

Прикладная биохимия и микробиология, 2022, T. 58, № 1, стр. 90-104
Методические аспекты исследования жирных кислот в биологических образцах
В. А. Зотов 1, *, В. В. Бессонов 1, Д. В. Рисник 1
1 Федеральный исследовательский центр питания, биотехнологии и безопасности пищи
109240 Москва, Россия
* E-mail: arkont-87@yandex.ru
Поступила в редакцию 10.01.2021
После доработки 07.07.2021
Принята к публикации 02.09.2021
- EDN: SKKQVR
- DOI: 10.31857/S0555109922010111
Аннотация
В обзоре рассмотрены основные современные методы определения жирных кислот (ЖК) в биологических объектах, а также менее распространенные, но перспективные методы. Описываются и сравниваются преимущества, недостатки и перспективы этих методов. Особое внимание уделено особенностям подготовки биологических образцов, таким как экстракция липидов и дериватизация жирных кислот. Рассматриваются методические аспекты определения жирных кислот методами газовой хроматографии (ГХ) и высокоэффективной жидкостной хроматографии (ВЭЖХ), особенности использования различных типов неподвижных фаз используемых для хроматографического разделения ЖК. Проанализированы возможности использования капиллярного электрофореза (КЭ) и спектроскопии ядерного магнитного резонанса (ЯМР) для анализа жирных кислот, особые требования и ограничения этих методов при работе с биоматериалом.
Жирные кислоты (ЖК) – ациклические одноосновные карбоновые кислоты, содержащиеся в жирах, маслах и восках биологического происхождения. В организме человека ЖК присутствуют в основном в виде продуктов этерификации, представленных триглицеридами, фосфолипидами и эфирами холестерина [1]. ЖК, как правило, содержат неразветвленную цепь из атомов углерода С4-С24, (включая углерод карбоксильной группы) и могут быть как насыщенными, так и ненасыщенными. Разветвленные жирные кислоты по сравнению с жирными кислотами с неразветвленной цепью в составе липидов растений и животных встречаются реже. В бактериях довольно часто встречаются монометил-разветвленные жирные кислоты [2].
По степени ненасыщенности ЖК подразделяют на насыщенные, в которых отсутствуют кратные связи, мононенасыщенные, содержащие одну двойную связь, и полиненасыщенные, в структуре которых содержится две и более кратные связи [3].
Ряд полиненасыщенных жирных кислот (ПНЖК), необходимых животным и человеку, но которые не образуются в ходе биосинтеза в их организмах, относят к незаменимым. К ним относятся 18-атомные кислоты семейств n-6 и n-3 (омега-6 и омега-3): линолевая кислота с двумя двойными связями 18:2n-6 и альфа-линоленовая кислота с тремя двойными связями 18:3n-3 [4].
Биологическая роль ЖК заключается в их участии в формировании клеточных мембран, внутриклеточных структур и органелл, а также в энергетическом обмене. Помимо этого, они участвуют в синтезе биологически активных соединений, таких как эйкозаноиды [5].
Изучение содержания ЖК и их метаболических профилей важно для лабораторно-клинической диагностики [6]. Например, изучение маркеров метаболизма ЖК применяется в диагностике рисков развития сердечно-сосудистых заболеваний и метаболического синдрома. Масштабные клинические и эпидемиологические исследования больших групп пациентов (более десяти тысяч человек) показали, что повышенное потребление омега-3 ПНЖК существенно снижает риск сердечно-сосудистых заболеваний у здоровых людей и на 35% снижает смертность среди людей, перенесших эти заболевания [7]. По современным представлениям влияние эйкозапентаеновой кислоты на сердечно-сосудистую систему связано с действием ее производных – эйкозаноидов, которые являются ингибиторами тромбообразования, способствуют расширению просвета сосудов, участвуют в регуляции иммунного ответа [8–10]. Возможные механизмы кардиопротекторных эффектов омега-3 ПНЖК заключаются в понижении уровней триглицеридов, ослаблении аритмий, снижении кровяного давления, предотвращении агрегации кровяных телец [11, 12].
Многие исследования связывают аномалии в метаболизме ЖК с развитием некоторых видов онкологических заболеваний. Однако общие тенденции изменений липидных профилей плазмы крови у больных имеют ряд противоречий, что связано с различием в механизмах синтеза и использования липидов при различных разновидностях раковых заболеваний [13, 14].
Нарушение нормальных процессов метаболизма ЖК может являться причиной хронических нервно-мышечных заболеваний, которые могут сопровождаться тяжелыми осложнениями, в том числе со смертельным исходом, включая миопатию, прогрессивную кардиомиопатию, рецидивирующую энцефалопатию и т.д.
В эпидемиологических исследованиях установлена прямая корреляция между недостаточной обеспеченностью омега-3 ПНЖК и развитием психических и нейродегенеративных заболеваний. Несмотря на то, что в настоящее время не найдено средств лечения одного из самых распространённых видов деменции – болезни Альцгеймера [7, 15], имеются достоверные данные, свидетельствующие о возможности снижения риска развития и даже замедления развития этого заболевания при дополнительном употреблении докозагексаеновой кислоты [16].
Чтобы раскрыть биологическую значимость ЖК в заболеваниях, необходимо дополнительное изучение роли и механизмов их метаболизма, совершенствование методик их количественного определения в биологических образцах, что позволит внести значительный вклад в практику лабораторно-клинической диагностики. Разработка и совершенствование надежных процедур определения ЖК в свободных и этерифицированных формах позволит усовершенствовать методы диагностики метаболического синдрома, наследственных и онкологических заболеваний, а также осуществить выявление отклонений у людей, находящихся в зоне риска на ранней стадии.
Изучение содержания ЖК в биологических объектах является сложной аналитической задачей из-за большого разнообразия веществ, в состав которых входят ЖК, и из-за сложного характера матрицы (посторонних компонентов образца). В настоящее время в основном используются хроматографические методы: газовая хроматография (ГХ), высокоэффективная жидкостная хроматография (ВЭЖХ), сверхкритическая флюидная хроматография (СФХ), а также комбинированные методы – масс-спектрометрия в сочетании с различными способами разделения, спектроскопия ядерного магнитного резонанса (ЯМР-спектроскопия) [17–19].
Газовая хроматография. Среди инструментальных методов анализа ЖК наиболее распространенным является ГХ с тем или иным способом детектирования. Наиболее часто используется пламенно-ионизационный детектор. Его основными преимуществами являются низкие пределы обнаружения (на уровне нг/мл) и широкий линейный диапазон [20].
Исторически это был первый хроматографический метод, использованный для анализа ЖК [17]. Известно, что основные требования, предъявляемые к аналитам при использовании этого метода разделения, – летучесть, термостабильность и инертность. Поскольку ЖК характеризуются термолабильностью, низкой летучестью, наличием полярных функциональных групп, а также легко поддаются полимеризации и дегидратации [1, 21, 22], что препятствует их прямому качественному и количественному определению методами ГХ [23]. Такие свойства обуславливают необходимость проведения предварительной дериватизации перед проведением анализа. Дериватизация позволяет повысить летучесть ЖК, а также улучшает эффективность их разделения и повышает чувствительность определения [24]. В литературе описано множество подходов к дериватизации ЖК. Чаще всего легколетучие производные ЖК получают путем одностадийной кислотной или щелочной переэтерификации.
Кислотно-катализируемые реакции. В случае кислотно-катализируемых реакций переэтерификации в прошлом широкое распространение получило использование растворов соляной (HCl) или серной (H2SO4) кислот в спиртах [25]. Обычно применяют 2–10% растворы в метаноле (CH3OH). Однако в современной практике чаще используют различные кислоты Льюиса: например, фторид бора (BF3) и хлорид алюминия (AlCl3) [26, 27]. Общая схема кислотно-катализируемых реакций дериватизации липидов представлена на рис. 1а.
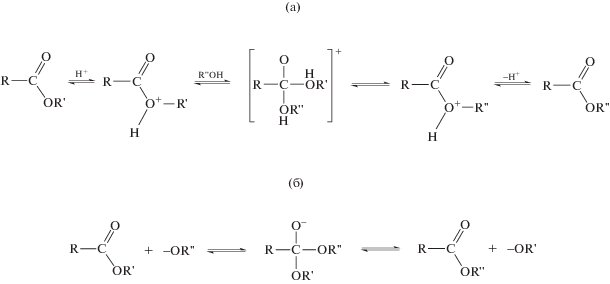
Применение трифторида бора для определения ЖК описано еще в 1964 г. [28]. В настоящее время есть множество данных о применении BF3 при анализе биологических образцов, в том числе для трансметилирования липидов в плазме крови человека [29], эритроцитах и сыворотке крови [23, 30], мышечной ткани коров и овец [31, 32], печени рыб [33], бактериях [34]. BF3 в метаноле позволяет конвертировать кислоты всех классов липидов (включая свободные ЖК) в метиловые эфиры [27]. Дериватизация проводится при 100°С. Этерификация свободных ЖК в таких условиях происходит за 2 мин, переэтерификация фосфолипидов – за 10 мин. Более длительного времени требует переэтерификация триглицеридов и сфигномиелина – 30 и 90 мин соответственно [28]. Выход реакции этерификации, выполненной таким образом для биологических объектов, достигает 99% [28, 35]. Применение BF3 для анализа ПНЖК плазмы крови и мембран эритроцитов описано в работе Вогнилда [36]. Экстракцию липидов проводили по методу Фолча (смесью хлороформа и метанола). Применение трифторида бора для этерификации свободных ЖК, полярных эфиров и триглицеридов возможно в комбинации с гидроксидом натрия. Метод продемонстрировал высокую эффективность при анализе общего содержания ЖК в плазме крови человека и тканях печени крысы. Эффективность этерификации превышает 85% для всех классов липидов [26]. Полученные метиловые эфиры экстрагируют с использованием н-гексана (или н-пентана) с последующей промывкой водой. Однако, такой способ является достаточно жестким. В целях предотвращения окисления ненасыщенных ЖК реакцию проводят в присутствии антиоксидантов.
В современной практике анализа ПНЖК в качестве антиоксиданта часто используется 2,6-дитретбутил-4-метилфенол (бутилгидрокситолуол или BHT). Одним из недостатков метода является ограничение по срокам и условиям хранения раствора фторида бора [22]. Фторид бора способен реагировать с холестерином с образованием метилового эфира, который мешает дальнейшему анализу компонентов. Необходимо отметить, что используемый в качестве антиоксиданта BHT, является весьма токсичным веществом и обладает ярко выраженным канцерогенным действием [37].
Хлорид алюминия (AlCl3) также используют при определении жирнокислотного состава липидов. Этот реагент может быть использован для прямого метилирования (без предварительной экстракции жиров). По способности переэтерификации AlCl3 сравним с фторидом бора. Преимуществом метода является меньшее содержание мешающих хроматографическому анализу продуктов. Недостаток – неспособность метилировать свободные ЖК [25].
Для прямого метилирования возможно также применение ацетилхлорида (CH3COCl). Его добавляют в пробу без предварительной подготовки с последующей экстракцией метиловых эфиров н-гексаном или н-пентаном. Система метанол-ацетилхлорид применима для дериватизации широкого спектра липидов, при этом ацетилхлорид отличается более высокой эффективностью дериватизации по сравнению с соляной кислотой и фторидом бора [38].
Растворы соляной или серной кислот в метаноле, как и BF3, быстро метилируют свободные ЖК и могут быть использованы для переэтерификации всех классов липидов, содержащихся в биологическом материале, в том числе, например, в плазме крови человека [39, 40], мышцах и печени животных [41]. Реакцию обычно проводят при 60–70°С в течение часа.
Одним из общих недостатков кислотных катализаторов является способность конвертировать плазмалогены, присутствующие в биологических образцах, в диметилацетали, которые могут затруднять хроматографическое разделение метиловых эфиров ЖК [42]. Следует отметить, что диметилацетали можно отличить от жирных кислот по масс-спектрам, поэтому их присутствие не мешает анализу при использовании масс-спектрометрического детектирования.
Основно-катализируемые реакции. Эти реакции (рис. 1б) проходят в более мягких условиях. Наиболее часто применяемыми способами дериватизации являются получение алкильных производных (метиловых, этиловых и изопропиловых) в результате взаимодействия растворов алкоголятов щелочных металлов в соответствующих спиртах. Классическим и наиболее распространенным способом в клинической практике, микробиологии и анализе пищевых продуктов является метилирование [40, 43].
Для щелочного трансметилирования наиболее часто используют растворы метилата натрия (CH3ONa) или гидроксида калия (KOH) в метаноле. Реакция с метилатом происходит достаточно быстро даже при комнатной температуре, поэтому нет необходимости в использовании антиоксидантов [31]. Особенностью этого основно-катализируемого метода дериватизации является отсутствие модификации свободных кислот и сфинголипидов в метиловые эфиры. Тем не менее, CH3ONa эффективно используется для трансметилирования липидов крови человека и мышечной ткани [31, 44]. Применение раствора KOH в метаноле для трансметилирования липидов предотвращает реакции изомеризации двойных связей в молекулах ненасыщенных кислот и образование метокси-артефактов [45, 46]. В обоих случаях не происходит высвобождение диметилацеталей из плазмалогенов, которые могут помешать хроматографическому определению.
Реже для щелочной этерификации используют производные аммония, например гидроксид тетраметиламмония (TMAH), гидроксид триметилфениламмония (TMPAH) и др. [47]. Связанные ЖК переходят в соли аммония, которые подвергаются пиролизу с получением метиловых эфиров в испарителе газового хроматографа. Преимущество этого способа заключается в том, что реакция проводится при комнатной температуре, в одну стадию без предварительной экстракции [48].
Известны способы метилирования ЖК с использованием диазометана (рис. 2) и алкилхлорформиатов [49]. Для метилирования ЖК может быть использован йодметан (CH3I). Этот метод был применен для определения состава сфинголипидов [50] и свободных ЖК в мозговой ткани. Кроме получения метильных производных липидных соединений применяют этерификацию до этил-, пропил-, изопропил-, бутил- и изобутилпроизводных [51].

Описаны методы этерификации ЖК до метиловых эфиров непосредственно в биоматериале (плазме крови, эритроцитах, тканях) без предварительной экстракции липидной фракции [30]. В этом случае не требуются такие стадии пробоподготовки, как, например, отгонка растворителя или центрифугирование, что позволяет избежать потерь в определяемых компонентах и значительно сократить время анализа. Производные, полученные после экстракции н-гексаном (или н-пентаном) и промывки водой, подвергают хроматографическому анализу.
В некоторых исследованиях приведены методы построения метаболических профилей ЖК с последовательным применением разных дериватизирующих агентов [52].
В целом, одним из главных недостатков метилирования является относительно длительное время реакции и высокая температура, что может способствовать изменению структуры определяемых кислот.
Разделение производных ЖК обычно проводят на капиллярных колонках с неподвижной фазой разной степени полярности. Среди неполярных неподвижных фаз распространены диметилполисилоксан и фенилметилполисилоксан. Часто для анализа метиловых эфиров жирных кислот (МЭЖК) используются полярные капиллярные колонки с неподвижной фазой на основе полиэтиленгликоля (ПЭГ) [18]. Это связано с тем, что капиллярные колонки с ПЭГ эффективно разделяют эфиры с изомерией углеродного скелета и кратных связей. Наиболее серьезным ограничением для ПЭГ-фазы является низкая разрешающая способность для пространственных изомеров, которые элюируются одновременно. Таким образом, эти колонки не пригодны, если требуется разделение специфичных цис-, транс-изомеров ЖК при использовании пламенно-ионизационного детектора. Дополнительную эффективность могут обеспечить капиллярные колонки с высокополярной (например, цианополисилоксановой) неподвижной фазой. Методика определения МЭЖК c использованием капиллярной колонки с цианопропильным покрытием предложена специалистами по ГХ компании “Agilent Technologies” (США). Методика позволила провести полное разделение 37-компонентной стандартной смеси МЭЖК, а использование водорода в качестве газа носителя позволило сократить время анализа до 18 мин [53].
Информацию о химической структуре анализируемых ЖК получают с помощью масс-спектрометрических (МС) детекторов. Предел обнаружения МС сравним с пределом обнаружения пламенно-ионизационного детектора. В практике анализа ЖК методом ГХ-МС помимо метилирующих агентов, для дериватизации ЖК используются и другие реагенты. Такими реагентами могут быть, например, N,O-бис(триметилсилил)трифторацетамид (BSTFA) [54], N-метил-N-(трет-бутилдиметилсилил) трифторацетамид (MTBSTFA) [55]. При реакции с MTBSTFA получают сложные эфиры трет-бутилдиметилсилила (TBDMS), преимущества которых состоят в высокой стабильности и образовании очень интенсивных пиков характеристических ионов при фрагментации и, следовательно, легко интерпретируемых масс-спектров. С использованием этого дериватизирующего агента разработан метод для одновременного определения ЖК и стеринов в биологических жидкостях. Методика была успешно применена для количественного определения 18 ЖК и некоторых стеринов включая прекурсоры холестерина в образцах слюны человека. Помимо этого методика подходит для анализа таких объектов, как моча, амниотическая жидкость, спинномозговая жидкость и может быть весьма перспективна в клинической практике [55]. Довольно мягкий способ дериватизации – реакция с бис(пентафторфенил)диметилсиланом – длится всего 15 мин. и проходит при комнатной температуре [56]. Для определения короткоцепочечных жирных кислот, являющихся маркерами многих метаболических заболеваний, предложена методика дериватизации пентафторбензилбромидом (PFBBr). При помощи этого метода определяли короткоцепочечные линейные и разветвленные жирные кислоты в фекалиях мышей. Оптимальное время реакции составило 90 мин при температуре 60°С и pH 7. Методика позволила одновременно обнаружить 8 основных короткоцепочечных ЖК с пределом обнаружения 0.244–0.977 мкмоль [57].
При определении ЖК методом ГХ-МС возможна дериватизация 3-пиридилкарбинолом с получением пиколиниловых эфиров. Спектры пиколиниловых эфиров ЖК весьма информативны, содержат интенсивные пики молекулярных ионов, позволяют достоверно определить структуру углеродной цепи кислот и положение кратных связей.
Применение этого способа дериватизации для экспрессного профилирования жирных кислот в бактериальных клетках описано в работе Куркевича с соавторами. Описанный ими метод интересен тем, что авторы использовали пиролизер в качестве термохимического микрореактора для дериватизации. Вкратце, аликвоту (5 мкл) суспензии бактериальных клеток в ацетоне помещали на термоэлемент пиролизера. Сразу после этого добавляли 1 мкл дериватизирующей смеси (10% водный раствор гидроксида натрия и 5% пиридилкарбинола), и растворители выпаривали в токе азота. Термоэлемент помещали в пиролизер по точке Кюри и проводили пиролитическую газовую хроматографию (метод исследования при котором образец подвергают пиролизу, а образовавшиеся летучие продукты разделяют в газовом хроматографе) с масс-спектрометрическим детектированием [58]. Температуры, при которых проводят разделение пиколиниловых эфиров, примерно на 50°С выше чем те, которые используют для разделения МЭЖК, так как они имеют более низкую летучесть. Необходимо отметить, что хроматографическое разделение этих производных характеризуется худшим разрешением по сравнению с методами разделения метиловых эфиров [59].
Высокоспецифичным масс-спектром обладают также производные ЖК 4.4-диметилоксазолина (DMOX). Способность к разделению у них сходна с метиловыми эфирами соответствующих кислот [60]. В случае применения пентафторфенилдиметилсилила для дериватизации предел обнаружения можно понизить до 0.05 нг/мл [61].
Жидкостная хроматография. Одним из альтернативных направлений в анализе липидов является ВЭЖХ [18]. Основное преимущество ВЭЖХ над ГХ заключается в большей чувствительности и более высокой селективности, что обусловлено разнообразием неподвижных фаз ВЭЖХ колонок. Недостатком ВЭЖХ для анализа липидов является длительность разделения и высокий расход органических растворителей.
Одна из сложностей анализа ЖК при использовании ВЭЖХ заключается в низком поглощении ими излучения в ближнем ультрафиолетовом (УФ) диапазоне, что затрудняет их прямое детектирование с помощью УФ детекторов или флуоресцентного детектора. В случае работы с этими наиболее распространенными в ВЭЖХ типами детекторов, перед анализом необходима дериватизация для введения сильных хромофорных УФ-поглощающих групп в молекулы ЖК. Дериватизирующими агентами для этого могут служить 2-бромацетофенон [62], 9-флуоренилметилхлорформиат [63], 2-нитрофенилгидразин [64].
Описан метод определения жирных кислот в семенах масличных культур с помощью флуоресцентного детектора с предколоночной дериватизацией 9-флуоренилметилхлороформиатом. Реакция дериватизации проводилась при 60°C в течение всего 10 мин. Хроматографическое разделение 14 жирных кислот (C10–C22) было достигнуто за 30 мин. Использовались длины волн возбуждения и излучения 265 и 315 нм соответственно. Метод показал хорошую чувствительность и воспроизводимость: пределы обнаружения – 0.01–0.05 мкг/мл, относительные стандартные отклонения – менее 0.27%. Этот метод был применен для количественного определения жирных кислот в кунжутном масле [63].
Дериватизация не требуется в случае применения детекторов, отклик которых не зависит от оптических свойств образца, например, детектора по светорассеянию (Evaporative Light Scattering Detector – ELSD). Этот детектор совместим с широким спектром растворителей, что обеспечивает некоторую гибкость при разработке методик. ELSD подходит для обнаружения C12C22 кислот. Кислоты с более короткой цепью слишком летучие для их уверенного детектирования таким способом [65].
Большинство разделений проводят на обращенной фазе с использованием колонок с октадецилсиликагелем (ODS, C18). Реже используются С30 и С8 фазы [50]. Фаза С8 в большей степени подходит для определения более полярных липидов, например фосфолипидов и короткоцепочечных насыщенных ЖК [66].
Хиральные неподвижные фазы, например, 3,5-динитробензоил фенилглицин (DNBPG) “Merck KGaA” (Германия), используют для разделения продуктов перекисного окисления липидов [67], разделения энантиомеров ЖК [68].
Широко распространенной группой методов в липидомике является ВЭЖХ в сочетании с масс-спектрометрией [69–71]. В последние десятилетия технический прогресс привел к существенному усовершенствованию ВЭЖХ-МС-систем, что позволило повысить чувствительность и селективность приборов. С помощью этих методов могут быть проанализированы все основные классы липидов крови человека (стерины, сложные эфиры холестерина, фосфохолины, фосфоэтаноламины, сфингомиелины, триацилглицерины, ЖК, лизофосфохолины и диацилглицерины), возросла скорость анализа, уменьшилось время на подготовку проб.
Среди методов ионизации, обычно используемых в ВЭЖХ-МС, наиболее популярны ионизация электрораспылением (ESI) и химическая ионизация при атмосферном давлении (APCI).
Для экстракции липидов из биологических матриц перед анализом методом ВЭЖХ-МС для экстракции обычно пользуются методом Фолча или Блайя–Дайера, которые считаются эталонными. Также для этой цели применяют смеси метил-трет-бутилового эфира (МТБЭ) и метанола, гексана и изопропанола, бутанола и метанола. Все эти методы экстракции демонстрируют различную степень эффективности извлечения для разных классов липидов. Известны методики анализа липидов с одностадийной экстракцией, например метанолом [72]. Метанол обладает высокой экстрагирующей способностью в отношении фосфолипидов и лизофосфохолинов. Помимо этого он вызывает денатурацию белков крови, которые затем легко удалить центрифугированием, а супернатант может быть непосредственно введен в ВЭЖХ-МС систему без дополнительной очистки. В силу своей простоты этот метод обладает очень высокой воспроизводимостью.
В литературных источниках описаны и более универсальные способы одностадийной экстракции, например с использованием смеси метанола, хлороформа и МТБЭ в соотношении 1.3 : 1 : 1 [73]. Соотношение объемов образца (100 мкл плазмы крови) и экстрагирующей смеси – 1 : 20. Экстракцию проводили в 5 мл пробирке Эппендорфа. Образцы встряхивали и выдерживали при комнатной температуре в течение 30 мин, затем повторно встряхивали и центрифугировали при 2000 оборотов в течение 10 мин. Верхний слой экстрагента подвергали хроматографированию без дополнительной очистки. Хроматографическое разделение проводили на хроматографе Agilent 1290 “Agilent Technologies” (США) на фазе С18 (колонка Zorbax Eclipse Plus RRHD 2.1 × × 150 мм, 1.8 мкм “Agilent Technologies” (США)). В качестве элюента применяли 5 мМ раствор формиата аммония в воде, в ацетонитриле и в изопропаноле. Разделение было выполнено при температуре 40°C. Детектирование проводили на квадруполь-времяпролетном масс-спектрометре Agilent 6540 “Agilent Technologies” (США) в режиме регистрации положительных ионов (напряжение на капилляре – 4000 В) с азотом в качестве газа-осушителя (температура – 250°C, скорость потока – 8.8 л/мин). Напряжение на фрагменторе, конусе, скиммере, и октаполе – 110, 500, 65, и 750 В, соответственно. Скорость сканирования – 2 ГГц. При таких условиях удалось обеспечить наилучшее соотношение сигнал/шум для стандартных образцов основных классов липидов, которые в режиме положительной ионизации были детектированы преимущественно в виде аддуктов состава [M + H]+. Еще более высокой чувствительности для ЖК (в виде аддуктов [M–HCOO]) удалось добиться в отрицательном режиме. Описанный метод экстракции обеспечивал близкую к 100% степень извлечения для девяти основных классов липидов крови человека [73].
Качественно нового уровня в нецелевой липидомике позволяет достичь сочетание ВЭЖХ с масс-спектрометрами высокого разрешения. В последнее время благодаря очень высокой разрешающей способности и точности для исследования липидных профилей все чаще используются масс-анализаторы ионно-циклотронного резонанса с преобразованием Фурье (ИЦР/ПФ). Например, в работе Ху с соавторами [74] предложен метод ВЭЖХ-ИЦР/ПФ для определения липидов в плазме крови человека и мыши. Хроматографическое разделение проводили на фазе C8 с поверхностно-пористыми частицами сорбента в градиентном режиме. В качестве элюента использовали смеси ацетонитрил–вода (60 : 40) и изопропанол–ацетонитрил (90 : 10). В одном цикле ВЭЖХ-ИЦР/ПФ было обнаружено более 160 липидов, относящихся к восьми различным классам. Метод был полностью валидирован. Предел обнаружения методики – 0.08–1.28 мкг/мл плазмы, стандартное отклонение воспроизводимости – 2.7–7.9%.
В работе Корц [75] для определения 6 ПНЖК, 14 эйкозаноидов и 3 окисленных метаболитов применяли осаждение белков и онлайн твердофазную экстракцию в сочетании с тандемным МС-детектированием. Разделение компонентов пробы проводили на фазе С18. Для точной идентификации соединений были подобраны специфические MRM-переходы (MRM – мониторинг множественных реакций). Предложенный метод отличается быстрой автоматизированной пробоподготовкой. Пределы обнаружения составили 200–1000 нг/мл для ПНЖК и 10–1000 пг/мл для их метаболитов.
В практике ВЭЖХ анализа липидов описаны методы дериватизации диметиламиноэтанолом. Например, в работе Петтинелла с соавторами [76] было предложено проводить определение ЖК в тканях атеросклеротических бляшек после их удаления с внутренней стенки артерии. Для этого сначала проводили экстракцию ЖК из матрицы с помощью смеси хлороформа и метанола, а затем полученную смесь гидролизовали с помощью 40% гидроксида калия. Далее проводили дериватизацию свободных ЖК деанолом (диметиламиноэтанол или DMEA), с добавлением йодметана. Полученные триметиламиноэтиловые эфиры ЖК определяли методом ВЭЖХ/МС-МС. Пределы обнаружения составили 4–40 нг/мл.
В настоящее время анализ липидов с помощью ВЭЖХ-МС – один из наиболее эффективных подходов для исследования самых разнообразных биологических объектов, в том числе плазмы крови и других биологических жидкостей. По совокупности таких характеристик, как чувствительность, селективность и точность определения, жидкостная хромато-масс-спектрометрия является непревзойденной техникой физико-химического анализа. Альтернативой является газовая хроматография-масс-спектрометрия (ГХ-МС), которая по-прежнему играет важную роль в липидомике. Однако методы липидного профилирования на основе ГХ-МС обычно требуют длительных процедур гидролиза и дериватизации, что может быть источником дополнительной погрешности при количественном определении. При этом для анализа неполярных липидов предпочтительнее использовать ГХ-МС.
Сверхкритическая флюидная хроматография. Сверхкритическая флюидная хроматография (СФХ) – вид хроматографии, в котором подвижной фазой служит сверхкритический флюид, – вещество при температуре и давлении выше критической точки. Свойства веществ в сверхкритическом состоянии являются промежуточными между их свойствами в газовом и жидком агрегатном состоянии. Например, при плотности близкой к плотности жидкости, они обладают гораздо более высокими коэффициентами диффузии и значительно меньшей вязкостью. Отсутствует поверхностное натяжение. В то же время их растворяющая способность намного выше, чем у газов. Контролируя температуру и давление флюида, можно в широком диапазоне изменять его свойства.
Первое упоминание об использовании сверхкритического флюида в хроматографии относится к 1962 г. Метод описан немецким химиком Клеспером [77]. Тогда он получил название “газовая хроматография высокого давления”. СФХ имеет ряд преимуществ перед ГХ и ВЭЖХ. Низкая вязкость позволяет использовать более длинные (и, соответственно, более эффективные) колонки и работать с более высокими скоростями потоков; хроматографы совместимы практически с любым типом детектора, включая, например, пламенно-ионизационный, что невозможно в ВЭЖХ. Немаловажным является экологичность, так как углекислый газ, который наиболее часто используется в качестве элюента в СФХ, не токсичен в отличие от таких растворителей, как метанол и ацетонитрил, обычно используемых в жидкостной хроматографии. В отличие от ГХ возможно разделение термолабильных и нелетучих веществ.
Такое сочетание свойств позволяет анализировать ЖК без дериватизации, поскольку не требуется обеспечивать летучесть образца, как в случае с ГХ, и не нужно вводить в молекулы аналитов хромофорные группы, как при ВЭЖХ с УФ-детектированием [78]. Следует отметить, что СФХ-систему гораздо проще сочетать с масс-селективными детекторами, так как поток подвижной фазы заметно меньше, чем в ВЭЖХ. Как и в случае ВЭЖХ-МС для ионизации предпочтительно применять электроспрей и химическую ионизацию при атмосферном давлении [79]. Также в отличие от ВЭЖХ СФХ позволяет использовать более высокие скорости потока при более низком давлении, что позволяет достичь большей эффективности и, соответственно лучшего разрешения, и одновременно сократить время анализа.
Современным трендом в СФХ-анализе биологических объектов и, в частности в липидомике, является развитие многомерной хроматографии. В многомерной хроматографии проба последовательно разделяется на двух колонках разной длины и с разными неподвижными фазами [18, 80]. Впервые применение двумерной СФХ для анализа жиров описано Хиратой в 2004 г. [81]. В работе выполнено разделение метиловых эфиров ЖК с помощью системы СКФ × СКФ с насадочными колонками и пламенно-ионизационным детектором. Первая колонка с силикагелем (150 × 4.6 мм, размер частиц сорбента 5 мкм) работала при постоянном давлении 13 МПа при 20°С и обеспечивала разделение по количеству кратных связей. Вторая колонка с октадецилсиликагелем (50 × × 4.6 мм, размер частиц сорбента 5 мкм) работала при постоянном расходе (3 мл/мин) при 50°C. Удерживание на второй колонке возрастало с ростом длины углеродной цепи, однако несколько сокращалось при увеличении количества кратных связей, что, вероятно, связано с некоторым увеличением полярности аналитов. Эта система позволяла добиться очень высокого разрешения и простой идентификации минорных компонентов пробы.
Высокой степени ортогональности позволяет добиться сочетание СФХ и обращено-фазовой жидкостной хроматографии ОФ-ВЭЖХ [82]. Применение подобной системы описано для анализа триглицеридов рыбьего жира [83]. На первом измерении разделение фенациловых эфиров ЖК происходило на фазе, модифицированной серебром. Фазы с закрепленными ионами переходных металлов, в частности серебра, чрезвычайно селективны по отношению к соединениям с кратными связями. В качестве элюента использовали углекислый газ с ацетонитрилом и изопропанолом в качестве модификаторов. На втором измерении разделение происходило на фазе С18 (колонка Zorbax SB C18 50 × 4.6 мм, с размером частиц сорбента 3.5 мкм) с градиентным элюированием водой и ацетонитрилом. Комбинация СФХ × × ОФ-ВЭЖХ обеспечила значительно более высокую пиковую емкость главным образом за счет высокой степени ортогональности, основанной на ненасыщенности и гидрофобности.
Несмотря на все преимущества, СФХ в настоящее время не получила широкого распространения. В первую очередь это связано со сложностью аппаратного исполнения СФХ систем. Поскольку жидкости обладают очень низкой сжимаемостью, их плотность почти постоянна независимо от давления. Сверхкритические флюиды, напротив, очень сжимаемы и их физические свойства существенно изменяются с изменением давления при движении через колонку. Для поддержания постоянного давления (даже при изменении расхода элюента) в системе требуются регуляторы обратного давления, которые очень сложны технически. Помимо этого для изготовления прокладок и уплотнительных колец необходимы специальные материалы, устойчивые к действию сверхкритической жидкости
Перспективы дальнейшего развития СФХ связаны с разработкой новых типов сорбентов, что позволит осуществлять разделению специфических аналитов и расширить область применения СФХ на ранее недоступные для этого метода задачи. Развитие методов динамического модифицирования может гибко регулировать свойства подвижной и стационарной фаз и обеспечить возможность одновременного анализа различных липидов с широким диапазоном полярностей. Большой потенциал в качестве комплексного и высокопроизводительного метода профилирования липидов имеет внедрение хроматографических систем с возможностью автоматической сверхкритической флюидной экстракции (СФЭ). Это позволит осуществлять извлечение аналитов из объектов исследования значительно эффективнее и быстрее, уменьшить расход реактивов и, следовательно, сократить время анализа и существенно снизить его стоимость.
Капиллярный электрофорез. Альтернативой хроматографическим методам анализа липидов является капиллярный электрофорез (КЭ). Метод основан на разделении заряженных компонентов пробы в кварцевом капилляре под действием приложенного электрического поля. Разделение происходит в кварцевом капилляре, предварительно заполненном электролитом (подходящий буферный раствор). После подачи высокого напряжения (до 30 кВ) к концам капилляра компоненты смеси начинают двигаться с разной скоростью, в зависимости от их заряда и ионного радиуса и, соответственно, в разное время достигают зоны детектирования. Разделение характеризуется очень высоким разрешением, для анализа требуется небольшое количество пробы – обычно объем анализируемого раствора составляет порядка 2 нл. Детектирование аналитов при капиллярном электрофорезе может осуществляться различными способами. Наиболее распространенно фотометрическое детектирование в ультрафиолетовой или видимой области и лазерная флуоресценция. Вполне допустимо сочетание систем капиллярного электрофореза с масс-спектрометрами.
Капиллярный электрофорез традиционно использовали для определения высокомолекулярных биомолекул, однако в последнее время возросла его роль в анализе низкомолекулярных органических веществ. В начале 21 века КЭ начали применять для исследования трансжиров в пищевых продуктах [84].
Для разработки эффективного метода КЭ необходимо принять во внимание несколько аспектов. Важным этапом анализа является оптимизация электрофоретических и аналитических параметров: концентрации и pH фонового электролита, типа и концентрации буферных добавок (поверхностно-активные вещества, органические растворители, циклодекстрины), приложенного напряжения, температуры системы. В первую очередь нужно подобрать pH. Поскольку значения констант кислотности (pKa) ЖК составляет приблизительно 5.0, обычно используют электролиты с pH выше 7.0, чтобы перевести аналиты в анионную форму.
Классическая разновидность КЭ – капиллярный зонный электрофорез – ограниченно применима для определения ЖК из-за плохой растворимости Аналитов в воде и низкого УФ-поглощения аналитов. Водные электролиты допустимо использовать для разделения кислот с длиной цепи C2–C14. Как и в случае ВЭЖХ, для фотометрического детектирования возможно введение в молекулы ЖК хромофорных групп, например, дериватизация ω-бромацетофеноном для получения фенациловых эфиров ЖК [85].
Для соединений, слабо поглощающих излучение в УФ-диапазоне, в практике КЭ существует возможность регистрации методом косвенного (непрямого) детектирования. В этом случае в фоновый электролит вводят небольшое количество вещества, хорошо поглощающего излучение требуемой длины волны, подвижность которого близка к подвижности разделяемых веществ. В случае определения анионов поглощающий ион также должен быть анионом, а при определении катионов – катионом. Чаще всего используют катионы ароматических аминов или гетероциклические соединения. Количество добавки должно быть мало для сохранения постоянной ионной силы ведущего электролита в процессе разделения. В зоне, где находится непоглощающий ион, уменьшается концентрация поглощающего иона, и на электрофореграмме регистрируются отрицательные пики, площади которых пропорциональны концентрациям определяемых ионов. Косвенное детектирование позволяет регистрировать все присутствующие в пробе компоненты, то есть является универсальным способом детектирования.
Наиболее распространено в практике анализа ЖК с помощью капиллярного электрофореза использование системы с неводным буферным электролитом и косвенным детектированием. Например, предложен метод разделения различных смесей насыщенных и ненасыщенных свободных ЖК с использованием аденозинмонофосфата (АМФ) в качестве хромофора. Преимущество этого реагента в том, что он имеет высокую молярную поглощающую способность, большое отношение фонового поглощения к фоновому шуму, растворим как в водных, так и в неводных средах [85].
В работе Хаддадиана с соавторами [86] разделение кислот C12–C31 было проведено с использованием электролита, содержащего N-метилформамид и диоксан в соотношении 3:2, 40 мМ 2-амино-2-гидроксиметил-пропан-1,3-диола (ТРИС), 2.5 мМ АМФ и 0.5% лаурилового эфира полиоксиэтилена (Brij 35) за 40 мин. Смесь 13 насыщенных и ненасыщенных изомеров C14–C22 ЖК была разделена до базовой линии за 37 мин с использованием смеси N-метилформамид–диоксан–вода в соотношении 5 : 4 : 1.
Косвенное детектирование было успешно применено для анализа фосфолипидов крови [87]. В качестве электролита использован 5 мМ раствор аденозинмонофосфата. Разделение проводили при температуре 25°C и приложенном напряжении 30 кВ. Кислоты разделяли в анионной форме и в виде цвиттер-ионов. Метод очень перспективен для определения фосфолипидов в биологических образцах.
Прямое УФ-детектирование возможно, если аналиты содержат несколько двойных связей в углеродной цепи, что обеспечивает возможность поглощения излучения в интервале от 200 до 250 нм [88]. Например, была предложена методика анализа содержания омега-3 ЖК в яйцах с прямым УФ-детектированием при 200 нм [89]. В исследовании применяли 12 мМ боратный буферный раствор, pH 9.2, содержащий 12 ммоль Brij 35, 17% ацетонитрила и 33% метанола. Рабочие условия: ввод пробы – 25 мбар в секунду, напряжение 27 кВ, температура 27°C, детектирование прямое при 200 нм. Параметры капилляра – 40 см × × 50 мкм × 375 мкм. Описанная в работе методика имела такие преимущества, как низкая себестоимость, короткое время анализа, отсутствие дериватизации в ходе подготовки проб и простота интерпретации данных.
Основные преимущества – высокая эффективность разделения, недоступная для ВЭЖХ, низкое расход образцов и реагентов, короткое время анализа, простота подготовки образцов (отсутствие дериватизации). К сожалению, КЭ обеспечивает гораздо более низкую чувствительность по сравнению с хроматографическими методами (ВЭЖХ или ГХ), а также худшую воспроизводимость. Таким образом, КЭ представляет собой интересную альтернативу для скрининга ЖК в образцах с высоким содержанием ЖК, например, в растительных маслах, продуктах питания, БАДах.
Спектроскопия ядерного магнитного резонанса. ЯМР-спектроскопия – метод исследования вещества, использующий явление ядерного магнитного резонанса. Это один из наиболее совершенных методов качественного анализа, дающий наиболее полную информацию о молекулярном строении химических веществ и реакционной способности молекул, допускающий также количественное определение. Сигнал пропорционален разности в заселенности уровней, которая определяется статистикой Больцмана. Достоинства метода – чувствительность к структурным изменениям, недеструктивность, высокая скорость анализа, возможность анализа смесей. Особые требования к подготовке образца для проведения ЯМР-анализа сокращают сферы применения или вообще делают невозможным его применение в работе с биоматериалом.
Однако возможен анализ биологических жидкостей и тканей с очень простой подготовкой (жидкость-жидкостная экстракция). Например, в случае липидного анализа плазмы крови, жиры могут быть выделены с помощью процедуры жидкостной экстракции по методу Фолча (смесь CHCl3 и CH3OH) и перерастворены в дейтерированном хлороформе (CDCl3) перед анализом. Образцы могут быть проанализированы непосредственно (без экстракции) после добавления равного объема D2O. ЯМР-анализы проводятся с использованием ЯМР 1H спектроскопии протонного магнитного резонанса (ПМР). Образец плазмы получают центрифугированием [90]. При этом ЯМР 1H является наиболее быстрым способом получения информации, поскольку в сравнении со спектроскопией на ядрах углерода (13С ЯМР-спектроскопия) для получения спектра нужно гораздо меньше времени.
Существуют исследования, в которых описано применение протонного магнитного резонанса для количественного определения ненасыщенных ЖК в тканях животных [91]. Свежие образцы тканей аорты, почек и сердца замораживали с помощью жидкого азота сразу после извлечения. Замороженные ткани взвешивали и гомогенизировали. Липиды экстрагировали смесью хлороформ-метанол (2:1) из расчета 20 об./ед. веса ткани (20 мл/г) в течение 20 мин. Затем образцы фильтровали через воронку из пористого стекла. Такой же объем раствора использовали для промывки остатков ткани на фильтре. Фильтрат смешивали с 0.74%-ным раствором KCl (1/5 общего объема фильтрата) для удаления всех нелипидных примесей и оставляли для разделения на ночь. Нижний слой (хлороформный) собирали, отгоняли растворитель в роторном испарителе при 30°C. Сухие экстракты перерастворяли в CDCl3 и использовали для анализа. Спектры ЯМР жирных кислот в экстрактах получены при 200 МГц на спектрометре Varian XL-200 с использованием ширины частотной полосы 5000 Гц, угол поворота вектора намагниченности 60°, время сбора данных 1.64 с, задержка 4 с.
Ввиду сложности матриц биологических объектов, ЯМР часто применяют в сочетании с каким-либо методом разделения, например, ВЭЖХ или ГХ. ЯМР в сочетании с ВЭЖХ незаменим при структурном анализе неизвестных соединений в биологических матрицах. Нужно отметить, что по сравнению с другими методами детектирования (такими как масс-спектрометрия), обычно применяемыми в хроматографии, ЯМР характеризуется более низкой чувствительностью [92]. Низкая чувствительность всегда была основным ограничением ЯМР-спектроскопии. Теоретически ЯМР не имеет предела чувствительности, поскольку чувствительность и разрешение связаны с силой магнитного поля (отношение сигнал-шум увеличивается пропорционально 1.5 степени вектора магнитной индукции). Кроме того, качество полученного спектра вещества может быть улучшено за счет увеличения числа сканирований (времени накопления сигналов). Однако для коммерчески доступных в настоящее время спектрометров пределы обнаружения все еще находятся в диапазоне от низких микромолярных до высоких наномолярных. Помимо этого проблема анализа биологических объектов связана с тем, что чем сложнее молекула органического соединения, тем больше сигналов содержит ее протонный спектр. При этом неизбежно наложение сигналов с близкими химическими сдвигами, что затрудняет расшифровку полученных данных. Увеличение рабочей частоты спектрометра разделяет мультиплетные спектральные линии взаимодействующих протонов, что позволяет получить более информативный спектр, но в то же время удорожает используемое оборудование.
Метод ЯМР является единственно возможным вариантом прямого подтверждения строения молекулы вещества, выделенного из биологического объекта и первоначально идентифицированного методами ГЖХ или ВЭЖХ. ЯМР спектросокопия позволяет наиболее простым способом провести анализ конформаций молекул веществ и типов химических связей всех классов липидов. Другие варианты подтверждения структуры менее информативны. Например, масс-спектрометрия имеет дело с фрагментами молекул, а ИК-спектроскопия позволяет оценить общие представления о наличии в веществе определенных функциональных групп или фрагментов структур молекулы. Таким образом, не смотря на то, что ЯМР спектросокопия определеяет строение не измененной молекулы, этот метод является в основном, подтверждающим.
Малая распространенность ЯМР обусловлена высокой эксплуатационной стоимостью по сравнению с масс-спектрометрами и другими распространенными аналитическими инструментами. Помимо этого, метод ЯМР требует очень высокой квалификации операторов, использования растворителей со специальным изотопным составом. Также высокие требования предъявляются к лабораториям, в которых эксплуатируются ЯМР-спектрометры (отсутствие вибраций, изоляцией от магнитных и радиочастотных помех и т.д.). Эти факторы, в дополнение к первостепенной проблеме низкой чувствительности сдерживают применение ЯМР в анализе биологических объектов.
В табл. 1 приведено сравнение рассмотренных в работе методов анализа ЖК.
Таблица 1.
Сравнение основных методов анализа содержания жирных кислот в биологических образцах
| Показатель | Вид анализа | ||||
|---|---|---|---|---|---|
| ГХ | ВЭЖХ | СФХ | КЭ | ЯМР | |
| Предел обнаружения | 0.1 пг/мл–1 нг/мл | 10 пг/мл–1000 нг/мл | ~1 пг/мл | 0.1 мкг/мл | 0.1–1 мкг/мл |
| Чувствительность | Высокая | Высокая | Высокая | Низкая | Низкая |
| Селективность | Средняя | Высокая | Высокая | Удовлетворительная (в случае непрямого детектирования) | Высокая |
| Продолжительность анализа | Длительная | Длительная | Короткая | Короткая (~40 мин) |
Короткая |
| Пробоподготовка | Экстракция, дериватизация | Экстракция | Экстракция | Экстракция | Экстракция, перерастворение в дейтерированном хлороформе; добавление равного объема D2O |
| Необходимость дериватизации | + (требуется) | + – (требуется для оптических детекторов) | + – (требуется для оптических детекторов) | + – (требуется для оптических детекторов в случае прямого детектирования) | – (не требуется) |
| Дериватизирующие агенты | Кислоты в метаноле (фторид бора, хлорид алюминия, соляная кислота, серная кислота); ацетилхлорид; метилат натрия; гидроксид калия в метаноле; гидроксид тетраметиламмония, гидроксид триметилфениламмония; диазометан и его производные; алкилхлороформиаты; йодметан; бис-триметилсилил-трифторацетамид; N-метил-N-(трет-бутилдиметилсилил) трифторацетамид; бис-(пентафторфенил) диметилсилан; пентафторбензилбромид | Хлорангидрид нафталинкарбоновой кислоты; фталевый ангидрид; дифеновый ангидрид; 2-сульфобензойный ангидрид; диметиламиноэтанол | Кислоты в метаноле; ω-бромацетофенон | ω-бромацетофенон | – |
| Продолжительность дериватизации | 2–90 мин | 30–120 мин | 2–90 мин | 15 мин | – |
| Температура при дериватизации | 25–100°С | 25–100°С | 60–100°С | 100°С | – |
| Температура в процессе анализа | 50–280°С | Комнатная | 20–50°С | Комнатная | Комнатная |
| Детекция | Пламенно-ионизационный детектор, масс-селективный детектор | Ультрафиолетовый детектор; флуоресцентный детектор; детектор по светорассеянию; масс-селективный детектор | Любой вид детектора, в т.ч. пламенно-ионизационный | Фотометрический детектор в ультрафиолетовой или видимой области; лазерная флуоресценция; масс-спектрометрия; косвенная детекция | – |
| Объем пробы | Маленький | Маленький | Маленький | Очень маленький (~2 нл) |
Большой |
| Расход реагентов | Низкий | Высокий | Низкий | Очень низкий | Высокий |
Обзор литературы, посвященный применению различных методов анализа ЖК в биологических объектах, показал, что все представленные хроматографические методы пригодны для качественного и количественного определения этих соединений. Выбор конкретной методики зависит от многих факторов – типа анализируемого образца, характера матрицы и т.п.
ГХ и ВЭЖХ в сочетании с различными детекторами являются доминирующими по распространенности методами анализа ЖК. Разнообразие детекторов увеличивает гибкость и расширяет сферу применимости этих групп методов к различным типам образцов. Очень перспективен для анализа липидов метод СФХ. Интересную альтернативу хроматографии для скрининга ЖК в различных матрицах представляет собой КЭ.
Применение ЯМР для анализа липидов ограничивается высокой эксплуатационной стоимостью и очень высокими требованиями к квалификации операторов, а также сравнительно низкой чувствительностью.
Работа проведена при финансовой поддержке Российского научного фонда по проекту № 19-76-30014 “Фундаментальные исследования паттернов питания человека как основа перспективных технологий производства пищевых продуктов заданного состава и свойств для реализации стратегии здорового питания и профилактики социально значимых заболеваний”.
Список литературы
Vance J.E., Vance D.E. Biochemistry of Lipids, Lipoproteins and Membranes. Amsterdam: Esevier, 2008. 639 p.
Köfeler H.C. // Encyclopedia of Lipidomics / Ed. M. Wenk. Dordrecht: Springer, 2016. https://doi.org/10.1007/978-94-007-7864-1_21-1
van Meer G., Voelker D.R., Feigenson G.W. // Nat. Rev. Mol. Cell Biol. 2008. V. 9. № 2. P. 112–124.https://doi.org/10.1038/nrm2330
Гладышев М.И. // Журн. СФУ. Биология. 2012. Т. 4. № 5. С. 352–386.
SanGiovanni J.P., Chew E.Y. // Prog. Retin. Eye Res. 2005. V. 24. № 1. P. 87–138. https://doi.org/10.1016/j.preteyeres.2004.06.002
Valera B., Suhas E., Counil E., Poirier P., Dewailly E. // J. Am. Coll. Nutr. 2014. V. 33. № 4. P. 288–296. https://doi.org/10.1080/07315724.2013.874913
Harris W.S., Mozaffarian D., Rimm E., Kris-Etherton P., Rudel L.L., Appel L.J., Engler M., Engler M., Sacks F. // Circulation. 2009. V. 119. № 6. P. 902–907. https://doi.org/10.1161/CIRCULATIONAHA.108.191627
Синицкая Е.Н., Кокоричева Л.В., Манык Ф.М. // Молодой ученый. 2018. № 50. С. 97–99.
Phang M., Lazarus S., Wood L., Garg M. // Semin. Thromb. Hemost. 2011. V. 37. № 03. P. 199–208. https://doi.org/10.1055/s-0031-1273084
Plourde M., Cunnane S. // Appl. Physiol. Nutr. Metab. 2007. V. 32. № 4. P. 619–634. https://doi.org/10.1139/H07-034
Hegele R.A. // Nat. Rev. Genet. 2009. V. 10. № 2. P. 109–121.https://doi.org/10.1038/nrg2481
Simopoulos A.P. // Exp. Biol. Med. 2008. V. 233. № 6. P. 674–688. https://doi.org/10.3181/0711-MR-311
Lv W., Yang T. // Clin. Biochem. 2012. V. 45. № 1-2. P. 127–133. https://doi.org/10.1016/j.clinbiochem.2011.10.011
Munir R., Usman H., Hasnain S., Smans K., Kalbacher H., Zaidi N. // Biochimie. 2014. V. 102. P. 9–18.https://doi.org/10.1016/j.biochi.2014.03.010
Wall R., Ross R.P., Fitzgerald G.F., Stanton C. // Nutr. Rev. 2010. V. 68. № 5. P. 280–289.https://doi.org/10.1111/j.1753-4887.2010.00287.x
Thomas J., Thomas C., Radcliffe J., Itsiopoulos C. // Biomed Res. Int. 2015. V. 2015. Article ID 172801. 13 p. https://doi.org/10.1155/2015/172801
Bielawska K., Dziakowska I., Roszkowska-Jakimiec W. // Toxicol. Mech. Methods. 2010. V. 20. № 9. P. 526–537. https://doi.org/10.3109/15376516.2010.515081
Dołowy M., Pyka A. // J. Chem. 2015. V. 2015. Article ID 120830. 20 p. https://doi.org/10.1155/2015/120830
Wu Z., Zhang Q., Li N., Pu Y., Wang B., Zhang T. // J. Sep. Sci. 2017. V. 40. № 1. P. 288–298. https://doi.org/10.1002/jssc.201600707
Tedone L., Costa R., De Grazia S., Ragusa S., Mondello L. // Phytochem. Anal. 2014. V. 25. № 5. P. 468–475. https://doi.org/10.1002/pca.2518
Murray R.K., Granner D.K., Mayes P.A., Rodwell V.W. Harper’s Illustrated Biochemistry. N.Y.: Lange Medical Books/McGraw-Hill, 2003. 693 p. doi.org/10.6.20.12:80/handle/123456789/48617
Shantha N.C., Napolitano G.E. // J. Chromatogr. A. 1992. V. 624. № 1–2. P. 37–51. https://doi.org/10.1016/0021-9673(92)85673-H
Cruz-Hernandez C., Thakkar S.K., Masserey-Elmelegy I., Buosi W., Fontannaz P., Giuffrida F. // J. Sep. Sci. 2017. V. 40. № 16. P. 3289–3300. https://doi.org/10.1002/jssc.201700030
Casal S., Oliveira B. // Encyclopedia of Chromatography. 3 ed. / Ed. J. Cazes. N.Y.: CRC Press, 2009. P. 833–845.
Eder K. // J. Chromatogr. B. Biomed. Sci. Appl. 1995. V. 671. № 1–2. P. 113–131. https://doi.org/10.1016/0378-4347(95)00142-6
Ostermann A.I., Müller M., Willenberg I., Schebb N.H. // Prostaglandins Leukot. Essent. Fatty Acids. 2014. V. 91. № 6. P. 235–241. https://doi.org/10.1016/j.plefa.2014.10.002
Seppänen-Laakso T., Laakso I., Hiltunen R. // Anal. Chim. Acta. 2002. V. 465. № 1–2. P. 39–62https://doi.org/10.1016/S0003-2670(02)00397-5
Morrison W.R., Smith L.M. // J. Lipid Res. 1964. V. 5. № 4. P. 600–608.
Bondia-Pons I., Castellote A.I., López-Sabater M.C. // J. Chromatogr. B. 2004. V. 809. № 2. P. 339–344. https://doi.org/10.1016/j.jchromb.2004.07.002
Araujo P., Nguyen T.T., Frøyland L., Wang J., Kang J.X. // J. Chromatogr. A. 2008. V. 1212. № 1–2. P. 106–113. https://doi.org/10.1016/j.chroma.2008.10.006
Murrieta C.M., Hess B.W., Rule D.C. // Meat Sci. 2003. V. 65. № 1. P. 523–529. https://doi.org/10.1016/S0309-1740(02)00244-9
Thomas M.L., Brown Jr A.H., Kellogg D.W., Rule D.C., Baublits R.T., Johnson Z.B., Anschutz K.S., Murrieta C.M. // J. Food Qual. 2008. V. 31. № 2. P. 189–204. https://doi.org/10.1111/j.1745-4557.2008.00190.x
Mondello L., Tranchida P.Q., Dugo P., Dugo G. // J. Pharm. Biomed. Anal. 2006. V. 41. № 5. P. 1566–1570. https://doi.org/10.1016/j.jpba.2006.01.027
Ichihara K., Fukubayashi Y. // J. Lipid Res. 2010. V. 51. № 3. P. 635–640. https://doi.org/10.1194/jlr.D001065
Lands W.E.M., Libelt B., Morris A., Kramer N.C., Prewitt T.E., Bowen P., Schmeisser D., Davidson M.H., Burns J.H. // Biochim. Biophys. Acta Mol. Basis. Dis. 1992. V. 1180. № 2. P. 147–162. https://doi.org/10.1016/0925-4439(92)90063-S
Vognild E., Elvevoll E.O., Brox J., Olsen R.L., Barstad H., Aursand M., Østerud B. // Lipids. 1998. V. 33. № 4. P. 427–436. https://doi.org/10.1007/s11745-998-0224-8
Bauer A.K., Dwyer-Nield L.D. // Methods in Cell Biology. V. 163. Chapter 10 /Eds. L. Galluzzi, A. Buqué. N.Y.: Academic Press, 2021. P. 151–173. https://doi.org/10.1016/bs.mcb.2020.07.003
Vičkačkaitė V., Lingytė A., Kasperovičienė J., Bugelytė B., Koreivienė J., Savadova K. // Chemija. 2016. V. 27. № 4.
Burdge G.C., Wright P., Jones A.E., Wootton S.A. // Br. J. Nutr. 2000. V. 84. № 5. P. 781–787. https://doi.org/10.1017/S0007114500002154
Ichihara K., Kohsaka C., Tomari N., Kiyono T., Wada J., Hirooka K., Yamamoto Y. // Anal. Biochem. 2016. V. 495. P. 6–8. https://doi.org/10.1016/j.ab.2015.11.009
Sampels S., Pickova J. // Food Chem. 2011. V. 128. № 3. P. 811–819https://doi.org/10.1016/j.foodchem.2011.03.089
Cruz-Hernandez C., Destaillats F. // Encyclopedia of Lipidomics. / Ed. M. Wenk. Dordrecht: Springer, 2016. https://doi.org/10.1007/978-94-007-7864-1_66-1
Liu Z., Ezernieks V., Rochfort S., Cocks B. // Food Chem. 2018. V. 261. P. 210–215. https://doi.org/10.1016/j.foodchem.2018.04.053
Akinyemi O., Bruckner G., Johnson J., Lennie T.A., Hildebrand D. // Open Nutr. J. 2017. V. 11. № 1. https://doi.org/10.2174/1874288201711010017
Delmonte P., Kramer J.K.G. // Reference Module in Food Science. Amsterdam: Elsevier, 2016. https://doi.org/10.1016/B978-0-08-100596-5.03051-1
Juárez M., Polvillo O., Contò M., Ficco A., Ballico S., Failla S. // J. Chromatogr. A. 2008. V. 1190. № 1-2. P. 327–332. https://doi.org/10.1016/j.chroma.2008.03.004
Drechsel D., Dettmer K., Engewald W. // Chromatographia. 2003. V. 57. № 1. P. S283–S289. https://doi.org/10.1007/BF02492117
Tsai C.J., Liu C.C., Hung L.B., Pan B.S. // J. Am. Oil Chem. Soc. 2012. V. 89. № 1. P. 9–16. https://doi.org/10.1007/s11746-011-1880-2
Gutnikov G. // J. Chromatogr. B. Biomed. Sci. Appl. 1995. V. 671. № 1–2. P. 71–89. https://doi.org/10.1016/0378-4347(95)00116-Z
Johnson S.B., Brown R.E. // J. Chromatogr. A. 1992. V. 605. № 2. P. 281–286. https://doi.org/10.1016/0021-9673(92)85248-R
Brondz I. // Anal. Chim. Acta. 2002. V. 465. № 1–2. P. 1–37.https://doi.org/10.1016/S0003-2670(01)01467-2
Dai L., Gonçalves C.M.V., Lin Z., Huang J., Lu H., Yi L., Liang Y., Wang D., An D. // Talanta. 2015. V. 135. P. 108–114https://doi.org/10.1016/j.talanta.2014.12.039
Zou Y., Wu H. // Agilent Technologies Application Note. 2018. № 5991-8706EN. https://www.agilent.com/cs/library/applications/5991-8706EN_37fattyacid_FAME_application.pdf
Tammekivi E., Vahur S., Kekišev O., Van der Werf I.D., Toom L., Herodes K., Leito I. // Anal. Methods. 2019. V. 11. № 28. P. 3514–3522. https://doi.org/10.1039/C9AY00954J
Moon J.Y., Kong T., Jang H., Kang H.C., Cho Y.Y., Lee J., Lee H. // J. Chromatogr. B 2018. V. 1093–1094. P. 82–90. https://doi.org/10.1016/j.jchromb.2018.06.059
Yang Y.J., Choi M.H., Paik M.J., Yoon H.R., Chung B.C. // J. Chromatogr. B. Biomed. Sci. Appl. 2000. V. 742. № 1. P. 37–46. https://doi.org/10.1016/S0378-4347(00)00098-0
He L., Prodhan M.A.I., Yuan F., Yin X., Lorkiewicz P.K., Wei X., Feng W., McClain C., Zhang X. // J. Chromatogr. B. 2018. V. 1092. P. 359–367. https://doi.org/10.1016/j.jchromb.2018.06.028
Kurkiewicz S., Dzierzewicz Z., Wilczok T., Dworzanski J. // J. Am. Soc. Mass Spectrom. 2003. V. 14. P. 58–62. https://doi.org/10.1016/S1044-0305(02)00817-6
Wetzel D.L., Reynolds J.E., Christie W.W., Budge I.S., Iverson S., Koopman H. // Mar. Mamm. Sci. 2007. V. 23. № 4. P. 989–992. https://doi.org/10.1111/j.1748-7692.2007.00158.x
Garrido J.L., Medina I. // Anal. Chim. Acta. 2002. V. 465. № 1–2. P. 409–416https://doi.org/10.1016/S0003-2670(02)00207-6
Choi M.H., Chung B.C. // Anal. Biochem. 2000. V. 277. № 2. P. 271–273. https://doi.org/10.1006/abio.1999.4407
Bodoprost J., Rosemeyer H. // Int. J. Mol. Sci. 2007. V. 8. P. 1111–1124. https://doi.org/10.3390/i8111111
Majnooni M.B., Mohammadi B., Jalili R., Babaei A., Bahrami G. // J. Liq. Chromatogr. Relat. Technol. 2016. V. 39. P. 877–881. https://doi.org/10.1080/10826076.2016.1275000
Shrestha R., Miura Y., Hirano K., Chen Z. Okabe H., Chiba, H., Hui S. // Anal. Sci. 2018. V. 34. P. 575–582. https://doi.org/10.2116/analsci.17P557
Bravi E., Perretti G., Montanari L. // J. Chromatogr. A. 2006. V. 1134. № 1–2. P. 210–214. https://doi.org/10.1016/j.chroma.2006.09.007
Suchocka Z., Gronostajska D., Suchocki P., Pachecka J. // J. Pharm. Biomed. Anal. 2003. V. 32. № 4–5. P. 859–865. https://doi.org/10.1016/S0731-7085(03)00188-2
Garscha U., Nilsson T., Oliw E.H. // J. Chromatogr. B. 2008. V. 872. № 1–2. P. 90–98. https://doi.org/10.1016/j.jchromb.2008.07.013
Lee S.H., Blair I.A. // BMB Rep. 2009. V. 42. № 7. P. 401. https://doi.org/10.5483/BMBRep.2009.42.7.401
Ahern K.W., Serbulea V., Wingrove C.L., Palas Z.T., Leitinger N., Harris T.E. // Sci. Rep. 2019. V. 9. № 1. P. 1–13. https://doi.org/10.1038/s41598-019-47693-5
Godzien J., Ciborowski M., Martínez-Alcázar M.P., Samczuk P., Kretowski A., Barbas C. // J. Proteome Res. 2015. V. 14. № 8. P. 3204–3216. https://doi.org/10.1021/acs.jproteome.5b00169
Reis A., Rudnitskaya A., Blackburn G.J., Fauzi N.M., Pitt A.R., Spickett C.M. // J. Lipid Res. 2013. V. 54. № 7. P. 1812–1824. https://doi.org/10.1194/jlr.M034330
Zhao Z., Xu Y. // J. Lipid Res. 2010. V. 51. № 3. P. 652–659. https://doi.org/10.1194/jlr.D001503
Pellegrino R.M., Di Veroli A., Valeri A., Goracci L., Cruciani G. // Anal. Bioanal. Chem. 2014. V. 406. № 30. P. 7937–7948. https://doi.org/10.1007/s00216-014-8255-0
Hu C., Van Dommelen J., Van Der Heijden R., Spijksma G., Reijmers T.H., Wang M., Slee E., Lu X.Xu G., van der Greef J., Hankemeier T. // J. Proteome Res. 2008. V. 7. № 11. P. 4982–4991. https://doi.org/10.1021/pr800373m
Kortz L., Dorow J., Becker S., Thiery J., Ceglarek U. // J. Chromatogr. B. 2013. V. 927. P. 209–213.https://doi.org/10.1016/j.jchromb.2013.03.012
Pettinella C., Lee S.H., Cipollone F., Blair I.A. // J. Chromatogr. B. 2007. V. 850. № 1–2. P. 168–176. https://doi.org/10.1016/j.jchromb.2006.11.023
Klesper E., Corwin A.H., Turner D.A. // J. Org. Chem. 1962. V. 27. P. 700–701. https://doi.org/10.1021/jo01049a069
Taylor L.T. // J. Supercrit. Fluids. 2009. V. 47. № 3. P. 566–573. https://doi.org/10.1016/j.supflu.2008.09.012
Hartmann A., Ganzera M. // Planta Med. 2015. V. 81. № 17. P. 1570–1581. https://doi.org/10.1055/s-0035-1545911
Bernal J.L., Martín M.T., Toribio L. // J. Chromatogr. A. 2013. V. 1313. P. 24–36. https://doi.org/10.1016/j.chroma.2013.07.022
Hirata Y., Sogabe I. // Anal. Bioanal. Chem. 2004. V. 378. № 8. P. 1999–2003. https://doi.org/10.1007/s00216-003-2487-8
Svan A., Hedeland M., Arvidsson T., Pettersson C.E. // Anal. Chim. Acta. 2018. V. 1000. P. 163–171. https://doi.org/10.1016/j.aca.2017.10.014
François I., Sandra P. // J. Chromatogr. A. 2009. V. 1216. № 18. P. 4005–4012. https://doi.org/10.1016/j.chroma.2009.02.078
de Oliveira M.A.L., Solis V.E., Gioielli L.A., Polakiewicz B., Tavares M.F. // Electrophoresis. 2003. V. 24. № 10. P. 1641–1647. https://doi.org/10.1002/elps.200305394
de Oliveira M.A.L., Porto B., Faria I., Lopes P., Barra P., Castro R., Sato R. // Molecules. 2014. V. 19. № 9. P. 14094–14113. https://doi.org/10.3390/molecules190914094
Haddadian F., Shamsi S.A., Warner I.M. // J. Chromatogr. Sci. 1999. V. 37. № 4. P. 103–107. https://doi.org/10.1093/chromsci/37.4.103
Gao F., Dong J., Li W., Wang T., Liao J., Liao Y., Liu H. // J. Chromatogr. A. 2006. V. 1130. № 2. P. 259–264. https://doi.org/10.1016/j.chroma.2006.03.070
Gabriel H., Amelia T.V., Cristina F., Aura R. // Acta Marisiensis – Seria Medica. 2015. V. 61. № 4. https://doi.org/10.1515/amma-2015-0103
Porto B.L.S., de Souza M.V.N., de Oliveira M.A.L. // Anal. Sci. 2011. V. 27. № 5. P. 541–541. https://doi.org/10.2116/analsci.27.541
Correia B.S.B., Torrinhas R.S., Ohashi W.Y., Tasic L. // Advances in Lipid Metabolism. Chapter 2. /Ed. R.V. Baez. IntechOpen, 2018. https://doi.org/10.5772/intechopen.81523
Chi Y., Gupta R.K. // Am. J. Hypertens. 1998. V. 11. № 3. P. 340–348. https://doi.org/10.1016/S0895-7061(97)00456-1
Emwas A.H., Roy R., McKay R.T., Tenori L., Saccenti E., Gowda G.A.N. et al. // Metabolites. 2019. V. 9. № 7. P. 123. https://doi.org/10.3390/metabo9070123
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология