 Да и менее. В обзоре рассмотрено влияние различных факторов на абиотическую и биотическую стадии деструкции ПЛ. Особое внимание уделено особенностям биоразложения полимера в условиях промышленного компостирования как наиболее перспективного способа его переработки в составе пищевых отходов. Приведены микроорганизмы, участвующие в деструкции ПЛ, а также их ферменты.
Ежегодно образуется практически 400 млн. т отходов из пластика, и только 18% в среднем по миру из них перерабатывается и 24% сжигается [1]. Остальные 58% либо отправляются на свалки, либо попадают в естественную среду [2, 3]. Хорошо известное так называемое “большое тихоокеанское мусорное пятно”, которое в основном состоит из пластика (около 80 000 тонн), занимает площадь более 1.6 млн км2 и является ярким свидетельством масштабов глобальной проблемы [4]. Ситуация усугубляется тем, что основная масса пластиковой упаковки продуктов питания после использования становится практически неотделима от потока пищевых отходов и соответственно должна утилизироваться совместно. Согласно последнему исследованию ООН в 2019 г. примерно 931 млн. т продуктов питания было выброшено в мусорные баки домашних хозяйств, пунктов розничной торговли и предприятий общественного питания (https://www.unep.org/ru/resources/doklad/doklad-yunep-ob-indekse-pischevykh-otkhodov-za-2021-god).
Поиск биоразлагаемых, получаемых из возобновляемых ресурсов материалов привлекает все больше внимание для решения глобальной угрозы загрязнения отходами из трудноразлагаемых полимеров и, в частности, формируемых частиц, так называемого, микропластика [5, 6]. Разработка методов создания биодеградируемых полимерных материалов, которые обычно называют биопластиками, и изучение особенностей процессов их деструкции остаются актуальными [7].
Мировое производство биопластика в настоящее время составляет более 2 млн. т ежегодно с тенденцией увеличения [8]. Переход изготовления одноразовой посуды и упаковки продуктов питания на биоразлагаемые материалы хорошо укладывается в концепцию устойчивого развития.
Среди биоразлагаемых полимеров особое внимание привлекают термопластичные сложные полиэфиры алифатического ряда, такие как полилактид (ПЛ), который может разлагаться микроорганизмами как в окружающей среде, так и в условиях промышленного компостирования [9–13]. Отсутствие токсичности позволяет широко применять ПЛ для разработки различных упаковочных материалов [14]. Молочная кислота, необходимая для производства ПЛ путем поликонденсации, в основном получается в результате микробной ферментации отходов производства таких сельскохозяйственных культур, как пшеница и кукуруза [15].
В настоящее время ПЛ является одним из наиболее коммерчески производимых биопластиков, на долю которого приходится 25% мирового производства [10], а также самым распространенным биоразлагаемым упаковочным материалом для пищевых продуктов [8, 16]. Потребление ПЛ за 2015–2019 гг. выросло на 63%: со 168 до 273 тыс. т (https://marketing.rbc.ru/articles/11659/). Рост потребления ПЛ связан с повышением спроса на биоразлагаемые материалы на основе возобновляемого сырья.
Полимерный материал должен обладать стабильными характеристиками в течение всего срока службы, но достаточно быстро разлагаться при попадании в категорию отходов. Тем не менее использование изделий из ПЛ имеет некоторые ограничения, поскольку полимер характеризуется довольно продолжительными сроками деградации [17]. Так, в работе [18] сообщается, что для его естественного разложения требуется до 2 лет. Медленная деградация ПЛ в окружающей среде является значимой проблемой и может ограничивать его применение.
В связи с общей рекомендацией выбрасывать биопластик совместно с твердыми коммунальными отходами концентрация этих продуктов в потоке отходов быстро увеличивается и, как ожидается, достигнет высоких значений в ближайшие годы: по прогнозу, в 2030 г. концентрация составит 8–10% по массе [19]. Таким образом, оценка влияния биопластиков на процесс и качество продуктов компостирования становится актуальной задачей.
В то же время постоянное улучшение технических свойств ПЛ, в том числе термостабильности и механических характеристик, приводит к значительному снижению его способности к микробному разложению. Таким образом, возникает противоречие между необходимостью улучшения потребительских свойств ПЛ и сохранением его способности к биоразложению.
Целью настоящего обзора является систематизация данных о механизмах биодеградации ПЛ, в том числе при компостировании в составе других органических отходов.
Деградации ПЛ в окружающей среде. В природных условиях сосуществуют как биотические, так и абиотические факторы разложения ПЛ [20]. На скорость деградации ПЛ в окружающей среде влияют: надмолекулярная структура, молекулярная масса, химический состав (содержание D-изомера), степень кристалличности, температуры стеклования Tg и плавления Tm, пористость, размеры изделия, наличие наполнителя и др., а также факторы окружающей среды, такие как влажность, температура, рН, микроорганизмы, среда разложения (водный раствор буферов, почва, компост, биологические жидкости, in vivo) [20, 21].
Обычно биоразложению предшествует частичная деструкция полимера в результате различных физико-химических воздействий (температура, влажность, УФ-излучение, др.) [18, 22]. Механизмы разложения ПЛ в окружающей среде можно формально разделить на: 1) физические, относящиеся к таким изменениям, как растрескивание, охрупчивание, отслаивание, др.; 2) химические, относящиеся к изменениям на молекулярном уровне, таким как разрыв связей в основной цепи или их окисление, что зачастую приводит к снижению молекулярной массы полимера [3]. Как правило, химическое разложение при температурах, близких к температуре окружающей среды, происходит через гидролиз или окисление, которые могут быть ускорены микробной активностью, теплом, светом или их комбинацией. ПЛ может подвергаться гидролитическому, термическому, окислительному, бактериальному, ферментативному расщеплению и фотодеградации [4, 23]. В зависимости от вида воздействия, вызывающего процессы деградации, их можно разделить на небиотические (гидролиз, термическая деградация, окисление, фотолиз) и биотические процессы (потребление микроорганизмами).
Гидролитическая деструкция ПЛ включает в себя стадии водопоглощения, расщепления сложноэфирных связей и диффузии водорастворимых олигомеров из объема материала в окружающую среду [13, 16]. В процессе гидролиза вода действует как пластификатор ПЛ, тем самым повышая подвижность полимерных цепей, что приводит к кристаллизации и потере прозрачности полимера. Отмечается, что этот процесс более характерен для ПЛ с молекулярной массой менее 100 кДа. Более высокомолекулярный ПЛ в меньшей степени подвержен деградации и дольше сохраняет свою прочность и прозрачность.
В естественных условиях гидролитическая абиотическая деградация сменяется биодеградацией, при которой происходит разложение растворимых олигомеров с молекулярной массой 10 000–20 000 Да микроорганизмами (биотический процесс) [10, 13]. При этом на поверхностях изделия из пластика образуются биопленки, сформированные микроорганизмами [24], плотность которых может значительно возрастать при наличии оптимальных условий [18, 22]. Низкомолекулярные продукты деградации ПЛ, образующиеся при действии ферментов, относящихся к классу гидролаз (липаза, кутиназа, эстераза, др.) могут использоваться микроорганизмами с выделением CO2, H2O и других продуктов метаболизма [18, 22]. Исследования Ришерт и Домбровской [24] показали, что консорциумы микроорганизмов, присутствующие в компосте и иле, достаточно активно участвуют в биодеградации полимерных материалов.
Абиотическая деградация. Необходимо отметить, что именно абиотический гидролиз считается лимитирующей стадией деградации ПЛ [4, 29]. Разрыв химических связей в макромолекулах является лишь начальной стадией разрушения (эрозии) полимера, которое в итоге приводит к его полному “исчезновению” в окружающей среде. По способу разрушения выделяют поверхностную (гетерогенная) и объемную (гомогенная) эрозию в зависимости от соотношения скоростей химической деструкции, протекающей на поверхности и в объеме полимерного изделия. При гидролитической деструкции поверхностная эрозия имеет место, когда скорость реакции гидролиза выше, чем диффузия воды в объем материала, например, в присутствии катализаторов – сильных кислот и оснований. В этом случае полимер теряет массу на границе с окружающей средой, со временем уменьшается его объем, но форма образца сохраняется. При объемной эрозии происходит быстрая диффузия воды в структуру полимера, и реакция гидролиза происходит одновременно во всем объеме образца примерно с одинаковой скоростью. Для этого случая обычно значительно изменяется молекулярная масса полимера, но масса и размер образца обычно остаются постоянными в течение значительного времени. Механизм эрозии определяется размером и формой деградируемого изделия, а именно соотношением его объема и поверхности, контактирующей с окружающей средой. В этом случае поверхностная эрозия происходит, когда толщина полимерного изделия больше её критической величины Lкр (для ПЛ Lкр = 7.4 см [27]), если же размеры меньше Lкр, то преобладает объемная эрозия.
Как правило, абиотическая деградация полиэфиров состоит из двух последовательных процессов: сначала происходит диффузия воды в объем полимера, а затем его гидролитическая деструкция через разрыв сложноэфирных связей макромолекул по реакции, рис. 1.
Рис. 1.
Гидролитическая деструкция полимера через разрыв сложноэфирных связей макромолекул: 1 и 4 – ПЛ со степенью полимеризации n и (n – 1) соответственно, 2 – промежуточный продукт реакции, 3 – молочная кислота.
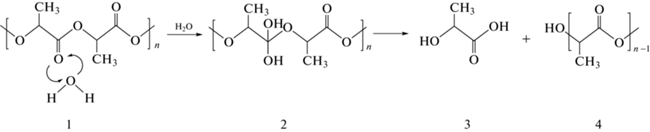
Скорость этого процесса в значительной степени зависит от молекулярной массы полимера. Так, срок разложения высокомолекулярного частично кристаллического поли(L-лактида) составляет не менее двух лет, тогда как низкомолекулярного – всего несколько недель. Кале с соавт. [13] сообщают о снижении скорости разложения ПЛ в компосте при увеличении молекулярной массы (Mм) полимера.
Важнейшими факторами, оказывающими влияние на скорость разложения, являются степень кристалличности и размер кристаллитов [25], поскольку доступ молекул воды в более плотные кристаллические области материала затруднен, и деструкция в них проходит значительно медленнее, чем в менее плотных аморфных областях. В первую очередь разлагаются аморфные области, степень кристалличности материала по мере его деструкции сначала растет и только, когда начинают деградировать кристаллические области, она падает [26]. На ранних этапах разложения, пока процесс происходит в аморфной фазе, размер кристаллитов не оказывает значительного влияния на скорость процесса. На поздних этапах разложения во время деструкции кристаллической фазы размер кристаллитов приобретает важную роль: чем размер больше, тем более она затруднена [28].
Влияние таких характеристик как стереорегулярность и оптическая чистота на скорость разложения значительно меньше, чем молекулярной массы. При введении звеньев D- или D, L-лактида в ПЛ деструкция полимера ускоряется. Это связано с нарушением упорядоченности цепей и уменьшением степени кристалличности, что приводит к ускорению диффузионных процессов в аморфных областях [29–31]. Например, для деструкции аморфного высокомолекулярного поли(D, L-лактида) требуется не более 12–16 мес [17].
Температура играет решающую роль в гидролизе ПЛ. Сообщается Кастро-Агирре с соавт. [15], что этот процесс активизируется при температуре выше 30°С. Значительное увеличение скорости гидролитической деструкции происходит при достижении температуры стеклования (около 60°С) [32]. В работах Иньигес-Франко и соавт. (2016) и Тейшейра и соавт. (2021) отмечается, что скорость гидролиза ПЛ выше в смесях воды с этанолом или акрилонитрилом, что связывают с пластифицирующим действием органических растворителей и ускорением диффузии воды в объем полимера [4, 33]. При этом при повышенной температуре (130–200°С) и действии микроволнового излучения в присутствии спиртов (этанол, бутанол) разрыв сложноэфирных связей ПЛ может происходить в результате реакции алкоголиза, без участия молекул воды [34].
Известно, что гидролиз сложноэфирных связей ускоряется в присутствии кислот или щелочей, а его механизм зависит от рН среды [21]. В сильнокислой среде преобладает механизм разложения, включающий отщепление концевого звена цепи, а в щелочной среде происходит отщепление линейного димера молочной кислоты с последующим разложением его до мономера. В слабокислой или нейтральной средах имеет место случайное расщепление сложноэфирных групп, постепенная олигомеризация исходного полимера с последующим образованием растворимых продуктов [21, 35–37]. Накопление продуктов разложения в матрице вызывает эффект автокатализа, что в свою очередь влияет на кинетику и характер деструкции: могут образовываться полости внутри образца, так как скорость разложения в объеме оказывается выше, чем на поверхности [38].
По теории Питта гидролиз ПЛ описывается кинетическим уравнением реакции третьего порядка, скорость которой зависит от концентрации сложноэфирных связей, количества адсорбированной воды и концентрации кислых продуктов гидролиза [4]. Эта теория предсказывает линейную зависимость между логарифмом молекулярной массы полимера и временем разложения, но не учитывает явления переноса в процессе деструкции. В присутствии сильнокислой среды кинетика гидролиза ПЛ достаточно хорошо описывается процессом псевдопервого порядка, когда скорость процесса зависит только от концентрации сложноэфирных связей и определяется степенью полимеризации полимера.
Твердофазная деструкция. Процесс разложения ПЛ в почве происходит с существенно более низкими скоростями по сравнению с компостом из-за более низких значений температуры и влажности. В работах Караманлиоглу с соавт., а также Калмон с соавт. [39, 40] обнаружили, что через 1 г. изменения в образце ПЛ были незначительными, а через 2 г. образец ПЛ только имел некоторые признаки деградации, хотя за это же время образец поли(ε-капролактона) разложился полностью. Это связывают с низкими скоростями гидролитической деструкции ПЛ при низких температурах – в таких условиях потребовалось бы много десятилетий, чтобы полимер разрушился до олигомерных продуктов, пригодных для утилизации микроорганизмами. Часто в результате деятельности мезо- и микрофауны, например, дождевых червей, происходит механическая деградация и фрагментация полимерных отходов, что приводит к включению их в состав почвы.
В этой связи становятся актуальными исследования, посвященные поиску подходов изменения скорости биодеградации ПЛ, которые бы позволили контролировать протекание этого процесса в том числе в окружающей среде и в условиях компостирования.
Методы изменения скорости разложения ПЛ. Для увеличения скорости гидролитической деструкции наиболее часто используют подход, связанный с изменением геометрии цепи ПЛ. Например, введение звездообразного ПЛ (представляет собой разветвленный полимер, в котором разветвления как “лучи” выходят из одного центра) в смесь с линейным ПЛ (макромолекула, не имеющая разветвлений) приводит к снижению способности такого материала к кристаллизации. В работе [41] было показано, что с увеличением количества звездообразного ПЛ в смесевых образцах происходило значительное ускорение их деградации: до 50% (по массе) вместо 10% – на основе линейного ПЛ. В качестве других причин увеличения скорости разложения в присутствии звездообразного ПЛ указывается его достаточно низкая молекулярная масса и большая концентрация концов с полярными ОН-группами.
Для получения материалов с контролируемыми сроками разложения часто проводят сополимеризацию исходных мономеров (лактидов) с другими лактонами [42] или аспарагиновой кислотой [43]. Например, меняя соотношение звеньев лактида и гликолида в сополимере, можно регулировать сроки разложения в диапазоне от нескольких месяцев до нескольких лет. Сополимер лактида с аспарагиновой кислотой показывает достаточно высокую скорость деградации при 40°С в широком диапазоне рН (3.4–12.0) даже в растворах с высокой концентрацией соли NaCl вплоть до полного разложения за 30–40 сут (рН 10.4) [43].
Другой подход – синтез привитого сополимера полиакриловой кислоты на ПЛ. Для подобного сополимера уже через 7 сут при 37°С наблюдали интенсивные процессы деструкции, которые сопровождались выделением олигомерных и низкомолекулярных веществ, хотя для исходного непривитого ПЛ подобные явления наблюдали только спустя 130 сут [44]. Ускорение деградации ПЛ происходит также при введении в него соевого масла, которое химически подшивают к основной цепи полимера как небольшие ответвления [45]. Оригинальный подход, позволяющий не только контролировать и варьировать сроки разложения ПЛ от 2 нед. до 2 лет, но и места его деструкции, предложен в работе [46]. Для этого при синтезе полимера в основную цепь включаются звенья, содержащие легко гидролизующуюся фосфоэфирную связь, которые становятся точками разрыва макромолекулы. Необходимо отметить, что синтез сополимеров строго определенного состава представляет собой достаточно сложную задачу, решение которой требует привлечения сложного оборудования и значительных экономических затрат.
Эффективный путь к регулированию кинетики деградации – введение в полимерную матрицу функциональных добавок, ускоряющих или замедляющих разложение [47]. Обычно введение гидрофильных наполнителей (частицы металлов и их оксидов, древесный уголь, водорастворимые полимеры, слоистые силикаты, др.) повышает гидрофильность полимерной матрицы, увеличивает площадь ее поверхности, контактирующей с водой и тем самым приводит к ускорению процессов гидролитической деструкции [48–52]. Важно отметить, что контакт микроорганизмов с полимером в компосте или почве происходит на границе раздела сред, поэтому свойства поверхности играют значительную роль. Так, шероховатая поверхность с большим количеством полярных гидрофильных функциональных групп гораздо более подвержена биоразложению, чем гладкая и гидрофобная. Действительно, добавление в ПЛ ацетата целлюлозы приводило к резкому росту потери массы образцом при выдерживании в почве [51]. Влияние на деструкцию ПЛ гидрофильных частиц неорганической природы в значительной мере определяется их дисперсностью и качеством смеси. Так, в работе [53] было отмечено, что введение компатибилизатора в смесь полимера с частицами слоистого силиката (монтмориллонит) дополнительно приводит к ускорению биоразложения ПЛ в компосте и почве. В работе [54] для получения волокнистых материалов из ПЛ с ускоренными сроками разложения авторы предложили использовать молекулярный йод в качестве функциональной добавки. Исследование процесса гидролитической деструкции подобных волокон показало, что такие образцы деградируют быстрее по сравнению с незаполненными матрицами.
Важно отметить, что введение наполнителей щелочной природы, например, карбонатов натрия, кальция или цинка в сополимер L-лактида и гликолида замедляет его разложение вследствие нейтрализации карбоксильных групп кислот, образующихся в ходе гидролиза, а также устранению автокаталитического эффекта продуктов разложения [55]. Наполнители, снижающие чувствительность ПЛ к влаге, также препятствуют его быстрой деградации. Например, добавление 30–40% (по массе) карбида кремния SiC приводит к увеличению срока разложения композита в нейтральной среде при 50°С до 140 сут (для исходного ПЛ этот срок составлял около 60 сут) [56], что авторы связывали с уменьшением влагопоглощения и коэффициента диффузии для наполненных образцов. Похожее действие наблюдали при добавлении частиц наноглин и графеновых нанопластинок [26]. Другой механизм, позволяющий снизить скорость биоразложения ПЛ, основан на введении в него веществ, обладающих антибактериальными свойствами, например, наночастиц серебра [57], что препятствует образованию на поверхности такого материала биопленки из микроорганизмов, осуществляющих биотическую деградацию.
Пленки из ПЛ, содержащие наночастицы лигнина, были предложены в качестве пищевых упаковочных материалов [58]. Сообщается, что добавление до 3% (по массе) наночастиц лигнина придавало пленкам ПЛ УФ-защитные, антиоксидантные и антибактериальные свойства, при этом термическая стабильность ПЛ не изменялась, и пленки разлагались при лабораторном компостировании до уровня распада выше 90% за 17 сут.
Важным способом изменения эксплуатационных свойств ПЛ и увеличения его теплостойкости является введение в полимер зародышеобразователей, которые ускоряют процесс кристаллизации [15]. Например, Corbion Purac® (Нидерланды) производит ударопрочную и термостойкую посуду на основе ПЛ (Puralact®), которую можно использовать в микроволновой печи. Ожидается, что термостойкая посуда для пищевых продуктов из ПЛ станет более доступной в ближайшие годы. В то же время необходимо помнить, что при достижении улучшенной прочности и увеличения степени кристалличности модифицированных ПЛ необходимо сохранять способность к биологическому разложению [59].
Компостирование ПЛ. Компостирование используется как универсальный подход к обработке биоразлагаемых твердых отходов [13]. В связи с недавней коммерциализацией ПЛ для продуктов с коротким сроком хранения его деградация в условиях промышленного компостирования все чаще становится предметом исследований. Однако механизмы деградации ПЛ и понимание роли микроорганизмов все еще недостаточно изучены [20]. Компостирование – экзотермический процесс биологического окисления, в котором органический субстрат подвергается биодеградации смешанной популяцией микроорганизмов, включающей бактерии, археи и грибы [60]. При этом происходит повышение температуры в компостируемом материале до 50–60°С и выше при относительно низкой массовой доли влаги: около 60% в начале процесса и 40–50% в конце созревания компоста.
Механизм деградации. При компостировании необходимо учитывать, что на субстрат оказывают воздействие как биотические, так и абиотические факторы [13]. Предполагается, что микроорганизмы могут разлагать полимер только после его предварительного химического гидролиза и уменьшения его молекулярной массы до 10 000 Да и менее [20]. Это отличительная особенность полученных промышленно полимеров, поскольку природные биоразлагаемые полимеры деградируют под воздействием микроорганизмов за одну стадию.
Возможный процесс разложения ПЛ при компостировании включает следующие стадии: адсорбция воды, расщепление сложноэфирных связей с образованием водорастворимых олигомерных продуктов и мономеров, разложение растворимых олигомеров с молекулярной массой до 10 000 Да микроорганизмами [13, 20]. Биоразложение начинается с адгезионного закрепления на поверхности ПЛ микробных клеток, которые выделяют экзоферменты, разрушающие материал [23].
Реакцию биодеградации ПЛ при компостировании можно описать следующим образом [61]:
(1)
${{{\text{С}}}_{{{\text{ПЛ}}}}} + {{{\text{О}}}_{2}} \to {{\,}^{{\left( a \right)}}}~{{{\text{C}}}_{{{\text{биомасса}}}}} \to {{\,}^{{\left( b \right)}}}{\text{С}}{{{\text{О}}}_{2}} + {{{\text{Н}}}_{{\text{2}}}}{\text{О}}$CПЛ превращается в биомассу Cбиомасса с кинетикой (a), а затем Cбиомасса превращается в CO2 с кинетикой (б).
Лучший способ мониторинга и количественной оценки биоразложения состоит в измерении реагента (O2) или конечного продукта (CO2) энергетического метаболизма. Процент биодеградации представляет собой отношение между выделенным диоксидом углерода CСО2 и теоретическим СО2, то есть количеством СО2, ожидаемым в случае полного окисления углерода, присутствующего в образце ПЛ (СПЛ)
(2)
${\text{Биодеградация}}\,\,\% = \frac{{{{{\text{С}}}_{{{\text{С}}{{{\text{О}}}_{{\text{2}}}}}}}}}{{{{{\text{С}}}_{{{\text{ПЛ}}}}}}} \times 100.$Продукты гидролиза и пути гидролиза, влияние продуктов гидролиза на процесс компостирования, глубина гидролиза. Первоначально биодеградация ПЛ происходит на поверхности и в конечном итоге распространяется внутри материала, способствуя полной его деградации [23]. Поверхностный гидролиз приводит к увеличению количества трещин на поверхности образцов ПЛ. Далее, за диффузией воды внутрь материала следует рост микроорганизмов и секреция ферментов.
Наличие сложноэфирных связей в основной цепи ПЛ делает полимер чувствительным к гидролизу как химическому, так и ферментативному [62]. В зависимости от рН среды механизм гидролиза различный: ПЛ разлагается быстрее в щелочных условиях, поскольку во время гидролиза расщепление сложноэфирных групп катализируется гидроксид-ионами [20]. Также сообщается, что при разложении пластиков смешанными культурами возможно воздействие на полимер агрессивных продуктов метаболизма (например, кислот), продуцируемых компонентами сообщества, растущими на доступных источниках углерода и энергии, и не образующих ферментов прямого воздействия на полимер [63]. Так при компостировании, в результате метаболизма молочнокислых бактерий, дрожжей и плесневых грибов рН отходов может снижаться от 7 до 4–5 и затем повышаться до 8–9 рН в результате разложения органических кислот и образования аммиака [64, 65].
Значимым является то, что побочные продукты разложения ПЛ могут изменять компостную среду и микробную популяцию [13]. Аурас и соавт. [66] было продемонстрировано, что не более 10% (по массе) образца ПЛ следует вносить в компостируемый материал, чтобы избежать подкисления из-за образования молочной кислоты в процессе гидролиза. По другим данным, при увеличении содержания ПЛ в компосте до 30% (по массе) происходило замедление разложения полимера вследствие снижения pH компоста с 6.0 до 4.0 через 4 нед. [67]. Таким образом, следует учитывать общее количество ПЛ, вносимого при компостировании, а также рН отходов.
Скорость гидролиза заметно возрастает с ростом температуры, особенно при превышении температуры стеклования [68]. Ранее было показано, что при компостировании как органической фракции твердых коммунальных отходов (ОФТКО), так и пищевых отходов, температура саморазогрева, превышающая температуру стеклования аморфного ПЛ (~60°C), наблюдалась в течение 10–12 сут [64, 65].
Микроорганизмы, участвующие в деградации ПЛ. На сегодняшний день опубликовано достаточно большое количество работ, посвященных микроорганизмам – деструкторам ПЛ, табл. 1. В лабораторных экспериментах исследованы и описаны, главным образом, монокультуры бактерий и грибов, разлагающих ПЛ. Вместе с тем, относительно мало работ по изучению активности бинарных культур и более сложных микробных сообществ, синергии и взаимодействия разных микроорганизмов при деградации ПЛ, кворум-сенсингу в таких сообществах. Все перечисленные вопросы станут новыми важными направлениями в исследованиях микробной деградации этого полимера [23, 69].
Способность к деградации ПЛ могут проявлять микробные сообщества из различных мест обитания, с разной степенью антропогенной нагрузки. В лабораторных условиях наибольшую эффективность демонстрируют микроорганизмы сообщества компоста и активного ила [24]. В сообществе компоста, включающего как бактерии, так и микромицеты, при разложении ПЛ наблюдалась синергия между этими группами микроорганизмов [70]. Микроорганизмы способны осуществлять деструкцию даже модифицированного ПЛ, включающего различные антибактериальные добавки. Возможность последующей деструкции такого полимера позволит создавать, к примеру, упаковки для пищевых продуктов с антибактериальными свойствами для пролонгирования сроков хранения [57, 71].
Большинство известных видов бактерий, разрушающих ПЛ, относятся к филумам Actinomycetota, Pseudomonadota, Bacillota и Bacteroidota, виды грибов – в основном относятся к Basidiomycota и Ascomycota [11, 62, 70, 72–80].
Сообщается, о выявленной способности к деградации ПЛ некоторых представителей таких родов, как Bacillus sp. (B. smithii, B. licheniformis), Pseudomonas sp. (P. geniculate), Pseudonocardia sp., Serratia sp. (S. plymuthica), Stenotrophomonas sp. (S. maltophilia), Brevibacillus, Geobacillus и др. [72–74]. Некоторые виды грибов: Aspergillus niger, Chaetomium globosum, Paecilomyces variotti, Penicillium pinophilium и Trichoderma viride, были использованы для увеличения скорости биодеградации ПЛ, в том числе за счет синергетического взаимодействия с бактериями из почвы или компоста [70].
Использование консорциума Penicillium chrysogenum, Cladosporium sphaerospermum, Serratia marcescens и Rhodotorula mucilaginosa позволяло достичь 44%-ной потери массы ПЛ за 30 сут компостирования [74]. С действием гриба Thermomyces lanuginosus из компоста связывают быстрое разложение композитов на основе ПЛ при 50°C [11].
Семейство Bacillaceae, в том числе бактерии родов Brevibacillus, Geobacillus и Bacillus, является одним из основных в прокариотическом сообществе микроорганизмов компостируемых пищевых отходов, а также ОФТКО, составляя 16–44% в термофильную стадию [64, 65]. Актиномицеты, а именно представители рода Actinomadura, представлены примерно в той же степени, что и бациллы. Основными представителями родов грибов, доминирующих при компостировании пищевых отходов – Penicillium и Cladosporium, а при высокотемпературном компостировании ОФТКО – Thermomyces. Таким образом, как прокариоты, так и грибы в составе микробиоты пищевых отходов и ОФТКО потенциально могут быть активными деструкторами ПЛ.
Обнаружено, что большинство актинобактерий, разлагающих ПЛ, принадлежат к семейству Pseudonocardiaceae. Из тайской лесной почвы был выделен и исследован штамм актинобактерий Actinomadura keratinilytica T16-1, который показал способность к разложению ПЛ при температуре 50°C в жидкой среде с добавлением пленки полимера в качестве источника углерода [76].
Сообщается также о трех бактериальных штаммах, выделенных из компоста и обладающих способностью разлагать ПЛ при температуре окружающей среды, – Pseudomonas aeruginosa S3 и Sphingobacterium S2, а при повышенных температурах – Geobacillus EC-3 [62]. Эти штаммы были способны использовать полимер в качестве единственного источника углерода при температуре ~30°C или 58°C соответственно. Все изоляты образовывали биопленку на ПЛ, что, по-видимому, сопровождалось оптимизацией секреции ферментов на поверхности полимера. Были также идентифицированы катаболические гены, ответственные за биодеградацию различных соединений (ксенобиотиков), гены транспорта и утилизации лактата, а также нескольких ферментов, которые, возможно, принимают участие также в деградации многих органических загрязнителей.
Как было указано выше, некоторые микромицеты принимают участие в разложении ПЛ, хотя работ, посвященных роли грибов в данном процессе, меньше, чем посвященных бактериям [69]. Среди микромицетов, способных к деградации ПЛ, можно отметить представителей родов Aspergillus, Penicillium и Parengyodontium (Tritirachium) из микробных сообществ почвы и компоста [70, 81]. Степень и скорость деградации ПЛ грибами во многом определяется температурой среды, а также кристалличностью самого полимера [82, 83].
В ряде работ изучали инокуляцию отдельными микроорганизмами для ускорения разложения ПЛ в компосте [75, 76]. Geobacillus thermoleovorans был выделен и идентифицирован как микробный штамм из компоста, способный разлагать ПЛ при 58°C [75]. В работе Паттанасуттичонлакул с соавт. [76] микробный консорциум осадка сточных вод молочного производства (преобладали представители рода Actinomadura) и чистая культура Pseudomonas geniculata при совместной инокуляции и по отдельности ускоряли разложение ПЛ при выдерживании в почве.
Калита и соавт. [10] показали, что разложение ПЛ ускоряется в условиях компостирования при увеличении содержания источников азота для роста микробного сообщества. В работе Бунлуксир и соавт. [77] показано значительное ускорение разложения ПЛ: потеря массы ПЛ до 99% наблюдалась после 60 сут выдерживания в почве при использовании штамма Pseudomonas geniculata и внесении соевого экстракта в качестве добавки. Быстрое разложение поли(L-лактида) смешанной культурой микроорганизмов компоста наблюдали авторы в работе [78]: после 5 недель пленки фрагментировались до мелкого порошка.
Ферменты, участвующие в деструкции ПЛ. Биодеградация сложных биополимеров может осуществляться двумя типами ферментов: внеклеточными ферментами, которые расщепляют длинноцепочечные полимеры на короткие олигомеры или мономеры, которые впоследствии могут потребляться клетками микроорганизмов, и внутриклеточными ферментами, которые дополнительно расщепляют небольшие олигомерные транспортируемые в клетку единицы субстрата [62].
К микробным ферментам, потенциально участвующим в деградации ПЛ, относят карбоксиэстеразы, кутиназу, липазы, сериновые протеазы (Tritirachium album, Amycolatopsis orientalis и Actinomadura sp.), табл. 1. Для деструкции полимеров микроорганизмы выделяют в окружающую среду экзоферменты – деполимеразы, обладающие гидролитической активностью, для разрушения сложноэфирной связи в структуре ПЛ. С помощью внутриклеточных ферментов происходит деградация микроорганизмами олиго- и мономерных продуктов деполимеризации ПЛ с образованием в качестве конечных продуктов воды, углекислого газа или метана [82]. Процесс индуцируется такими веществами, как желатин, эластин, фиброин, а также некоторыми белками, пептидами и аминокислотами [20]. Большинство индукторов имеют субъединицу L-аланина, схожую с субъединицей L-молочной кислоты в стереохимической позиции хирального углерода. Активность ферментов, разрушающих ПЛ, зависит от значений температуры и рН среды, кристалличности материала, стереохимии полимерной цепи [23].
Таблица 1.
Ключевые микроорганизмы и ферменты, разрушающие ПЛ
| Вид, штамм | Ссылка | Фермент |
|---|---|---|
| Бактерии | ||
| Amycolatopsis orientalis | [79, 84] | Сериновая протеаза |
| Actinomadura keratinilytica T16-1 | [76, 80] | ПЛ-разрушающие протеазы |
| Pseudomonas aeruginosa S3 | [62] | Н.о. |
| Pseudomonas geniculata | [72, 76, 77] | Н.о. |
| Brevibacillus sp. | [62] | Н.о. |
| Serratia plymuthica | [72] | Н.о. |
| Stenotrophomonas maltophilia | [73] | Н.о. |
| Bacillus smithii | [74] | Эстераза |
| Bacillus licheniformis | [11, 74] | Н.о. |
| Geobacillus thermoleovorans, Geobacillus EC-3 | [62, 74, 75] | Н.о. |
| Sphingobacterium S2 | [62] | Н.о. |
| Cryptococcus sp. штамм S-2 | [84] | Кутиназа (от 12 до 20%, гомологи с белками, принадлежащими к семейству кутиназ) |
| Грибы | ||
| Engyodontium album (Tritirachium album) |
[70, 79, 81, 84] | Протеиназа К (сериновая протеаза) |
| Aspergillus niger | [70, 72] | Липаза |
| Paecilomyces variotti | [70] | Н.о. |
| Thermomyces lanuginosus | [11, 20] | Н.о. |
| Trichoderma viride | [70] | Н.о. |
| Chaetomium globosum | [70] | Н.о. |
| Penicillium pinophilium | [70] | Н.о. |
Первым описанным в литературе ферментом, разлагающим ПЛ, была протеиназа K (сериновая протеаза), выделенная из микромицета Engyodontium album (Tritirachium album) [85]. Сообщается также, что у актинобактерий рода Amycolatopsis идентифицирована сериновая протеаза, которая может осуществлять гидролиз ПЛ в качестве субстрата и расщеплять сложноэфирную связь [79]. В геноме Pseudomonas aeruginosa S3, разрушающего ПЛ, было обнаружено 75 различных типов протеиназ, 50 эстераз и 25 различных типов липаз [62].
В работе [84] было показано, что фермент (от 12 до 20%, гомологи с белками, принадлежащими к семейству кутиназ), экстрагированный из дрожжей Cryptococcus sp. штамм S-2, значительно ускорял разложение высокомолекулярного ПЛ. Так, при концентрации фермента 0.8 мкг мл–1 полное разложение высокомолекулярного ПЛ (Mм 140 кДа) происходило за 60 ч, тогда как протеиназа K полностью гидролизовала ПЛ при концентрации 400 мкг мл–1 за 88 ч.
Эффективность компостирования. Компостирование начинается с мезофильной фазы, в которой температура постепенно повышается до 45°С. При этом, при компостировании ОФТКО, в первые 7 сут из-за деятельности молочнокислых микроорганизмов происходит закисление среды до рН 4–5 [64]. За этим этапом следует термофильная фаза, которая протекает при температурах, оптимальных для проявления микробной активности (между 45 и 70°С) и сопровождается образованием аммиака в газовых порах до 6 мг NH3 сут–1 кг–1 сухого органического вещества. К окончанию активной фазы разложения ОФТКО (первые 28 сут) рН и электропроводность повышаются до 8–9 и 2000 мкСм см–1 соответственно. Такая динамика, возможно, обеспечивает сравнительно быструю деградацию ПЛ в составе ОФТКО.
Обычно на разложение упаковки из ПЛ в компосте требуется не более 90 дней [13]. Сравнительные результаты биодеградации ПЛ при компостировании представлены в табл. 2 [13, 15, 58, 86–90]. Подавляющее большинство испытаний на биоразлагаемость ПЛ проведено в лабораторных условиях при постоянной температуре 58°С, рН ~ 7 и использовании в качестве субстрата стабильного компостного материала из растительных и животноводческих отходов. Несмотря на близкие условия результаты биоразложения сильно разнятся – от 30 до 100% за время от 17 до 98 сут. В настоящее время отсутствуют результаты исследования биодеградации ПЛ в составе пищевых отходов в контролируемых условиях промышленного компостирования.
Таблица 2.
Сравнительные результаты биодеградации ПЛ при компостировании
| Вид ПЛ, размер образца | Субстрат | Условия, метод | Время компости-рования, сут | Температура, массовая доля влаги, рН | Разложение | Источник |
|---|---|---|---|---|---|---|
| Аморфный ПЛ, нд | нд | Лабораторные, без перемешивания | 60 | 58 ± 5°C, нд | 75% | [15] |
| Аморфный ПЛ, 200 × 65 × 1 мм | Компост (3 мес) из коровьего навоза, древесной стружки и отходов корма | Лабораторные ASTM D5338, перемешивание 2 раза в неделю | 58 | 58 ± 5°C, нд | 77.8–84.2% | [13] |
| Производственные, без перемешивания | 30 | 65 ± 5°C, 63 ± 5% по массе, рН 8.5 ± 0.5 |
снижение Mм до 4100 Дa | |||
| Аморфный ПЛ, 25 × 25 × 1 мм | Компост (нет данных о составе) |
Лабораторные ISO 20200, без перемешивания | 90 | 58 ± 2°С, н.д. | 100% | [86] |
| 80 ± 2°С, н.д. | 100% | |||||
| Аморфный ПЛ + галлуазит (нанотрубки),
1–5% по массе, 10 × 5 × 1 мм |
Смесь листьев, древесной щепы, травы, почвы, компоста |
Лабораторные, модель бурта, перемешивание каждые 7 сут в первые 42 сут | 90 | 34–65 °С (max 72°C), 50–60% по массе, 6.7 рН |
90% | [87] |
| 34–80°С (max 87°C), 50–60% по массе, 6.7 рН |
100% | |||||
| Аморфный ПЛ + водоросли, 5% по массе, пластины толщиной 0.45мм |
Пищевые отходы | Лабораторные ASTM D5338, без перемешивания | 98 | 58 ± 5°C, нд | 90% | [88] |
| Аморфный ПЛ + лигнин 1–3% по массе, пленка 25 × 25мм | Компост, опилки, корм для кроликов, крахмал, сахара, масла и мочевина | Лабораторные ISO 20200, с перемешиванием | 17 | 58 ± 5°C, 50% по массе, нд |
90% | [58] |
| Аморфный ПЛ + тальк/мел, 30 × 60 × 2 мм | Компост (нд. о составе) | Лабораторные ISO 16929, с перемешиванием | 84 | 60°C, нд, >5 рН |
30–40%* | [89] |
| Аморфный ПЛ + LLDPE**, 1–5% по массе, пластины толщиной 0.45мм | Шлам бумажной фабрики, листья, навоз коровий, опилки | Лабораторные ASTM D5338, с перемешиванием | 90 | 58 ± 5°C, 60% по массе, нд |
50–80% | [90] |
Наряду с так называемым термофильным компостированием, нередки случаи дальнейшего повышения температуры саморазогрева до 80°С и выше в течение 5–7 сут и более, что является критерием отнесения процесса к гипертермофильному компостированию (ГТК) [91]. Некоторые исследователи считают, что ГТК способствует более быстрому созреванию компоста, удержанию азота в системе, а также в большей степени снижает содержание антибиотиков и микропластика в отходах [92–94]. Запуск такого процесса с внесением культур гипертермофильных микроорганизмов показан в работе [95].
Компостирование приводит к значительному снижению молекулярной массы ПЛ (рис. 2). Гил-Кастелл с соавт. [86] показали уменьшение Mм ПЛ с 57 000 до 5000–10 000 Да. Причем это снижение было обнаружено уже на 15 сут нахождения ПЛ в компосте как при 58°C, так и при 80°C. Последующая выдержка не приводила к дальнейшему снижению Mм вплоть до полного разрушения образцов. Такое поведение характерно для материалов на основе ПЛ, подвергающихся биодеградации при температуре выше температуры стеклования, которая также усиливается за счет более высокой активности молекул воды. Температура является решающим фактором для снижения Mм. Более высокие температуры увеличивают как подвижность полимерных цепей, так и активность молекул воды, что приводит к усиленному гидролизу полимеров за счет разрыва сложноэфирной связи. Между тем, если такую сверхвысокую температуру (>80°С) отходы приобретают при компостировании в результате естественного микробного экзотермического разложения, то это может быть действенным инструментом ускорения биодеградации ПЛ.

ASTM D5338 и ISO 14855 являются основными стандартами, которые используют для измерения аэробного биоразложения пластиковых материалов в условиях компостирования путем подсчета, выделяющегося CO2 [15]. Определение степени дезинтеграции пластиковых материалов в лабораторных моделируемых условиях компостирования проводятся согласно ISO 20200. Согласно этому методу, используемый компост должен быть от 2 до 4 мес зрелости с рН 7–9. При испытании поддерживается постоянная температура 58 ± 2°С. В то же время условия при реальном компостировании значительно отличаются от условий испытаний [64, 65]: сукцессия микробного сообщества, рН меняется от 4–5 до 8–9, температура от 20 до 75°С. Так, по мнению Кале с соавт. [13], время биодеградации в смоделированных и реальных условиях промышленного компостирования может сильно различаться. По сведениям Тосин и соавт. [96], методы испытаний с использованием зрелого компоста в качестве субстрата могут не соответствовать скорости биодеградации, происходящей в свежих отходах.
Следует учитывать также, что разложение термоустойчивых кристаллических ПЛ протекает медленнее, чем аморфных [20, 97]. При этом скорость ферментативной деградации в 7 раз ниже для высококристаллического ПЛ по сравнению с аморфными образцами. Таким образом, улучшение физических свойств ПЛ, с одной стороны, расширяет области применения этого материала, с другой стороны, ухудшает биоразлагаемость и перспективу утилизации отработанной массы полимера.
***
В обзоре обобщены результаты современных исследований по биодеградации ПЛ. Анализ литературных данных показал, что изменение или химическая модификация ПЛ его молекулярных и структурных характеристик и материалов на его основе, природы вводимых функциональных добавок и изменения условий разложения обуславливают возможность изменять скорость деградации биопластика в широких пределах. При разработке новых подходов, которые направлены на улучшение потребительских свойств биопластика и их сохранение в течение всего срока службы в условиях окружающей среды необходимо находить баланс между обеспечением сохранения и улучшения свойств материала и необходимостью быстрой деградации после его использования, когда биопластик становится отходом.
Детальное изучение процесса деградации структурно различных ПЛ и композитов на его основе представляется актуальным, поскольку открывает возможности регулирования скорости деградации изделий, изготовленных из ПЛ, и расширения областей их применения. Разработка биопластиков с контролируемым периодом разложения остается приоритетной задачей.
Компостирование является наиболее действенным способом быстрой деградации ПЛ в составе пищевых отходов. Динамика изменения физико-химических и биологических свойств пищевых отходов при компостировании обуславливает достаточно высокие скорости гидролиза и микробной деградации ПЛ. В отличие от стандартных, в реальных условиях ПЛ претерпевает воздействие меняющихся величин рН (от 3–5 до 8–9), высоких температур 65–75°С, высоких концентраций аммиака и широкого спектра различных микроорганизмов и синтезируемых ими ферментов. Все эти факторы влияют на деструкцию полимера и обуславливают совершенно другие скорости процесса и возможные механизмы. Также необходимы дополнительные исследования влияния увеличения содержания ПЛ в составе отходов на биологические и физико-химические параметры процесса и качество компоста, получаемого в коммерческих потоках компостирования.
Список литературы
Kumagai Sh., Nakatani J., Saito Y., Fukushima Y., Yoshioka T. // J. Japan Petroleum Institute. 2020. V. 63. № 6. P. 345–364.
Geyer R., Jambeck J.R., Law K.L. // Sci. Adv. 2017. V. 3. № 7. Art. 1700782. https://doi.org/10.1126/sciadv.1700782
Chamas A., Moon H., Zheng J., Qiu Y., Tabassum T., Jang J.H., Abu-Omar M., Scott S.L., Suh S. // ACS Sustainable Chem. Eng. 2020. V. 8. P. 3494–3511.
Teixeira S., Eblagon K.M., Miranda F., R. Pereira M.F., Figueiredo J.L. // J. Carbon Research. 2021. V. 7. P. 42.
Arpia A.A., Chen W.H., Ubando A.T., Naqvi S.R., Culaba A.B. // J. Hazardous Materials. 2021. V. 418. Art. 126381. https://doi.org/10.1016/j.jhazmat.2021.126381
Ivleva N.P. // Chem. Rev. 2021. V. 121. P. 11886–11936.
Friedrich D. // Sustainable Development. 2021. V. 29. P. 915–929.
Shaikh S., Yaqoob M., Aggarwal P. // Current Research in Food Science. 2021. V. 4. P. 503–520.
Tsuji H., Daimon H., Fujie K. // Biomacromolecules. 2003. V. 4. P. 835–840.
Kalita N.K., Damare N.A., Hazarika D., Bhagabati P., Kalamdhad A., Katiyar V. // Environmental Challenges. 2021. V. 3. Art. 100067. https://doi.org/10.1016/j.envc.2021.100067
Al Hosni A.S., Pittman J.K., Robson G.D. // Waste Manag. 2019. V. 97. P. 105–114.
Narancic T., Verstichel S., Reddy Chaganti S., Morales-Gamez L., Kenny S.T., De Wilde B., Babu Padamati R., O’Connor K.E. // Environ. Sci. Technol. 2018. V. 52. P. 10441–10452.
Kale G., Auras R., Singh S.P. // Packag. Technol. Sci. 2007. V. 20. P. 49–70.
Mysiukiewicz O., Barczewski M., Skórczewska K., Matykiewicz D. // Polymers. 2020. V. 12. № 6. Art. 1333. https://doi.org/10.3390/polym12061333
Castro-Aguirre E., Iñiguez-Franco F., Samsudin H., Fang X., Auras R. // Adv. Drug Delivery Rev. 2016. V. 107. P. 333–366.
Musioł M., Sikorska W., Adamus G., Janeczek H., Richert J., Malinowski R., Jiang G., Kowalczuk M. // Waste Manag. 2016. V. 52. P. 69–76.
Cameron R.E., Kamvari-Moghaddam A. // In: Durability and Reliability of Medical Polymers. / Eds. M. Jenkins, A. Stamboulis. Woodhead Publishing, 2012. P. 96–118. ISBN: 9780857096517.
Kaushal J., Khatri M., Arya Sh.K. // Cleaner Engineering and Technology. 2021. V. 2. Art. 100083. https://doi.org/10.1016/j.clet.2021.100083
Cucina M., de Nisi P., Tambone F., Adani F. // Bioresour. Technol. 2021. V. 337. Art. 125459. https://doi.org/10.1016/j.wasman.2021.08.016
Karamanlioglu M., Preziosi R., Robson G.D. // Polym. Degrad. Stab. 2017. V. 137. P. 122–130.
Van Nostrum C.F., Veldhuis T.F.J., Bos G.W., Hennink W.E. // Polymer. 2004. V. 45. P. 6779–6787.
Tokiwa Y., Calabia B.P., Ugwu C.U., Aiba S. // Int. J. Mol. Sci. 2009. V. 10. P. 3722–3742.
Zaaba N.F., Jaafar M. // Polym. Eng. Sci. 2020. V. 60. P. 2061–2075.
Richert A., Dąbrowska G.B. // Int. J. Biol. Macromol. 2021. V. 176. P. 226–232.
Miao Yu., Cui H., Dong Zh., Ouyang Yi, Li Yi., Huang Q., Wang Z. // ACS Omega. 2021. V. 6. P. 29254–29266.
Tripathi N., Misra M., Mohanty A.K. // ACS Eng. 2021. V. 1. P. 7–38.
Von Burkersroda F., Schedlb L., Gӧpferich A. Biomaterials. 2002. V. 23. P. 4221–4231.
Tsuji H., Ikarash K., Fukuda N. // Polym. Degrad. Stab. 2004. V. 84. P. 515–523.
Lee W.K., Gardella J.A. // Langmuir. 2000. V. 16. P. 3401–3406.
Tsuji H., Suzuyoshi K. // Polym. Degrad. Stab. 2002. V. 75. P. 347–355.
Tsuji H., Suzuyoshi K. // Polym. Degrad. Stab. 2002. V. 75. P. 357–365.
Mitchell M.K., Hirt D.E. // Polymer Engineering & Science. 2014. V. 55. № 7. P. 1652–1660.
Iñiguez-Franco F., Auras R., Burgess G., Holmes D., Fang X., Rubino M., Soto-Valdez H. // Polymer. 2016. V. 99. P. 315–323.
Hirao K., Nakatsuchi Y., Ohara H. // Polym. Degrad. Stab. 2010. V. 95. № 6. P. 925–928.
Elsawy M.A., Kim K.-H., Park J.-W., Deep A. // Renew. Sust. Energ. Rev. 2017. V. 79. P. 1346–1352.
De Jong S., Arias E., Rijkers D.T., van Nostrum C., Hennink W. // Polymer. 2001. V. 42. P. 2795–2802.
Yoon S.-D., Kwon Y.-S., Lee K.-S. // Int. Neurourol. J. 2017. V. 21. P. 48–54.
Rezwan K., Chen Q.Z., Blaker J.J., Boccaccini A.R. // Biomaterials. 2006. V. 27. № 18. P. 3413–3431.
Karamanlioglu M., Robson G.D. // Polym. Degrad. Stab. 2013. V. 98. P. 2063–2071.
Calmon A., Guillaume S., Bellon-Maurel V., Feuilloley P., Silvestre F. // J. Polym. Environ. 1999. V. 7. P. 157–166.
Scoponi G., Guzman-Puyol S., Caputo G., Ceseracciu L., Athanassiou A., Heredia-Guerrero J.A. // Polymer. 2020. V. 193. Art. 122371. https://doi.org/10.1016/j.polymer.2020.122371
Middleton J.C., Tipton A.J. // Biomaterials. 2000. V. 21. P. 2335–2346.
Oyama H.T., Kimura M., Nakamura Y., Ogawa R. // Polym. Degrad. Stab. 2020. V. 178. Art. 109216. https://doi.org/10.1016/j.polymdegradstab.2020.109216
Höglund A., Hakkarainen M., Edlund U., Albertsson A.-C. // Langmuir. 2010. V. 26. P. 378–383.
Li P., Zhu X., Kong M., Lv Y., Huang Y., Yang Q., Li G. // Intern. J. Biol. Macromol. 2021. V. 183. P. 222–234.
Rheinberger T., Wolfs J., Paneth A., Gojzewski H., Paneth P., Wurm F.R. // J. Am. Chem. Soc. 2021. V. 143. P. 16673–16681.
Raquez J.-M., Habibi Y., Murariu M., Dubois Ph. // Progress in Polymer Science. 2013. V. 38. P. 1504–1542.
Rincón-Iglesias M., Salado M., Lanceros-Mendez S., Lizundia E. // Polymer. 2022. V. 249. Art. 124804. https://doi.org/10.1016/j.polymer.2022.124804
Haryńska A., Janik H., Sienkiewicz M., Mikolaszek B., Kucińska-Lipka J. // ACS Sustainable Chem. Eng. 2021. V. 9. P. 6923–6938.
Zhang H.Y., Jiang H.B., Kim Ji-E., Zhang Sh.X., Kim K.-M., Kwon J.-S. // J. Mech. Behav. Biomed. Mater. 2020. V. 112. Art. 104061. https://doi.org/10.1016/j.jmbbm.2020.104061
El Assimi T., Blažic R., Vidović E., Raihane M., El Meziane A., Baouab M.H. V., Khouloud M., Beniazza R., Kricheldorf H., Lahcini M. // Progress in Organic Coatings. 2021. V. 156. Art. 106255. https://doi.org/10.1016/j.porgcoat.2021.106255
Chen H., Wang Y., Chen J., Yang J., Zhang N., Huang T., Wang Y. // Polym. Degrad. Stab. 2013. V. 98. № 12. P. 2672–2679.
Olewnik-Kruszkowska E., Burkowska-But A., Tarach I., Walczak M., Jakubowska E. // International Biodeterioration & Biodegradation. 2020. V. 147. Art. 104840. https://doi.org/10.1016/j.ibiod.2019.104840
Khavpachev M., Trofimchuk E., Nikonorova N., Garina E., Moskvina M., Efimov A., Demina V., Bakirov A., Sedush N., Potseleev V., Cherdyntseva T., Chvalun S. // Macromolecular Materials and Engineering. 2020. V. 305. № 7. P. 2000163.
Zhang Y., Zale S., Sawyer L., Bernstein H. // J. Biomed. Mater. Res. 1997. V. 34. № 4. P. 531–538.
Kakanuru P., Pochiraju K. // Additive Manufacturing. 2020. V. 36. Art. 101529. https://doi.org/10.1016/j.addma.2020.101529
Gorrasi G., Sorrentino A., Pantani R. // J. Polymers and the Environment. 2015. V. 23. № 3. P. 316–320.
Cavallo E., He X., Luzi F., Dominici F., Cerrutti P., Bernal C., Foresti M.L., Torre L., Puglia D. // Molecules. 2021. V. 26. Art. 126. https://doi.org/10.3390/molecules26010126
Su S., Kopitzky R., Tolga S., Kabasci S. // Polymers. 2019. V. 11. № 7. Art. 1193. https://doi.org/10.3390/polym11071193
Ножевникова А.Н., Миронов В.В., Бочкова Е.А., Литти Ю.В., Русскова Ю.И. // Прикл. биохимия и микробиология. 2019. Т. 55. № 3. С. 211–221.
Chinaglia S., Tosin M., Degli-Innocenti F. // Polym. Degrad. Stab. 2018. V. 147. P. 237–244.
Satti S.M., Castro-Aguirre E., Shah A.A., Marsh T.L., Auras R. // International J. Molecular Sciences. 2021. V. 22. № 14. Art. 7385. https://doi.org/10.3390/ijms22147385
Котова И.Б., Тактарова Ю.В., Цавкелова Е.А., Егорова М.А., Бубнов И.А., Малахова Д.В., Ширинкина Л.И., Соколова Т.Г., Бонч-Осмоловская Е.А. // Микробиология. 2021. Т. 90. № 6. С. 627–659.
Mironov V., Vanteeva A., Sokolova D., Merkel A., Nikolaev Y. // Microorganisms. 2021. V. 9. Art. 1877. https://doi.org/10.3390/microorganisms9091877
Mironov V., Vanteeva A., Merkel A. // Agronomy. 2021. V. 11. Art. 928. https://doi.org/10.3390/agronomy11050928
Auras R., Harte B., Selke S. // Macromol. Biosci. 2004. V. 4. P. 835–864.
Ghorpade V.M., Gennadios A., Hanna M. // Bioresour. Technol. 2001. V. 76. P. 57–61.
Trofimchuk E.S., Moskvina M.A., Nikonorova N.I., Efimov A.V., Garina E.S., Grokhovskaya T.E. et al. // European Polymer J. 2020. V. 139. Art. 110000. https://doi.org/10.1016/j.eurpolymj.2020.110000
Qi X., Ren Y., Wang X. // International Biodeterioration & Biodegradation. 2017. V. 117. P. 215–223.
Saadi Z., Rasmont A., Cesar G., Bewa H., Benguigui L. // J. Polymers and the Environment. 2011. V. 20. № 2. P. 273–282.
Richert A., Kalwasińska A., Brzezinska M.S., Dąbrowska G.B. // Int. J. Mol. Sci. 2021. V. 22. № 19. Art. 10228. https://doi.org/10.3390/ijms221910228
Janczak K., Dąbrowska G.B., Raszkowska-Kaczor A., Kaczor D., Hrynkiewicz K., Richert A. // International Biodeterioration & Biodegradation. 2020. V. 155. Art. 105087. https://doi.org/10.1016/j.ibiod.2020.105087
Yuan J., Ma J., Sun Y., Zhou T., Zhao Y., Yu F. // Science of the Total Environment. 2020. V. 715. Art. 136968. https://doi.org/10.1016/j.scitotenv.2020.136968
Nair N.R., Sekhar V.C., Nampoothiri K.M. // Indian J Microbiol. 2016. V. 56. P. 59–63.
Castro-Aguirre E., Auras R., Selke S., Rubino M., Marsh T. // Polym. Degrad. Stab. 2018. V. 154. P. 46–54.
Pattanasuttichonlakul W., Sombatsompop N., Prapagdee B. // International Biodeterioration & Biodegradation. 2018. V. 132. P. 74–83.
Boonluksiri Y., Prapagdee B., Sombatsompop N. // Polym. Degrad. Stab. 2021. V. 188. Art. 109562. https://doi.org/10.1016/j.polymdegradstab.2021.109562
Hakkarainen M., Karlsson S., Albertsson A.-C. // Polymer. 2000. V. 41. P. 2331–2338.
Hedstrom L. // Chem. Rev. 2002. V.102. № 12. P. 4501–4524.
Sukkhum S., Tokuyama S., Tamura T., Kitpreechavanich V. // J Gen Appl Microbiol. 2009. V. 55. № 6. P. 459–467.
Tertyshnaya Y.V., Shibryaeva L.S., Levina N.S. // Fibre Chemistry. 2020. V. 52. № 1. P. 43–47.
Lipsa R., Tudorachi N., Darie-Nita R.N., Oprică L., Vasile C., Chiriac A. // Int. J. Biol. Macromol. 2016. V. 88. P. 515–526.
Antipova T.V., Zhelifonova V.P., Zaitsev K.V. et al. // J. Polym. Environ. 2018. V. 26. P. 4350–4359.
Masaki K., Kamini N.R., Ikeda H., Iefuji H. // Appl. Environ. Microbiol. 2005. V. 71. № 11. P. 7548–7550.
Williams D.F. // Engineering in Medicine. 1981. V. 10. № 1. P. 5–7.
Gil-Castell O., Andres-Puche R., Dominguez E., Verdejo E., Monreal L., Ribes-Greus A. // Polym. Degrad. Stab. 2020. V. 180. Art. 109288. https://doi.org/10.1016/j.polymdegradstab.2020.109288
Czarnecka-Komorowska D., Tomasik M., Thakur V.K., Kostecka E., Rydzkowski T., Jursa-Kulesza J., Bryll K., Mysłowski J., Gawdzińska K. // Industrial Crops and Products. 2022. V. 183. Art. 114974. https://doi.org/10.1016/j.indcrop.2022.114974
Kalita N.K., Hazarika D., Kalamdhad A., Katiyar V. // Bioresource Technology Reports. 2021. V. 15. Art. 100705. https://doi.org/10.1016/j.biteb.2021.100705
Tolga S., Kabasci S., Duhme M. // Polymers. 2021. V. 13. № 1. Art. 10. https://doi.org/10.3390/polym13010010
Kalita N.K., Bhasney S.M., Kalamdhad A., Katiyar V. // J. Environmental Management. 2020. V. 261. Art. 110211. https://doi.org/10.1016/j.jenvman.2020.110211
Wang S., Wu Y. // Processes. 2021. V. 9. Art. 675. https://doi.org/10.3390/pr9040675
Chen X.-H., Huang Y.-H., Lü H., Mo C.-H., Xiang L., Feng N.-X., Zhao H.-M., Li H., Li Y.-W., Cai Q.-Y. // Bioresour. Technol. 2022. V. 347. Art. 126702. https://doi.org/10.1016/j.biortech.2022.126702
Liu X., Hou Y., Li Z., Yu Z., Tang J., Wang Y., Zhou S. // Waste Manag. 2020. V. 103. P. 342–351.
Liao H., Zhao Q., Cui P., Chen Z., Yu Z., Geisen S., Friman V.-P., Zhou S. // Environment International. 2019. V. 133. Art. 105203. https://doi.org/10.1016/j.envint.2019.105203
Yu Z., Tang J., Liao H., Liu X., Zhou P., Chen Z., Rensing C., Zhou S. // Bioresour. Technol. 2018. V. 265. P. 146–154.
Tosin M., Degli-Innocenti F., Bastioli C. // J. Environ. Polym. Degr. 1996. V. 4. P. 55–63.
Saeidlou S., Huneault M.A., Li H., Park C.B. // Progress in Polymer Science. 2012. V. 37. P. 1657–1677.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология