

Прикладная биохимия и микробиология, 2020, T. 56, № 5, стр. 419-427
Вакцины против туберкулеза: проблемы и перспективы (обзор)
Н. И. Надолинская 1, *, Д. С. Карпов 2, А. В. Гончаренко 1
1 Институт биохимии им. А.Н. Баха, Федеральный исследовательский центр
“Фундаментальные основы биотехнологии” Российской академии наук
119071 Москва, Россия
2 Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
* E-mail: nioriss@gmail.com
Поступила в редакцию 16.03.2020
После доработки 13.04.2020
Принята к публикации 22.04.2020
Аннотация
Несмотря на усилия мирового медицинского и научного сообщества, туберкулез остается основной причиной смерти от инфекционных заболеваний. Ожидания положительного эффекта от применения разработанных новых противотуберкулезных препаратов не оправдались, и внимание исследователей в значительной степени обращено на создание новых микобактериальных штаммов для вакцинопрофилактики туберкулеза. Предлагаемый обзор содержит современную информацию о существующих вакцинных штаммах и разработке новых генно-инженерных штаммов для профилактики туберкулеза, а также для профилактики и лечения других заболеваний. В обзор включена актуальная информация о корреляции между вакцинацией БЦЖ и частотой и тяжестью протекания COVID-19.
Туберкулез – инфекционное заболевание, вызываемое Mycobacterium tuberculosis (МТБ), остается основной причиной смерти от инфекционных заболеваний во многих странах [1]. На сегодняшний день лечение туберкулеза представляет собой комбинированную антибиотикотерапию в течение 6–9 мес., вызывающую значительные побочные эффекты. В настоящее время стратегия Всемирной организации здравоохранения по борьбе с туберкулезом направлена на усиление профилактики заболевания [2] и по всему миру ведется активная работа по разработке улучшенных противотуберкулезных вакцин, некоторые из которых вступили в стадию клинических исследований.
Бацилла Кальмета–Герена (БЦЖ), Bacillus Calmette-Guérin (BCG) на сегодняшний день остается единственной вакциной против туберкулеза, успешно защищает от туберкулеза (ТБ) детей, но обеспечивает только частичную защиту от респираторных форм ТБ подростков и взрослых. Альберт Кальмет и Камиль Герен получили БЦЖ от возбудителя туберкулеза крупного рогатого скота Mycobacterium bovis путем непрерывного пассирования в течение нескольких лет, что привело к ослаблению штамма. БЦЖ применяется для вакцинации с 1921 г. и приводит также к уменьшению общей детской смертности за счет неспецифического иммуномодулирующего действия микобактериальной вакцинации. Хотя БЦЖ внесла значительный вклад в профилактику ТБ [3], потеря нескольких доминирующих антигенов и ключевых молекулярных признаков патогенных микобактерий может объяснить ее ограничения в профилактике туберкулеза взрослого населения. Предлагаемый обзор содержит современные данные о существующих штаммах БЦЖ, о разработке генетически модифицированных вакцинных штаммов, возможно, способных заменить БЦЖ, оптимальном пути доставки вакцины в организм, а также в нем обсуждаются возможности использования БЦЖ для борьбы с другими заболеваниями.
Штаммы БЦЖ и их генеалогия. Основное отличие БЦЖ от исходного штамма M. bovis – отсутствие нескольких сегментов генома, в частности, так называемого региона различия (region of difference 1, RD1) [4]. Он кодирует иммунодоминантные антигены, такие как ESAT6, CFP10 и уникальную микобактериальную систему секреции ESX-1 типа II (T7SS) [5, 6]. Потеря ESX-1 привела к утрате способности БЦЖ проникать из фагосомы в цитозоль, что явилось мощным фактором аттенуации штамма.
В настоящее время выделяется довольно большое количество штаммов БЦЖ, что обусловлено особенностями условий поддержания линий в разных странах [7]. Интересно отметить, что первый дочерний штамм БЦЖ, полученный от исходной культуры, был зарегистрирован в России в 1940 г. [8].
Бер с соавт. [9] исследовали генеалогию распространения БЦЖ и объединили штаммы в группы на основе информации о количестве копий гена IS6110 (инсерционный элемент, характерный только для группы микобактерий, вызывающих туберкулез) и наличию или отсутствию гена mpt64 (консервативный антиген МТБ, используемый для диагоностики). Исследование обнаружило синонимичные штаммы под разными названиями и было проведено их объединение для упрощения типирования [10].
Говоря о генетических особенностях штаммов БЦЖ, интересно рассмотреть один из них более подробно. При изучении штамма BCG-Prague была обнаружена мутация гена phoP в виде однонуклеотидной вставки, которая приводила к разрушению С-концевого ДНК-связывающего домена [11, 12]. Эта мутация специфична для BCG-Prague и не обнаруживается в других штаммах БЦЖ [13]. PhoP является регулятором ответа двухкомпонентной системы PhoP-PhoR, которая позитивно регулирует более 40 генов в МТБ, включая два важных протективных антигена (Ag85A, PPE18), которые используются для конструирования субъединичных вакцин [14, 15]. Таким образом, возникло предположение, что низкая иммуногенность BCG-Prague может быть результатом мутации phoP. Показано, что комплементация мутации аллелем M. bovis phoP действительно восстанавливает иммуногенность BCG-Prague. Также сверхэкспрессия аллеля M. bovis phoP-phoR в BCG-Japan с полноценной копией phoP-phoR еще более повышает иммуногенность и защитную эффективность этого вакцинного штамма. Вакцинация мышей C57BL/6 рекомбинантным штаммом rBCG-Japan/PhoPR индуцировала более высокие уровни продукции интерферона-γ (IFN-γ) CD4+ Т-клетками, чем при использовании BCG-Japan. Морские свинки, вакцинированные rBCG-Japan/PhoPR, были лучше защищены от заражения МТБ, чем те, которые иммунизированы BCG-Japan, демонстрируя значительно более длительное время выживания, сниженные бактериальные нагрузки и менее тяжелую патологию. В результате этих исследований идентифицировали генетическую модификацию, которая может применяться для создания новых рекомбинантных вакцин БЦЖ.
Способы защиты микобактерий от иммунной системы инфицированного хозяина. После проникновения в организм клетки МТБ сталкиваются с первым эшелоном защиты в виде фагоцитирующих клеток. Фагоцитированная бактерия подвергается кислотному стрессу, воздействию активных форм кислорода и азота (ROS и RNS), гидролитических ферментов и катионных антимикробных пептидов. Низкий рН внутри созревающей фагосомы активирует ферменты, которые разлагают бактериальные липиды и белки [16, 17]. Однако микобактерии выработали различные пути адаптации к бактерицидной среде созревающей фагосомы. В частности, они уходят от действия бактерицидных механизмов, блокируя протонный “насос”, подкисляющий содержимое фагосом, и ингибируют слияние фагосом и лизосом [18, 19]. С понижением pH МТБ может изменить свой метаболизм и использовать вместо цикла Кребса глиоксилатный путь, что уменьшает синтез НАДН и высвобождение CO2. Таким образом, дополнительный углерод затем может быть использован для синтеза липидов или других метаболитов для дальнейшего снижения преодоления стресса [20]. Так, согласно исследованиям, синтез гиперфосфорилированных гуаниновых нуклеотидов (p)ppGpp необходим для выживания микобактерий в неблагоприятных условиях, включая и окислительно-восстановительный стресс [21]. Другой механизм ответа на закисление среды – выработка аммиака для повышения pH. МТБ используют для этого оперон ompATb (необходим для функционирования при пониженном pH), источником аммиака служит аспарагин [22]. Уреазный оперон также вносит вклад в защелачивание среды [23]. Следует добавить, что микобактериальная уреаза участвует в предотвращении нормальной экспрессии молекул МНС класса II, которые участвуют в связывании антигенов и презентации их Т-клеткам на клеточной поверхности [24]. Кроме того, в ответе на окислительный стресс МТБ также задействует эрготионеиновые, тиоловые и тиоредоксиновые системы, и описанную выше систему PhoP-PhoR [25].
Блокировав образование фаголизосомы, микобактерия разрушает мембрану фагосомы с помощью системы секреции ESX-1, что позволяет туберкулезным антигенам и самим бактериям выходить в цитозоль [19, 26]. Напротив, БЦЖ не имеет ESX-1 и остается внутри фагосомы хозяина, поэтому антигены и бактериальная ДНК не попадают в цитозоль [5, 6, 27]. Исследования показали, что иммунный ответ инфицированных микобактериями макрофагов характеризуется сниженной экспрессией генов МНС класса II и других генов, индуцируемых IFN-γ. Одним из механизмов подавления экспрессии MHC II является специфическое ингибирование путем деацетилирования гистонов, что приводит к снижению ответов CD4+ T-клеток [28, 29]. CD4+ Т-клетки дифференцируются в Т-клетки центральной памяти (TCM), которые, в свою очередь, дифференцируются в клетки эффекторной памяти TEM, эффекторные клетки Th1 или Th17, далее они мигрируют и осуществляют свои эффекторные функции в инфицированных тканях. Когда CD4+ T-клетки прибывают в место инфекции, они сталкиваются с агрегатами МТБ-содержащих макрофагов и других иммунных клеток и вместе образуют плотную клеточную структуру, называемую гранулемой. CD4+ Т-клетки секретируют цитокины, которые активируют инфицированные макрофаги для контроля роста бактерий и привлечения большего количества иммунных клеток к гранулеме [30]. Присутствие микобактериальной ДНК в цитозоле приводит к активации инфламмосом NLPR3 и AIM-2, высвобождению интерферонов и увеличению аутофагии и апоптоза [20, 31, 32], что в конечном итоге может привести к лучшей индукции ответов Т-клеток [33].
Во время развития заболевания увеличивается количество MТБ-специфичных CD8+ Т-клеток, но их роль в защитном иммунитете не ясна. Было показано, что истощение CD8+ клеток приводит к снижению протективности вакцинации БЦЖ [34]. В то же время даже высокий уровень индуцированных вакцинацией CD8+-клеток не влияет на пролиферацию MТБ и не приводит к распознаванию MТБ-инфицированных макрофагов [35].
Роль гуморального иммунитета при инфекции МТБ остается спорной, однако показано, что индивидуумы с латентной туберкулезной инфекцией и активным заболеванием имеют различные МТБ-специфические гуморальные ответы и обладают различными паттернами гликозилирования антител [36].
Пути повышения протективности БЦЖ. BCG::ESX-1. Несколько кандидатов в рекомбинантные вакцины БЦЖ были сконструированы так, чтобы обеспечить разрушение мембраны фагосомы клетками вакцинного штамма (рис. 1) [37]. Как обсуждалось выше, у штамма БЦЖ утеряны гены, необходимые для секреции типа VII (T7S) ESX-1, предназначенные для секреции белков, играющих ключевую роль во взаимодействиях хозяина с патогеном [38, 39].
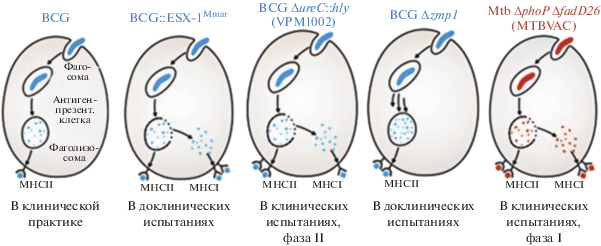
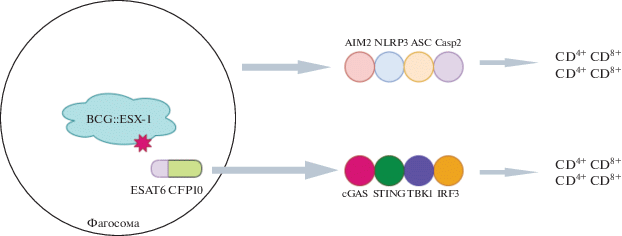
Основная система ESX-1 состоит из АТФаз EccB-D, которые образуют трансмембранную структуру в виде канала. Цитозольные компоненты системы ESX-1 представлены АТФазой EccA, шаперонами EspD-H и секретируемыми субстратными белками EsxAB, PE35-PPE68, EspA-C и EspE. Другие важные эффекторы, которые секретируются этой системой – антигенная мишень раннего секретирования 6 кДа (early secreted antigenic target of 6 kDa, ESAT-6) и белок культурального фильтрата 10 кДа (culture filtrate protein of 10 kDa, CFP-10). Белки секретируются в виде гетеродимера, связываясь с ядерными компонентами системы ESX-1 [40], что вызывает каскад неспецифических иммунных реакций [41–43]: активацию AIM-2 и NLRP3, повышение секреции интерлейкина-1b (IL-1b) и/или IL-18 [30, 31, 44] и активацию циклической синтазы GMP-AMP (cGas), стимулятора генов интерферона (STING) и TANK-связывающей киназы 1 (TBK1). Последний сигнальный каскад приводит к образованию интерферонов типа I (IFN) [17]. Помимо неспецифической иммунной активации, секретируемые эффекторы ESX-1 также индуцируют специфические реакции Th1-клеток с сильным защитным потенциалом [45].
Посредством гетерологичной экспрессии области esx-1 Mycobacterium marinum в БЦЖ была создана рекомбинантная БЦЖ, обладающая ESX-1-активностью (BCG::ESX-1Mmar), которая индуцирует цепочку cGas/STING/TBK1/IRF-3/интерферон I и усиливает воспалительную активность AIM2 и NLRP3, что стимулирует образование эффекторных CD8+ и CD4+ Th1-клеток (рис. 2) [46]. Важно отметить, что штамм BCG::ESX-1Mmar характеризовался низкой вирулентностью и обеспечивал более эффективную защиту по сравнению с исходным штаммом БЦЖ при заражении высоковирулентным MТБ.
BCG ΔureC::hly (VPM1002) и производные. VPM1002 представляет собой рекомбинантный штамм rBCG ΔureC::hly, в котором ген уреазы С был заменен геном hly, кодирующим листериолизин O (LLO), из Listeria monocytogenes [47]. Уреаза С препятствует закислению фагосом, содержащих микобактерии, путем образования аммиака, и тем самым ингибирует созревание фаголизосом и способствует выживанию микобактерий внутри макрофагов [48, 49]. Уменьшение продукции уреазы С приводит к подкислению фагосом, что способствует образованию фаголизосом [50]. LLO представляет собой холестеринзависимый цитолизин, который образует трансмембранные поры в фаголизосоме, позволяя L. monocytogenes выйти в цитозоль [50, 51]. Его экспрессия в VPM1002 приводила к высвобождению антигенов и бактериальной ДНК в цитозоль, вызывая аутофагию, активацию воспаления и апоптоз. VPM1002 продемонстрировал существенное повышение иммуногенности, эффективности и безопасности по сравнению с БЦЖ в доклинических исследованиях, успешно прошел клинические испытания I и II фаз. Продолжаются клинические испытания II/III фазы в Индии. Вакцина прошла I фазу клинических испытаний в Германии и Южной Африке, продемонстрировав ее безопасность и иммуногенность у молодых людей. Она также был успешно опробована в фазе IIa рандомизированного клинического исследования на здоровых новорожденных в Южной Африке и в настоящее время проходит фазу IIb исследования на ВИЧ-инфицированных новорожденных [52].
Производные второго поколения rBCG ΔureC:-:hly секретируют цитокины для усиления иммуногенности [53] и ауксотрофны по витамину B6 [54]. Кроме того, получен штамм rBCG ΔureC::hly ΔnuoG, делетированный по гену антиапоптотической вирулентности nuoG, предотвращающего апоптоз инфицированных клеток [55]. Для мышей вакцинация rBCG ΔureC::hly ΔnuoG оказалась более безопасной, чем вакцинация немодифицированной БЦЖ, и более протективной по сравнению с rBCG ΔureC::hly, значительно снижала МТБ-нагрузку в легких мышей, уменьшала патологические проявления в легких и усиливала иммунные реакции. Транскриптомный анализ дренирующих лимфатических узлов после вакцинации либо rBCG ΔureC::hly, либо rBCG ΔureC::hly ΔnuoG продемонстрировал более раннюю и более сильную индукцию иммунных ответов, чем при использовании немодифицированной БЦЖ. Таким образом, rBCG ΔureC::hly ΔnuoG является многообещающим кандидатом на вакцину с улучшенной эффективностью и безопасностью [55].
БЦЖ, экспрессирующая перфринголизин О. Недостатком rBCG ΔureC::hly является активность LLO в узком диапазоне pH. Был создан штамм БЦЖ, экспрессирующий перфринголизин О (Pfo) – цитолизин Clostridium perfringens [56]. Чтобы уменьшить его цитотоксический эффект, использовали мутантную форму этого белка, содержащую замену G137Q (PfoAG137Q). Мутация значительно уменьшила период полураспада белка, что привело к устранению его цитотоксичности. Для полученного штамма rBCG, обозначенного AERAS-401 (BCG1331 ΔureC::ΩpfoAG137Q), было подтверждено отсутствие активности уреазы. Секреция биологически активного гемолизина была показана с помощью лизиса эритроцитов овцы в присутствии супернатанта рекомбинантного штамма. Кроме того, было показано, что AERAS-401 оказался менее вирулентным для мышей с иммунодефицитом по сравнению с родительским штаммом БЦЖ.
BCGΔzmp1. Было установлено, что удаление гена металлопротеазы цинка zmp1 ослабляет МТБ и способствует усилению неспецифического иммунитета, воспалительных и IL-1-зависимых иммунных механизмов при инфицировании мутантным штаммом. Аналогичная делеция была получена у БЦЖ, и эксперименты по иммунизации in vitro и in vivo на мышах и морских свинках показали, что делеционный мутант BCG Δzmp1 является более иммуногенным, и также усиливает презентацию антигенов в составе МНСII [57]. Кроме того, один из ингибиторов Zmp1 был способен снижать выживаемость МТБ в первичных макрофагах человека [58]. Таким образом, BCG Δzmp1 ожидает дальнейшей оценки и клинических испытаний.
rBCG-SOCS1DN. Среди разнообразных механизмов ухода МТБ от иммунной системы хозяина – индукция экспрессии супрессора цитокинов SOCS1 клетками хозяина, что приводит к подавлению передачи сигналов цитокинов и нарушению передачи сигналов JAK/STAT, участвующих в врожденном иммунитете и последующем адаптивном иммунитете. Реализация этого механизма вакцинным штаммом БЦЖ может быть причиной недостаточной протективности вакцины. Был получен штамм БЦЖ (rBCG-SOCS1DN), секретирующий доминантно-негативный мутантный вариант SOCS1. Показано, что иммунизация rBCG-SOCS1DN усиливала активацию дендритных клеток костного мозга и активацию Т-клеток по сравнению с клетками контрольного штамма БЦЖ, повышала уровень продукции цитокинов IFN-γ, TNF-α и IL-6. Кроме того, у мышей, иммунизированных rBCG-SOCS1DN, было отмечено значительное снижение количества КОЕ МТБ в легких и селезенке по сравнению с таковыми у контрольных мышей, иммунизированных БЦЖ, после инфицирования высокопатогенным штаммом МТБ. Из полученных данных вытекает новая концепция рекомбинантной вакцины БЦЖ как инструмента для иммуномодулирования цитокинов в клетках-хозяевах [59].
Вакцины на основе МТБ. Осуществляются попытки получить вакцинный штамм из МТБ путем его аттенуации с помощью удаления RD1 локуса в результате нарушения синтеза кофактора (panCD) и биосинтеза аминокислот (leuD, lysA) [60]. Исследованные штаммы с различными комбинациями этих мутаций показали протективность и иммуногенность в моделях с мышами, морскими свинками и приматами.
Наиболее успешным оказался вакцинный штамм MTBVAC – MTB ΔphoP ΔfadD26, производное штамма МТБ MT103. Он был сконструирован путем создания двух независимых стабильных генетических делеций в генах phoP и fadD26, кодирующих два основных фактора вирулентности [61]. Уже упоминавшийся ген phoP, который кодирует фактор транскрипции двухкомпонентной системы вирулентности PhoPR, и ген fadD26, участвующий в биосинтезе и экспорте димикоцерозатов фтиоцерола (PDIM), – основные связанные с вирулентностью липидов клеточной стенки МТБ [62, 63]. В доклинических оценках MTB ΔphoP ΔfadD26 имел сравнимую безопасность с БЦЖ, но лучшую иммуногенность и защитную эффективность [61, 64].
На настоящий момент MTBVAC является единственной живой аттенуированной вакциной на основе человеческого патогена, которая после почти 20 лет исследований успешно прошла клинические испытания в качестве профилактической вакцины у новорожденных (вместо БЦЖ) и в качестве профилактической вакцины у подростков и взрослых (получивших прививку БЦЖ при рождении). MTBVAC сохраняет большинство Т-клеточных эпитопов, описанных для ТБ, включая основные иммунодоминантные антигены ESAT-6 и CFP-10 RD1. Исследования показали, что MTBVAC-индуцированный иммунитет к ESAT-6 и CFP-10 коррелирует с лучшей протективностью по сравнению с БЦЖ [65].
Сравнение БЦЖ и MTBVAC показало, что ассоциированный с MTBVAC иммунитет длится дольше, чем иммунитет БЦЖ, когда вводится в виде однократной дозы [66].
Пути доставки БЦЖ в организм. В настоящее время вакцинация БЦЖ производится подкожно, однако существуют и другие способы [67]: через слизистые оболочки (перорально, интраназально, аэрозольно) или внутривенно. Ниже будет рассмотрено возможное влияние выбора пути вакцинации на ее эффективность.
Последние исследования показывают возрождение интереса к пероральному введению БЦЖ, в свое время от него отказались в пользу подкожного в связи с загрязнением вакцины туберкулезом в Любеке. Однако, некоторые данные демонстрируют повышение эффективности защиты, если вместе с традиционной подкожной вакцинацией использовать пероральную [68, 69].
Другой метод доставки БЦЖ – интраназальный. Данный вариант введения БЦЖ в организм демонстрировал достаточную эффективность в исследовании на мышах [70, 71]. Однако, по сравнению с другими путями, тоже использующими слизистую оболочку, интраназальный обладает определенными недостатками. Так, по некоторым данным, он сопряжен с опасностью развития паралича лицевого нерва (паралича Белла) [72].
Существует, кроме того, аэрозольное введение БЦЖ. Так, у макак и морских свинок было показано преимущества защитных свойств такого пути перед подкожным [73, 74]. Было проведено исследование, демонстрирующее повышение иммунного ответа именно при аэрозольном введении вакцины [75, 76]. У людей также проводилась аэрозольная вакцинация БЦЖ, данный вариант может быть перспективным [77].
Дарра с соавт. изучили влияние внутривенного способа введения БЦЖ на приматах [78]. В этом исследовании они показали способность БЦЖ давать очень высокую степень защиты при таком варианте доставки: у шести из десяти макак инфекция не была детектирована; девять из десяти показывали высокую защищенность. Внутривенная иммунизация индуцировала значительно более сильные Т-клеточные ответы в крови, селезенке, бронхоальвеолярном лаваже и лимфатических узлах легких, чем другие способы вакцинации.
Обобщая данные о различных способах доставки БЦЖ в организм, можно отметить, что при доставке через слизистые оболочки наблюдается повышение защиты от микобактерий в сравнении с принятым сейчас подкожным способом вакцинирования; в то же время, внутривенная вакцинация, по-видимому, обладает еще более сильным эффектом, но потенциально более опасна.
Использование БЦЖ для борьбы с другими заболеваниями. Известно, что вакцинация БЦЖ приблизительно вдвое снижает смертность в первые 6–12 мес. жизни [79], скорее всего, из-за стимуляции неспецифического иммунитета, приводящего к повышению устойчивости к респираторным заболеваниям и сепсису у новорожденных [80, 81]. Было показано, что вакцинация БЦЖ дает положительные неспецифические иммунные эффекты, приводящие к улучшению реакции на другие немикобактериальные патогенные микроорганизмы. В частности, вакцинация БЦЖ значительно увеличивает секрецию IL-1B, который играет важную роль в противовирусном иммунитете [82]. Вакцинированные БЦЖ мыши были устойчивее к вирусу коровьей оспы, и у них был зарегистрирован повышенный уровень продукции гамма-интерферона CD4+ клетками [83]. Феномен иммунного эффекта после гетерологичной вакцинации, получивший название “тренированный иммунитет”, предположительно вызван метаболическими и эпигенетическими изменениями, приводящими к стимулированию активности генетических областей, кодирующих провоспалительные цитокины [84].
Благодаря своим иммуностимулирующим свойствам БЦЖ является стандартной терапией для предотвращения рецидивов после операции в случае немышечно-инвазивного рака мочевого пузыря [85]. Инстилляция БЦЖ в мочевой пузырь способствует противоопухолевой активности, вероятно, за счет стимулирования рекрутирования CD4+ Т-клеток, нейтрофилов и лимфоцитов, а также активации иммунных клеток для устранения раковых клеток уротелия, инфицированных БЦЖ [85–87]. Канно с соавт. [88] получили штаммы rBCG, экспрессирующие Bordetella pertussis токсин (S1PT), для дальнейшего использования в качестве альтернативной иммунотерапии для модели рака мочевого пузыря у мышей и показали их большую эффективность в иммунотерапии [88].
Текущие исследования направлены на выявление клинических параметров и биомаркеров, которые могут предсказать индивидуальный ответ на терапию БЦЖ [89, 90]. Поскольку побочные эффекты приводят к прекращению терапии БЦЖ у некоторых пациентов, BCG ΔureC::hly в настоящее время тестируется в качестве замены в фазе II клинических испытаний.
Осуществляются попытки лечения меланомы с использованием рекомбинантных штаммов БЦЖ [91]. Наиболее успешными оказались варианты БЦЖ, экспрессирующие интерлейкин-2 (rBC-G-IL-2) и гранулоцитарно-макрофагальный колониестимулирующий фактор (rBCG-GM-CSF).
Другим возможным приложением БЦЖ может стать создание новых инструментов для борьбы с ВИЧ. На основе описанного выше вакцинного штамма MTBVAC, был создан рекомбинантный штамм MTBVAC.HIVA2auxo, в качестве двойной вакцины против туберкулеза и ВИЧ [92]. Эта вакцина против туберкулеза и ВИЧ обладает сходной протективностью с родительским штаммом MTBVAC при заражении МТБ у мышей и оказалась более безопасной, чем БЦЖ и MTBVAC для мышей с тяжелым комбинированным иммунодефицитом. Вакцина MTBVAC.HIVA2auxo, усиленная модифицированным вирусом коровьей оспы Ankara (MVA).HIVA, индуцировала специфичные для ВИЧ-1 и МТБ Т-клеточные ответы и ВИЧ-1-специфические CD8+ Т-клетки.
Был проведен анализ текущих эпидемиологических данных инфекции COVID-19, который выявил корреляцию между вакцинацией БЦЖ и снижением заболеваемостью и смертностью от COVID-19 во всем мире [93]. Было обнаружено, что страны, не практикующие вакцинацию БЦЖ (Италия, Нидерланды, США) пострадали от пандемии сильнее, чем страны с всеобщей и давней политикой вакцинации БЦЖ. Страны с поздним началом всеобщей вакцинации БЦЖ (Иран, 1984) имеют более высокую смертность, что согласуется с идеей о том, что БЦЖ защищает привитое пожилое население. Поскольку показано, что вакцинация БЦЖ обеспечивает широкую защиту от вирусных инфекций и сепсиса [94], вероятно, защитный эффект БЦЖ может быть не связан напрямую с защитой от COVID-19. Тем не менее, обнаружено, что вакцинация БЦЖ коррелировала с уменьшением числа зарегистрированных случаев COVID-19. Это позволяет предположить, что БЦЖ может предоставить некоторую защиту и непосредственно от COVID-19. Широкое использование вакцины БЦЖ в популяции может снизить количество носителей, и в сочетании с другими мерами может замедлить или остановить распространение COVID-19.
Масштабное исследование III клинической фазы покажет, эффективен ли рекомбинантный вариант БЦЖ VPM1002 против COVID-19. Исследование будет проведено на группах пожилых людей и работников здравоохранения, наиболее подверженных риску заболевания [95].
Таким образом, многочисленные исследования показывают, что БЦЖ остается многообещающей основой для генетических модификаций, способных повысить эффективность вакцины. Кроме того, адьювантные свойства БЦЖ, достаточная емкость ее генома и внутриклеточная локализация патогена позволяет рассматривать перспективы использования БЦЖ в качестве аттенуированного вектора для различных, в том числе вирусных антигенов. Неспецифические иммунотерапевтические эффекты БЦЖ делают вакцинацию ей потенциально новым инструментом в борьбе с COVID-19 и, вероятно, с другими инфекционными заболеваниями.
Работа выполнена при частичной поддержке РФФИ (грант № 1901500149A).
Список литературы
Global Tuberculosis Report. World Health Organization, 2017. Geneva. 147 p.
Uplekar M., Weil D., Lonnroth K., Jaramillo E., Lienhardt C., Dias H. et al. // Lancet. 2015. V. 385. № 9979. P. 1799–1801.
De Gijsel D., von Reyn C.F. // Int. J. Infect. Dis. 2019. V. 80. P. S6–S8.
Lewis K., Reiling L. Guinn K., Hickey M., Smith S., Behr M., Sherman D. // Bone. 2011. V. 23. № 1. P. 1–7.
Demangel C., Hinds J., Neyrolles O., Butcher P., Leclerc C., Cole, S., Brosch R. // Infect. Immun. 2006. V. 74. № 1. P. 88–98.
Simeone R., Bottai D., Brosch R. // Curr. Opin. Microbiol. 2009. V. 12. № 1. P. 4–10.
Леви Д.Т., Обухов Ю.И., Александрова Н.В., Волкова Р.А., Эльберт Е.В., Альварес Фигероа М.В., Прокопенко А.В., Луданный Р.И. // Биопрепараты. Профилактика, диагности, лечение. 2016. V. 16. P. 49–54.
Corran P., Griffiths E. // World Health Organization. WHO Technical Report Series № 980. Geneva, Switzerland. 2012. 509 p.
Behr M., Small P. // Vaccine. 1999. V. 17. № 7–8. P. 915–922.
Fomukong N., Dale J., Osborn T., Grange J. // J. Appl. Bacteriol. 1992. V. 72. № 2. P. 126–133.
Ahn S., Tran V., Leung A., Ng M., Li M., Liu J. // Mol. Ther. 2018. V. 26. № 12. P. 2863–2874.
Sun R., Skeiky Y., Izzo A., Dheenadhayalan V., Imam Z., Penn E., et al. // Vaccine. 2009. V. 27. № 33. P. 4412–4423.
Abdallah A., Hill-Cawthorne G., Otto T., Coll F., Guerra-Assunção J., Gao G., et al. // Sci. Rep. 2015. V. 5. № October P. 1–15.
Andersen P., Kaufmann S. // Cold Spring Harb Perspect Med. 2014. V. 4. № 6. pii: a018523.
Walters S.B., Dubnau E., Kolesnikova I., Laval F., Daffe M., Smith I. // Mol. Microbiol. 2006. V. 60. № 2. P. 312–330.
Flannagan R., Cosío G., Grinstein S. // Nat. Rev. Microbiol. 2009. V. 7. P. 355–366.
Stanley S., Johndrow J., Manzanillo P., Cox J. S. // J. Immunol. 2007. V. 178. № 5. P. 3143–3152.
Van der Wel N., Hava D., Houben D., Fluitsma D., van Zon M., Pierson J., Brenner M., Peters P. // Cell. 2007. V. 129. № 7. P. 1287–1298.
Kaufmann S. // Trends Immunol. 2012. V. 33. № 7. P. 373–379.
Gengenbacher M., Nieuwenhuizen N.E., Kaufmann S. // Curr. Opin. Immunol. 2017. V. 47. P. 8–16.
Baker J., Dechow S., Abramovitch R. // Trends Microbiol. 2019. V. 27. №. 11. P. 942–953.
Prusa J., Zhu D., Stallings C. // Pathog. Dis. 2018. V. 76. №. 5. P. 1–13.
Song H., Huff J., Janik K., Walter K., Keller C., Ehlers S. et al. // Mol. Microbiol. 2011. V. 80. P. 900–18.
Mukai T., Maeda Y., Tamura T., Miyamoto Y., Makino M. // FEMS Immunol. Med. Microbiol. 2008. V. 53. P. 96–106.
Sendide K., Deghmane A., Reyrat J., Talal A., Hmama Z. // Infect Immun. 2004. V. 72. P. 4200–9.
Gengenbacher M., Kaufmann S. // FEMS Microbiol. Rev. 2012. V. 36. № 3. P. 514–532.
Saiga H., Nieuwenhuizen N., Gengenbacher M., Koehler A., Schuerer S., Moura-Alves P. et al. // J. Infect. Dis. 2015. № 1. P. 1–25.
Wang Y., Curry H., Zwilling B., Lafuse W. // J. Immunol. 2005. V. 174. № 9. P. 5687–5694.
Andersen P., Scriba T. // Nat. Rev. Immunol. 2019. V. 19. № 9. P. 550–562.
Fulton, S., Reba S., Pai R., Pennini M., Torres M., Harding C., Boom W. // Infect. Immun. 2004. V. 72. № 4. P. 2101–2110.
Dorhoi A., Nouailles G., Jörg S., Hagens K., Heinemann E., Pradl L. et al. // Eur. J. Immunol. 2012. V. 42. № 2. P. 374–384.
Kupz C., Er U., Stäber M., Perdomo A., Dorhoi A., Brosch R., Kaufmann S. // J. Clin. Invest. 2016. V. 126. № 6. P. 2109–2122.
Tzelepis F., Verway M., Daoud J., Gillard J., Hassani-A-rdakani K., Dunn J. et al. // J. Clin. Invest. 2015. V. 125. № 2. P. 752–768.
Chen C., Huang D., Wang R., Shen L., Zeng G., Yao S. et al. // PLoS pathogens. 2009. V. 5. № 4. e1000392.
Lindenstrom T., Aagaar, C., Christensen D., Agger E., Andersen P. // Eur. J. Immunol. 2014. V. 44. P. 1699–1709.
Lu L., Chung A., Rosebrock T., Ghebremichael M., Yu W., Grace P. et al. // Cell. 20166. V. 167. № 2. P. 433–443.
Mavi P.S., Singh S., Kumar A. // Antioxid. Redox Signal. 2019. P. 1–59. https://doi.org/10.1089/ars.2019.7867
Gröschel M., Sayes F., Simeone R., Majlessi L., Brosch R. // Nat. Rev. Microbiol. 2016. V. 14. № 11. P. 677–691.
Augenstreich J., Arbues A., Simeone R., Haanappel E., Wegener A., Sayes F. et al. // Cell. Microbiol. 2017. V. 19. № 7. P. 1–19.
Tiwari S., Casey R., Goulding C., Hingleywilson, S., Jacobs W. // Microbiol Spectr. 2019. V. 7. https://doi.org/10.1128/microbiolspec
Collins A., Cai H., Li T., Franco L., Li X., Nair R. et al. // Cell Host Mycrobe. 2015. V. 17. № 6. P. 820–828.
Wassermann R., Gulen M., Sala C., Perin S., Lou Y., Rybniker J. et al. // Cell Host Microbe. 2015. V. 17. № 6. P. 799–810.
O. W., Bell S., MacDuff D., Kimmey J., Elie D., Olivas J. et al. // Cell Host Microbe. 2015. V. 176. № 1. P. 811–819.
Wong K., Jacobs W. // Cell. Microbiol. 2011. V. 13. № 9. P. 1371–1384.
Brodin P., Majlessi L. Brosch R., Smith D., Bancroft G., Clark S. et al. // J. Infect. Dis. 2004. V. 190. № 1. P. 115–122.
Gröschel M., Sayes F., Shin S., Frigui W., Pawlik A., Orgeur M. et al. // Cell Rep. 2017. V. 18. № 11. P. 2752–2765.
Grode L., Ganoza C., Brohm C., Weiner J., Eisele B., Kaufmann S. // Vaccine. 2013. V. 31. № 9. P. 1340–1348.
Reyrat J., Berthet F., Gicquel B.// Proc. Natl. Acad. Sci. USA. 1995. V. 92. № 19. P. 8768–8772.
Gordon M., D’Arcy Hart A., Young P. // Nature. 1980. V. 286. № 5768. P. 79–80.
Hamon M.A., Ribet D., Stavru F., Cossart P. // Trends Microbiol. 2012. V. 20. № 8. P. 360–368.
Shaughnessy L., Hoppe A., Christensen K., Swanson J. // Cell. Microbiol. 2006. V. 8. № 5. P. 781–792.
Nieuwenhuizen N., Kulkarni P., Shaligram U., Cotton M., Rentsch C., Eisele B., Grode L., Kaufmann S. // Front. Immunol. 2017. V. 8. № SEP. P. 1–9.
Rao M., Vogelzang A., Kaiser P., Schuerer S., Kaufmann S., Gengenbacher M. // PLoS One. 2013. V. 8. № 11. P. 1–10.
Gengenbacher M., Vogelzang A., Schuerer S., Lazar D., Kaiser P., Kaufmann S. // MBio. 2014. V. 5. № 3. P. 1–8.
Gengenbacher M., Nieuwenhuizen N., Vogelzang A., Liu H., Kaiser P., Schuerer S. et al. // MBio. 2016. V. 7. № 3. P. 1–10.
Sun R., Skeiky Y., Izzo A., Dheenadhayalan V., Imam Z., Penn E. et al. // Vaccine. 2009. V. 27. № 33. P. 4412–4423.
Sander P., Clark S., Petrera A., Vilaplana C., Meuli M., Selchow P. et al.// Vaccine. 2015. V. 3. № 11. P. 1353–1359.
Paolino M., Brindisi M., Vallone A., Butini S., Campiani G., Nannicini C. et al. // ChemMedChem. 2018. V. 13. № 5. P. 422–430.
Mizuno S., Soma S., Inada H., Kanuma T., Matsuo K., Yasutomi Y. // J. Immunol. 2019. V. 203. № 1. P. 188–197.
Sambandamurthy V., Derrick S., Jalapathy K., Chen B., Russell R., Morris S., Jacobs W. // Infect. Immun. 2005. V. 73. № 2. P. 1196–1203.
Arbues A., Aguilo J., Gonzalo-Asensio J., Marinova D., Uranga S., Puentes E. et al. // Vaccine. 2013. V. 31. № 42. P. 4867–4873.
Cox J., Chess B., McNeil M., Jacobs W. // Nature. 1999. V. 402. № 6757. P. 79–83.
Kirksey M., Tischler A., Siméone R., Hisert K., Uplekar S., Guilhot C., McKinney J. // Infect. Immun. 2011. V. 79. № 7. P. 2829–2838.
Aguilo N., Uranga S., Marinova D., Monzon M., Badiola J., Martin C. // Tuberculosis. 2016. V. 96. P. 71–74.
Gonzalo-Asensio J., Marinova D., Martin C., Aguilo N. // Front. Immunol. 2017. V. 8. № 12. P. 1–8.
Clark S., Lanni F., Marinova D., Rayner E., Martin C., Williams A. // J. Infect. Dis. 2017. V. 216. № 5. P. 525–533.
Tanner R., Villarreal-Ramos B., Vordermeier H., McShane H. // Front. Immunol. 2019. V. 10. № @. P. 5–7.
Hoft D., Brown R., Belshe R. // Clin. Infect. Dis. 2000. V. 30. № 3. P. S217–S222.
Monteiro-Maia R., de Pinho R. // Mem. Inst. Oswaldo Cruz. 2014. V. 109. № 6. P. 838–845.
Falero-Diaz G., Challacombe S., Banerjee D., Douce G., Boyd A., Ivanyi J.// Vaccine. 2000. V. 18. № 28. P. 3223–3229.
Lyadova I., Vordermeier H., Eruslanov E., Khaidukov S., Apt A., Hewinson R. // Clin. Exp. Immunol. 2001. V. 126. № 2. P. 274–279.
Mutsch M., Zhou W., Rhodes P., Bopp M., Chen R., Linder T., Spyr C., Steffen R. // N. Engl. J. Med. 2004. V. 350. № 9. P. 896–903.
Barclay W., Busey W., Dalgard D., Good R., Janicki B., Kasik J. et al. // Am. Rev. Respir. Dis. 1973. V. 107. № 3. P. 351–358.
Garcia-Contreras L., Wong Y., Muttil P., Padilla D., Sadoff J., De Rousse J., et al. // Proc. Natl. Acad. Sci. USA. 2008. V. 105. № 12. P. 4656–4660.
Dijkman K., Sombroek C., Vervenne R., Hofman S., Boot C., Remarque E. et al. // Nat. Med. 2019. V. 25. № 2. P. 255–262.
Scriba T., Nemes E. // Nat. Med. 2019. V. 25. № 2. P. 199–201.
Thomas Z., McShane H. // Trans. R. Soc. Trop. Med. Hyg. 2014. V. 109. № 3. P. 175–181.
Darrah P., Zeppa J., Maiello P., Hackney J., Wadsworth M., Hughes T. et al. // Nature. 2020. V. 577. № 7788. P. 95–102.
Higgins J., Soares-Weiser K., López-López J., Kakourou A., Chaplin K., Christensen H. et al. // BMJ. 2016. V. 355.
Aaby P., Whittle H., Benn C. // BMJ. 2012. V. 345. № 7864. P. 1–6.
Aaby P., Kollmann T., Benn C. // Nat. Immunol. 2014. V. 15. № 10. P. 895–899.
Kleinnijenhuis J., Quintin J., Preijers F., Benn C., Joosten L., Jacobs C. et al. // J. Innate Immun. 2014. V. 6. P. 152–158.
Mathurin K., Martens G., Kornfel H., Welsh R. // J. Virol. 2009. V. 83. P. 3528–3539.
Netea M., Joosten L., Latz E., Mills, K., Natoli G., Stunnenberg H., O’Neill L., Xavier R. // Science. 2016. V. 352. № 6284. aaf1098.
Zheng Y., Naguib Y., Dong Y., Shi Y., Bou S., Cui Z. // Expert Rev. Vaccines. 2015. V. 14. № 9. P. 1255–1275.
Pichler R., Fritz J., Zavadil C., Schäfer G., Culig Z., Brunner A. // Oncotarget. 2016. V. 7. № 26. P. 39 916–39 930.
Suttmann H., Riemensberger J., Bentien G., Schmaltz D., Stöckle M., Jocham D., Böhle A., Brandau S. // Cancer Res. 2006. V. 66. № 16. P. 8250–8257.
Kanno A., Goulart C., Leite L., Pagliarone A., Nascimento I. // Biomed Res. Int. 2019. V. 2019. P. 9 630 793.
Steinberg R., Thomas L., Mott S., O’Donnell M. // Bl. Cancer. 2016. V. 2. № 2. P. 215–224.
Zhang N., Jiang G., Liu X., Na R., Wang X., Xu J. // Biomed Res. Int. 2016. V. 2016. P. 9 859 021.
Benitez M., Bender C., Oliveira T., Schachtschneider K., Collares T., Seixas F. // Appl. Microbiol. Biotechnol. 2019. V. 103. № 19. P. 7903–7916.
Broset E., Saubi N., Guitart N., Aguilo N., Uranga S., Kilpeläinen A. et al. // Mol. Ther. – Methods Clin. Dev. 2019. V. 13. P. 253–264.
Miller A., Reandelar M., Fasciglione K., Roumenova V., Li Y., Otazu G. // medRxiv (preprint). 2020. https://doi.org/10.1101/2020.03.24.20042937
Moorlag S., Arts R., van Crevel R., Netea M. // Clin. Microbiol. Infect. 2019. V. 25. P. 1473–1478.
Max Planck Society, 2020. https://medicalxpress.com/ news/2020-03-vaccinebcg-immune-boost-coronavirus. html
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология