

Прикладная биохимия и микробиология, 2020, T. 56, № 4, стр. 327-335
Пероксидазы прокариот и их применение в биотехнологии (обзор)
Л. Е. Хмелевцова 1, *, И. С. Сазыкин 1, Т. Н. Ажогина 1, М. А. Сазыкина 1
1 Южный федеральный университет, Академия биологии и биотехнологии им. Д.И. Ивановского
344090 Ростов-на-Дону, Россия
* E-mail: lehmelevcova@sfedu.ru
Поступила в редакцию 07.10.2019
После доработки 06.12.2019
Принята к публикации 23.12.2019
Аннотация
В обзоре рассмотрены работы, посвященные разнообразию структуры и функций прокариотических пероксидаз. Описаны такие наиболее значимые группы пероксидаз, встречающиеся у бактерий, как каталазы (Kat), каталазы-пероксидазы (CP), дигемовые цитохром-с пероксидазы (DiHCcP) и краситель-обесцвечивающие пероксидазы (DyP). Приведены данные об их применении в биотехнологии. Обсуждается их участие в биодеградации трудно утилизируемых субстратов микроорганизмами и возможность использования в биоремедиации.
Пероксидазы катализируют восстановление пероксида водорода и окисляют различные субстраты. Эта биохимическая функция обусловливает их участие во многих таких важных биологических процессах, как защитные механизмы, иммунный ответ и патогенез. Эти белки присутствуют во всех царствах жизни и являются членами мультигенных семейств (насчитывают от двух до более ста изоформ) (http://peroxibase.toulouse.inra.fr/).
Согласно классификации ферментов пероксидазы относят к подклассу КФ.1.11.1.x (донор:Н2О2 оксидоредуктазы), в которую входят 15 различных пероксидаз с КФ 1.11.1.1 по 1.11.1.16 (за исключением КФ 1.11.1.4, которая в данный момент не рассматривается как пероксидаза). Из-за присутствия в их структуре двойных доменов некоторые пероксидазы отнесены к номерам КФ 1.13.11.44, КФ 1.14.99.1 и КФ 1.6.3.1, а также КФ 4.1.1.44.
Пероксидазы могут быть гем-содержащими или не содержать гем. Согласно филогенетическим деревьям, пероксидазы разделяют на 13 основных групп, которые, в свою очередь, подразделяются на более чем 60 классов (рис. 1) (http://peroxibase.toulouse.inra.fr/infos/documentation).
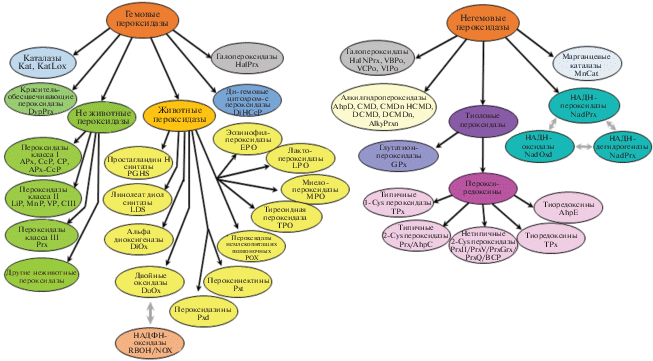
Среди гем-содержащих пероксидаз выделяют два крупных суперсемейства: первое из них встречается в основном у бактерий, грибов и растений (не животные пероксидазы) [1], а второе – главным образом у животных, грибов и бактерий (пероксидазы животных; пероксидазы-циклооксигеназы) [2, 3]. Члены первого суперсемейства пероксидаз были идентифицированы у большинства живых организмов за исключением животных. Суперсемейство включает три независимых класса структурно родственных пероксидаз (классы I, II и III), из которых у прокариот присутствуют только каталазы-пероксидазы (CP), принадлежащие к классу I. Все они обладают гемовым фрагментом (феррипротопорфирином IX), в качестве проксимального лиганда консервативным гистидином, а в качестве дистальных лигандов консервативными остатками аргинина и гистидина. Через общий пероксидантный цикл гемсодержащие пероксидазы используют в качестве акцептора H2O2, а в качестве доноров – различные субстраты, которые будут превращаться в радикалы. В этом цикле в гемовой группе пероксидазы FeIII превращается в FeIV [4].
Второе суперсемейство (пероксидазы-циклооксигеназы) включает 11 групп пероксидаз, из которых у прокариот описаны бактериальные пероксицины (Pxc) и короткие пероксидокерины (PxDo) из семейства пероксинектинов (Pxt). Семейство пероксицины, по-видимому, содержит самых старых представителей суперсемейства. Бактериальные ферменты состоят либо только из домена пероксидазы (короткие пероксицины), либо из нескольких доменов (длинные пероксицины). Физиологическая роль как пероксицинов, так и пероксдокеринов, пока неизвестна [5].
Помимо вышеописанных ферментов к гем-содержащим пероксидазам относят 4 суперсемейства каталаз (Kat), включающих ди-гемовые пероксидазы (семейство ди-гемовых цитохром-с пероксидаз (DiHCcP), белки утилизации метиламина (MauG), другие пероксидазы (DiHPOX) и краситель-обесцвечивающие пероксидазы (dye-decolorizing peroxidases, DyPs). Практически все из них представлены у прокариот.
Негемовые пероксидазы подразделяются на 5 групп: семейство алкилпероксидаз (включает 9 подсемейств), суперсемейство галопероксидаз (включает 2 семейства), семейство марганцевых каталаз (MnCat), семейство НАДН-пероксидаз (NadPrx) и суперсемейство тиоловых пероксидаз, подразделяющееся на семейства глутатионпероксидаз и пероксиредоксинов. Все группы негемовых пероксидаз описаны у прокариот.
Более подробно будут рассмотрены некоторые основные группы пероксидаз, встречающиеся у прокариотических организмов.
Каталазы. Каталаза или гидропероксидаза II (Kat или HPII) является гемсодержащим ферментом, способным дисмутировать H2O2. Это ключевой фермент, защищающий все типы организмов от активных форм кислорода. Контролируется как часть регулона sS у бактерий. Помимо перекиси водорода и алифатических перекисей эта каталаза может окислять определенные 2-электронные акцепторы. Фермент катализирует распад H2O2 по двухстадийному механизму.
Одна молекула пероксида водорода окисляет гем (Por-FeIII), а вторая молекула пероксида водорода используется в качестве восстановителя соединения I (Por+• – FeIV = O) для регенерации исходного состояния фермента :
Выделяют монофункциональные гем-каталазы (типичные или классические каталазы), каталазы-пероксидазы (атипичные), негемовые каталазы (псевдокаталазы) и минорные каталазы (на рис. 1 они разнесены в разные группы, но в этой части будет целесообразно рассмотреть их совместно).
Монофункциональные гем-каталазы являются исходным классом каталаз, встречающихся повсеместно у животных, растений и микроорганизмов. Структура каталаз варьируется в зависимости от количества и идентичности доменов у разных видов организмов. Обычно каталазы существуют в виде тетрамера, который имеет форму гантели и состоит из четырех одинаковых субъединиц. Молекулярная масса монофункциональных каталаз составляет 200–340 кДа, они содержат простетическую гемовую группу в каталитическом центре [6]. Ферменты разделены на три клады, которые, как полагают, образовались в результате дупликации соответствующих генов [7].
Негемовые каталазы являются минорным семейством бактериальных белков, у которых вместо гемовой группы присутствует димарганцевый активный сайт. Их также называют марганцевыми каталазами или псевдокаталазами. Эти ферменты имеют молекулярную массу в диапазоне 170–210 кДа. Структура марганцевой каталазы Lactobacillus plantarum в гомогексамерной форме содержит субъединицы размером около 30 кДа [8]. В димарганцевом активном сайте этого фермента каждый ион марганца координирован с тремя молекулами глутамата и одной молекулой гистидина (вместо классической координации Glu4His2), что имеет значение при катализе разложения H2O2. Псевдокаталазы дифференцированы в пять различных клад, что произошло, вероятно, благодаря латеральному переносу генов между различными бактериальными таксонами [9].
Минорные каталазы включают хлоропероксидазы, бромопероксидазы [10] и каталазы-фенолоксидазы [11]. Хлоропероксидаза встречается в виде мономера с молекулярной массой 42 кДа и одной гем-b-группой. Общая структура и активный сайт этого фермента отличаются от других каталаз и пероксидаз наличием цистеина в проксимальной части, выступающего в качестве пятого лиганда для гемового железа. Другим значительным отличием является наличие глутаминовой кислоты в виде единичного каталитического остатка на дистальной стороне гема, а не гистидина с аспарагином или аргинином как у других каталаз. Каталаза-фенолоксидаза представляет собой тетрамерный гемовый белок с молекулярной массой 320 кДа. Этот фермент обладает уникальной способностью окислять ряд фенольных соединений в отсутствие пероксида водорода [12].
Каталазы-пероксидазы. Данные ферменты (КФ 1.11.1.6; Н2О2:Н2О2 оксидоредуктазы или KatG) обнаружены у эубактерий, архей и низших эукариот (грибов и простейших). В настоящее время база PeroxiBase содержит данные об аминокислотных последовательностях 544 каталаз-пероксидаз, идентифицированных у таксономических групп Actinobacteria, Alphaproteobacteria, Bacteroidetes/Chlorobi, Betaproteobacteria, Chlamydiae/Verrucomicrobia, Cyanobacteria, Deltaproteobacteria, Firmicutes и Gammaproteobacteria.
Они являются мультифункциональными ферментами и, как правило, обладают двойной каталитической активностью, действуя как в качестве каталазы, так и в качестве пероксидазы. У бактерий они входят в состав регулона OxyR, который реагирует на активные формы кислорода. Впервые фермент, обладающий как каталазной, так и классической пероксидазной активностью, был выделен у E. coli в 1979 г. Первоначально он был описан как каталаза с пероксидазной активностью широкого спектра, что позволило его назвать “каталаза-пероксидаза” [13]. Позже по мере изучения нуклеотидной последовательности гена katG, кодирующего этот фермент, было показано его сходство с растительными пероксидазами [14], и в конечном итоге он был классифицирован как член семейства пероксидаз класса I, наряду с цитохром-с пероксидазой (CcP) и аскорбат-пероксидазой (APx) [15]. Однако в то время как классические пероксидазы обычно являются мономерами, KatG существуют в виде гомодимеров или гомотетрамеров субъединиц размером 80 кДа, каждая из которых состоит из двух доменов [16]. Хотя и из этого правила также есть исключения. Так, в работе [17] описана каталаза-пероксидаза KatG B. pumilis, которая стереоселективно превращает b-лактамы в их (R)-сульфоксиды. Она является мономером и содержит одну гемовую группу, в то время как KatG других бактерий являются димерами или тетрамерами и имеют низкое содержание гема (один на димер или два на тетрамер). N-концевая аминокислотная последовательность KatG B. pumilis не показала значительного сходства ни с одной из известных каталаз-пероксидаз KatG, присутствующих в базе данных [17].
Каталитическая реакция каталазы-пероксидазы протекает в две стадии, как и у каталазы. СР может использовать органические доноры электронов для восстановления соединения I с помощью двух одноэлектронных переносов до состояния покоя. Каталазная активность СР порой даже выше, чем у типичных каталаз, несмотря на то, что KatG не имеют с ними структурного сходства. Катализ осуществляется с помощью активного сайта, который обладает сильным сходством с сайтом типичной пероксидазы (например, цитохром-с пероксидазы). Для этого используются два взаимозависимых кофактора: гем и совершенно уникальный ковалентный аддукт Met-Tyr-Trp (MYW). Гемовая группа расположена в N-концевом домене, в котором предположительно и осуществляется катализ. Было высказано предположение, что его формирование произошло в результате дупликации гена. Гем необходим для создания кофактора MYW, который позволяет СР использовать гемовые промежуточные соединения для создания уникального механизма окисления H2O2 [18]. Несмотря на отличия KatG от каталаз и пероксидаз их общие структурные элементы представляют собой узкие каналы доступа к активным сайтам. Это оказалось благоприятным для каталитического окисления H2O2, поскольку другие гораздо более крупные доноры электронов лишены доступа к краю гема.
В работе Пассарди с соавт. [4] проведен масштабный филогенетический анализ последовательностей различных пероксидаз и предложена гипотеза о прокариотическом происхождении суперсемейства растительных/грибковых/бактериальных пероксидаз, присутствующих ныне у эукариот. Предполагается, что у бактерий и архебактерий неоднородное распределение каталаз-пероксидаз может быть связано с латеральным переносом генов. Последовательности генов каталаз-пероксидаз в эукариотических организмах не содержат интронов, и сходны между собой более чем на 70%. Это подтверждает гипотезу о том, что аминокислотные последовательности каталаз-пероксидаз грибов сформированы путем переноса бактериальных генов.
Изучение KatG имеет важное прикладное значение. Было обнаружено, что устойчивость к противотуберкулезному препарату изониазиду у Mycobacterium tuberculosis ассоциирована с мутациями в гене katG [19]. Изониазид используется для лечения туберкулеза с 1952 г., однако механизм его действия был выяснен только в 1990 гг. Каталаза-пероксидаза KatG активирует про-изониазид по реакции, которая включает замену гидразидной части молекулы НАД-фрагментом. Получающаяся в результате активированная форма изониазида, изоникотиноил-НАД, связывается с эноил-АСР-редуктазой InhA, которая ингибирует биосинтез миколовой кислоты и, следовательно, рост M. tuberculosis [20]. Мутантный фермент KatG проявляет меньшую комплементарность и снижает взаимодействие с изониазидом. Белково-лекарственный комплекс дестабилизируется таким образом [21].
Известно, что каталазная и пероксидазная функции фермента каталазы-пероксидазы необходимы для защиты бактерии от H2O2 и молекулярного кислорода, которые используются макрофагами во время фагоцитоза. КatG катаболизирует пероксиды, вырабатываемые НАДФН-оксидазой во время механизма защиты хозяина. Но предполагают, что в отсутствие этого внешнего давления KatG не имеет особого биологического смысла [22], и в большинстве случаев наличие мутаций в гене katG не обязательно приводит к образованию неполноценной бактериальной популяции [23]. Удивительно, что, несмотря на большое количество реакций, катализируемых этим ферментом, фактическая его роль in vivo, кроме удаления H2O2, не ясна, и субстрат in vivo для реакции пероксидазы остается неустановленным [16]. В работе [4] сообщается, что пероксидазы отсутствуют у большого количества бактерий, и возможно, они не настолько важны для их выживания, так как во многих бактериальных родах некоторые виды обладают СР, а другие − нет. Было даже отмечено, что в пределах одного вида некоторые штаммы содержат CP, а другие не содержат. В отсутствие пероксидаз другие белки (супероксиддисмутаза, каталаза и хлоропероксидаза, обнаруживаемые у бактерий, не содержащих СР) должны справляться с детоксификацией АФК [4].
Ди-гемовые цитохром-с пероксидазы (DiHCcP). Эти пероксидазы обнаружены у широкого спектра видов бактерий и у дрожжей, у которых они участвуют в детоксикации H2O2 путем двухэлектронного восстановления до воды и кислорода без выделения активных форм кислорода.
В качестве окисляемого субстрата эти ферменты, как и другие цитохром-с пероксидазы, используют гем с-типа. Фермент сформирован двумя доменами, каждый из которых содержит одну гемовую группу c-типа, а на границе раздела доменов расположен сайт связывания кальция. При этом обе гемовые группы ковалентно связаны с полипептидной цепью белка тиоэфирными связями с остатками цистеинов связывающего мотива Cys–X1–X2–Cys–His [24]. Вследствие присутствия двух окислительно-восстановительных центров восстановление H2O2 не требует стабилизации промежуточного радикала или образования оксоферрильного π-катионного радикала (соединение I). Молекулярная масса DiHCcP варьирует от 35 до 40 кДа. Ди-гемовые цитохром-с пероксидазы были описаны у таких микроорганизмов, как Methylococcus capsulatus, Nitrosomonas europaea, Paracoccus pantotrophus, P. aeruginosa, P. stutzeri и Rhodobacter capsulatus [25].
Было обнаружено, что экспрессия бактериальной цитохром-с пероксидазы усиливается в условиях гипоксии [26]. Н2О2 является продуктом дисмутации супероксида, активной формой кислорода, образующейся в условиях гипоксии в результате ограничения потока электронов в цепи переноса электронов. Это приводит к увеличению уровня восстановления пула кофермента Q и реакции семихинона с кислородом с образованием супероксида [27].
В работе Нобрега с соавт. [28] описан фермент YhjA E. coli, имеющий хинол-пероксидазную активность, первичная структура которого наиболее близка к классическим DiHCcP. Однако помимо двух С-концевых доменов, гомологичных обычным DiHCcP, он содержит дополнительный N-концевой домен, содержащий также гем c-типа, то есть обладает в общей сложности тремя гемовыми группами. В отличие от растворимых классических DiHCcP фермент является мембрано-связанным. Можно предположить, что YhjA в клетке E. coli представляет собой один из компонентов антиоксидантой защиты клетки при переходе от анаэробных к аэробным условиям вследствие способности использовать пул хинола, снижая таким образом уровень его автоокисления. Гомологи YhjA присутствуют и в клетках патогенных бактерий из родов Salmonella и Shigella. Предполагают, что они участвуют в механизмах инфицирования эпителиального слоя, поскольку в слизистой оболочке несколько более высокая концентрация кислорода, что приводит к индукции ключевых факторов вирулентности. В условиях инфекции у патогенных штаммов E. coli YhjA также может играть ключевую роль в подготовке к клеточной инвазии.
Краситель-обесцвечивающие пероксидазы (dye-decolorizing peroxidases, DyPs). Обесцвечивающие красители пероксидазы представляют собой семейство бактериальных и грибных пероксидаз, которые впервые были идентифицированы в 1999 г. у гриба Bjerkandera adusta (ранее Geotrichum candidum) [29]. Свое название эта группа пероксидаз получила после обнаружения их способности эффективно катализировать обесцвечивание широкого спектра промышленных красителей, в частности производных антрахинона, которые являются плохими субстратами для других пероксидаз [30]. Их характеристики, включая аминокислотную последовательность, третичную структуру, структуру каталитического центра, субстратную специфичность и оптимальный рН действия, отличаются от характеристик хорошо известных гемовых пероксидаз млекопитающих и растений [31]. Исследование структуры DyP выявило наличие двухдоменной ферредоксиноподобной складки α + β, которая отличается от всех α-спиральных складок других суперсемейств пероксидаз. В качестве каталитического аминокислотного остатка для реакции DyP с Н2О2 в отличие от других гемовых пероксидаз, содержащих гистидин, используется остаток аспарагиновой кислоты [32].
В 2006 г. аминокислотные последовательности, схожие с последовательностью DyP, были идентифицированы также у бактерий [33], что дало повод классифицировать их как новое семейство, названное пероксидазами DyP-типа. В настоящее время база PeroxiBase содержит информацию о 237 последовательностях этих пероксидаз. На основании данных об аминокислотных последовательностях и структуре они разделены на четыре филогенетически различных класса или типа DyP: A – D. Классы A – C содержат преимущественно последовательности бактериальных ферментов, в то время как класс D представлен в основном грибными DyP. Белки класса A и B имеют более низкую каталитическую активность, чем белки класса D [34]. Гены, кодирующие белки, принадлежащие к классу А и В, почти всегда обнаруживаются в бактериях. Аминокислотные последовательности белков класса A короче, чем у белков класса D, и длиннее, чем у белков класса B. Белки класса А (YcdB у E. coli, YwbN и BsDyP у B. subtilis, FepB у S. aureus, TfuDyP у Thermobifida fusca, DyPA у R. jostii RHA1) отличаются наличием специфической N-концевой последовательности Tat (twin arginine translocation).
Предполагают, что пероксидный цикл DyP, по существу, такой же, как у хорошо охарактеризованных классических пероксидаз [32]:
Все классы DyP обладают пероксидазной активностью. Помимо стандартных пероксидазных субстратов они способны окислять и ряд других соединений, 2,2'-азинобис[3-этилбензотиазолин-6-сульфоновой кислоты]-диаммониевая соль (ABTS), 2,6-диметоксифенол, синтетические красители (например Azure B и Reactive Blue 5), нефенольные метоксилированные ароматические соединения (например, вератриловый спирт), b-каротин, лигнин и Mn2+. В связи с этим DyP являются перспективным классом бактериальных пероксидаз для применения в области биотехнологии.
Деградация лигнина бактериальными пероксидазами. Большое количество исследований сосредоточено на изучении свойства DyP окислять лигнин, который относится к сложным ароматическим полимерам и, наряду с целлюлозой и гемицеллюлозой, представляет собой основной компонент клеточных стенок растений. Лигноцеллюлозная биомасса может эффективно использоваться благодаря огромному количеству и возобновляемости. Полагают, что в природе биодеградация лигнина протекает в две стадии, включающие деполимеризацию нативного лигнина и минерализацию образующихся гетерогенных ароматических соединений, при этом первую стадию осуществляют грибы, а вторую – бактерии [35]. Одними из основных деструкторов лигнина в природе являются базидиомицеты, например Phanerochaete chrysosporium и Ceriporiopsis subvermispora. Изучение деградации лигнина в основном было сосредоточено на изучении грибов, продуцирующих внеклеточные пероксидазы, которые могут атаковать лигнин. Гриб белой гнили P. chrysosporium продуцирует гемсодержащую лигнин-пероксидазу (LiP), способную катализировать расщепление модельных соединений лигнина, и гем-зависимую марганец-пероксидазу (MnP), которая катализирует быстрое окисление MnII до MnIII. Затем MnIII способен окислять ряд модельных соединений лигнина. P. chrysosporium и ряд других грибов также продуцируют внеклеточные медьзависимые ферменты, лакказы, которые способны окислять модельные соединения лигнина, используя окислительно-восстановительный потенциал. Однако несмотря на изучение в течение 25 лет грибных ферментов, разрушающих лигнин, это не привело к их широкому коммерческому использованию для его деградации [36].
До недавнего времени, несмотря на сообщения о том, что некоторые бактерии, например, Streptomyces viridosporus, могут разрушать лигнин [37], его ферментативная деградация бактериями была плохо изучена [38]. В 2011 г. в работе Ахмада с соавт. [36] было показано существование пероксидаз DyP-типа DypA и DypB у актинобактерии Rhodococcus jostii RHA1. DypB была определена у R. jostii RHA1 как новая марганец-зависимая пероксидаза, которая играет важную роль в деградации лигнина. Это была первая кинетически охарактеризованная рекомбинантная бактериальная лигнин-пероксидаза.
Позже появились сведения и о других бактериях, способных окислять лигнин. Соответствующие пероксидазы были выделены и описаны. В работе [39] сообщается об идентификации у Amycolatopsis sp. 75iv2 многофункциональной DyP2, которая обладала способностью к окислению модельных соединений лигнина и проявляла гораздо более высокую активность окисления MnII, чем другие бактериальные DyP.
Пероксидазы DyP-типа (TfuDyP) T. fusca, умеренной термофильной бактерии, проявляют краситель-обесцвечивающую активность, а также способны окислять такие субстраты, как гваякол, 2,6-диметоксифенол и вератриловый спирт. TfuDyP может катализировать энантиоселективное сульфоксидирование, то есть такой тип реакции, о котором ранее не сообщалось для пероксидаз DyP-типа [40]. Две новые бактериальные пероксидазы DyP-типа из B. subtilis и Pseudomonas putida MET94 были охарактеризованы кинетически и спектральными методами. Была идентифицирована пероксидаза Dyp1B P. fluorescens Pf-5, которая окисляла фенольные субстраты и полимерный лигнин (в частности, лигноцеллюлозу соломы пшеницы) в присутствии MnII [38]. Предполагается, что в качестве биокатализаторов для превращения лигнина в возобновляемые химические вещества бактериальные пероксидазы DyP-типа обладают большим потенциалом, который можно использовать, в частности, в целлюлозно-бумажном производстве и при производстве биотоплива [31].
В ходе микробиологической деградации лигнина обычно требуется длительное культивирование (от 10 до 100 сут) для того, чтобы достичь высокой степени его разложения. В связи с этим при разработке биотехнологий, направленных на использование лигнолитических ферментов, используются методы направленной эволюции белков. Например, в работе [41] этот метод применили для повышения эффективности бактериальной PpDyP у P. putida MET94 при действии на фенольные соединения. Так, с помощью подверженной ошибкам ПЦР с последующим высокопроизводительным скринингом использовали три раунда случайного мутагенеза гена ppDyP. Полученный рекомбинантный фермент демонстрировал каталитическую эффективность по отношению к 2,6-диметоксифенолу (DMP), повышенную в 100 раз, что было на уровне действия грибных лигнин-пероксидаз. Он был также эффективен по отношению к ряду фенольных соединений сирингильного типа, гваякола, ароматических аминов и крафт-лигнина [41].
Применение бактериальных пероксидаз для биоремедиации и деградации трудноутилизируемых соединений. В последние годы особое внимание уделялось разработке эффективных методов очистки окружающей среды от различных поллютантов. Одним из перспективных способов является ферментативная биодеградация с помощью микробных ферментов, в частности пероксидаз [42]. Пероксидаза относится к универсальным окислительным ферментам, которые могут генерировать свободные радикалы и катализировать разрушение углерод-углеродных связей в сложных органических соединениях. Биоразлагаемые интермедиаты могут затем быть использованы микроорганизмами в качестве источника углерода и энергии, что приводит к их дальнейшей деградации и, наконец, минерализации [43]. Сообщается об успешном опыте деградации различных углеводородов и ксенобиотиков в биореакторе при помощи бактериальных пероксидаз. В работе [44] описано полное разложение фенола пероксидазой в циклическом биореакторе. Для этого в биореактор добавляли Н2О2 с целью стимулирования производства внеклеточных пероксидаз микроорганизмами in situ. По такому же принципу в различного типа биореакторах осуществлялась деградация и других соединений, таких как бисфенол А [45], пары толуола [46], ацетаминофен [47] и углеводороды нефти [43].
В работе Бхарагава с соавт. [48] показано, что бактерия Aeromonas hydrophila способна деградировать краситель кристаллический фиолетовый вследствие продукции лигнинолитических ферментов – лигнин-пероксидазы и лакказы. A. hydrophila деградировала 99% красителя до фенола, 2,6-бис (1,1-диметилэтила), 2', 6'-дигидроксиацетофенона и бензола в течение 8 ч. Было показано также снижение токсичности кристаллического фиолетового. В связи с этим данный микроорганизм рассматривается как перспективный для использования в промышленности с целью эффективной очистки текстильных сточных вод.
В работе Кумар с соавт. [49] была изучена деградация меланоидинов бактериальным консорциумом, продуцирующим марганец-пероксидазу и лакказу. Меланоидины представляют собой сложные темно-окрашенные, аминокарбонильные полимерные соединения с высокой молекулярной массой, образующиеся в ходе реакции Майяра в ряде промышленных процессов [50, 51]. Они содержатся в отходах ликероводочных заводов, работающих на основе мелассы. Вследствие сложной химической структуры деградация этих соединений представляет проблему для промышленности, поэтому внимание исследователей привлекают биологические методы очистки. В условиях лабораторных экспериментов хорошие результаты были получены для различных грибов, продуцирующих лигнолитические ферменты. Показана их способность метаболизировать меланоидины в качестве единственного источника углерода и азота [52, 53]. Но промышленное применение грибов имеет свои ограничения из-за их медленного роста, образования огромного количества спор и низкого значения рН (3.0–5.0) среды, в которой происходит деградация [54]. В связи с этим перспективным методом является использование бактериальных штаммов, продуцирующих лигнолитические ферменты (марганец-пероксидазу и лакказу) для очистки сточных вод, загрязненных меланоидинами.
Еще одним важным применением бактериальных пероксидаз является деградация пластика. Известно, что биоразложение пластика крайне затруднено из-за отсутствия гидролизуемых функциональных групп в его основных цепях [55, 56]. В окружающей среде первоначальное разрушение пластика достигается путем сочетанного воздействия биотических и абиотических факторов, а затем уже он становится более доступным для микробной деградации. Было показано, что ферменты, способные разрушать лигнин (лакказа, марганец-пероксидаза и лигнин-пероксидаза), могут также участвовать в деградации пластика, в частности, полиэтилена [56]. В работе [57] сообщается о штамме B. cereus, разлагающем полиэтилен, облученный ультрафиолетом, своими внеклеточными лакказами и марганец-пероксидазами.
Одним из перспективных классов прокариотических пероксидаз, использующихся в биоремедиации (а также пищевой промышленности, медицине и фармацевтике) являются каталазы [58–60]. С целью экологической очистки окружающей среды используется микробиологическая биоремедиация, основанная на применении бактерий для разрушения поллютантов [61]. С помощью аэробной биоремедиации могут быть эффективно элиминированы, например, нефтяные углеводороды и некоторые кислородные оксиды топлива (например, метил-трет-бутиловый эфир (MTBE) [62, 63]. Бактериальные каталазы, разлагая пероксид водорода на кислород и воду, способствуют поступлению необходимого для окисления поллютантов кислорода, повышая эффективность ремедиации.
В работе Гоголевой с соавт. [64] была показана прямая зависимость между снижением каталазной активности углеводородокисляющих микроорганизмов Gordonia terrae, Rhodococcus rubropertinctus и R. erythropolis и интенсивностью деструкции нефтепродуктов. При выращивании этих бактерий в минеральной среде с дизельным топливом в качестве единственного источника углерода было отмечено накопление пероксидов в среде культивирования. Было также обнаружено, что у активных нефтедеструкторов в ходе деградации дизельного топлива увеличивалась численность бактерий и снижалась их каталазная активность, в то время как у штамма, не использующего углеводород, численность бактерий и активность каталазы практически не менялись в ходе эксперимента. На основании этого был сделан вывод о возможности оценки интенсивности процесса нефтедеструкции по снижению уровня каталазной активности. Сходные результаты получены в работах [65, 66]. Было исследовано изменение активности каталазы и супероксиддисмутазы (СОД) в ходе инкубации нефтеокисляющих микроорганизмов с нефтью и индивидуальными углеводородами в качестве единственного источника углерода и энергии. Показана высокая активность каталазы при росте культуры на богатой среде, и снижение ее при культивировании микроорганизмов в средах с углеводородами. При этом уровень СОД, напротив, возрастал при культивировании с углеводородами.
В ряде работ [67–70] описано использование каталазной активности почв в качестве индикатора деградации углеводородов в ходе биоремедиации. Было показано, что каталазная активность почв снижается сразу после внесения нефтепродуктов, однако затем возрастает при разложении углеводородов. Предполагают, что каталазная активность может быть полезным биомаркером для индикации начала процесса биодеградации и по ее снижению можно делать вывод о снижении скорости биодеградации. Однако необходимо отметить, что каталазная активность почв обеспечивается не только активностью ферментов аэробных микроорганизмов, но и каталаза-миметическим действием содержащихся в почве таких компонентов, как органические соединения, глинистые минералы и металлы переменной валентности [71]. Вопрос индикаторного значения и механизмов участия каталазы в процессе бактериальной биодеградации углеводородов требует более тщательного изучения.
Таким образом, пероксидазы представляют собой многообразную группу ферментов. Появившись на заре эволюции живых организмов, они выполняют важнейшую функцию – детоксикацию активных форм кислорода, защищая клетки от повреждений. Дальнейшая дивергенция пероксидаз привела к появлению многочисленных классов этих ферментов, обладающих не только пероксидазной, но и каталазной, циклооксигеназной, хлорит-дисмутазной и пероксигеназной активностью. Следует отметить, что, будучи ферментами, направленными на элиминацию активных форм кислорода, пероксидазы способны их производить при осуществлении каталитического цикла. Это является не только побочным эффектом, но и дает бактериям преимущества в виде способности неспецифически окислять различные субстраты, повышая их биодоступность и расширяя спектр возможных экологических ниш. Это свойство нашло широкое применение в биотехнологии и промышленности как экологически безопасная и эффективная альтернатива химическим методам очистки окружающей среды от поллютантов.
Работа выполнена при финансовой поддержке Министерства образования и науки РФ (проект № 6.2379.2017/ПЧ) и Российского Фонда Фундаментальных Исследований (проект № 17-04-00787 А).
Список литературы
Passardi F., Theiler G., Zamocky M., Cosio C., Rouhier N., Teixera F., Margis-Pinheiro M., Ioannidis V., Penel C., Falquet L., Dunand C. // Phytochemistry. 2007. V. 68. № 12. P. 1605–1611.
Daiyasu H., Toh H. // J. Mol. Evol. 2000. V. 51. № 5. P. 433–445.
Furtmuller P.G., Zederbauer M., Jantschko W., Helm J., Bogner M., Jakopitsch C., Obinger C. // Arch. Biochem. Biophys. 2006. V. 445. № 2. P. 199–213.
Passardi F., Bakalovic N., Teixeira F.K., Margis-Pinheiro M., Penel C., Dunand C. // Genomics. 2007. V. 89. № 5. P. 567–579.
Nicolussi A., Auer M., Sevcnikar B., Paumann-Page M., Pfanzagl V., Zámocký M., Hofbauer S., Furtmüller P.G., Obinger C. // Arch. Biochem. Biophys. 2018. V. 643. P. 14–23.
Lee D.H., Oh D.C., Oh Y.S., Malinverni J.C., Kukor J.J., Kahang H.Y. // J. Microbial. Biotechnol. 2007. V. 17. № 9. P. 1460–1468.
Klotz M.G., Klassen G.R., Loewen P.C. // Mol. Biol. Evol. 1997. V. 14. № 9. P. 951–958.
Barynin V.V., Whittaker M.M., Antonyuk S.V., Lamzin V.S., Harrison P.M., Artymiuk P.J., Whittaker J.W. // Structure. 2001. V. 9. № 8. P. 725–738
Zamocky M., Furtmuller P.G., Obinger C. // Antioxid. Redox. Signal. 2008. V. 10. № 9. P. 1527–1548.
Nicholls P., Fita I., Loewen P.C. // Adv. Inorg. Chem. 2001. V. 51. P. 51–106.
Vetrano A.M., Heck D.E., Mariano T.M., Mishin V., Laskin D.L., Laskin J.D. // J. Biol. Chem. 2005. V. 280. № 42. P. 35372–35381.
Kocabas D.S., Bakir U., Phillips S.E., McPherson M.J., Ogel Z.B. // Appl. Microbiol. Biotechnol. 2008. V. 79. № 3. P. 407–415.
Claiborne A., Fridovich I. // J. Biol. Chem. 1979. V. 254. № 10. P. 4245–4252.
Triggs-Raine B.L., Doble B.W., Mulvey M. R., Sorby P.A., Loewen P.C. // J. Bacteriol. 1988. V. 170. № 9. P. 4415–4419.
Welinder K.G., Mauro J.M., Nørskov-Lauritsen L. // Biochem. Soc. Trans. 1992. V. 20. № 2. P. 337–340.
Singh R., Wiseman B., Deemagarn T., Jha V., Switala J., Loewen P.C. // Arch. Biochem. Biophys. 2008. V. 471. № 2. P. 207–214.
Sangar S., Pal M., Moon L.S., Jolly R.S. // Bioresour. Technology. 2012. V. 115. P. 102–110.
Njuma O.J., Ndontsa E.N., Goodwin D.C. // Arch. Biochem. Biophys. 2014. V. 544. P. 27–39.
Zhang Y., Heym B., Allen B., Young D., Cole S. // Nature. 1992. V. 358. № 6387. P. 591–593.
Lei B., Wei C.-J., Tu S.-C. // J. Biol. Chem. 2000. V. 275. № 4. P. 2520–2526.
Singh A., Singh A., Grover S., Pandey B., Kumari A., Grover A. // Gene. 2018. V. 641. P. 226–234.
Singh R., Wiseman B., Deemagarn T., Donald L.J., Duckworth H.W., Carpena X., Fita I., Loewen P.C. // J. Biol. Chem. 2004. V. 279. № 41. P. 43098–43106.
Pym A.S., Saint-Joanis B., Cole S.T. // Infect. Immun. 2002. V. 70. № 9. P. 4955–4960.
Fulop V., Ridout C.J., Greenwood C., Hajdu J. // Structure. 1995. V. 3. № 11. P. 1225–1233.
Hoffmann M., Seidel J., Einsle O. // J. Mol. Biol. 2009. V. 393. № 4. P. 951–965.
Otten M.F., Stork D. M., Reijnders W.N., Westerhoff H.V., van Spanning R.J. // Eur. J. Biochem. 2001. V. 268. № 8. P. 2486–2497.
Pettigrew G.W., Echalier A., Pauleta S.R. // J. Inorg. Biochem. 2006. V. 100. № 4. P. 551–567.
Nóbrega C.S., Devreese B., Pauleta S.R. // BBA. Bioenergetics. 2018. V. 1859. № 6. P. 411–422.
Kim S.J. Shoda M. // Biotechnol. Bioeng. 1999. V. 62. № 1. P. 114–119.
Yoshida T., Sugano Y. //Arch. Biochem. Biophys. 2015. V. 574. P. 49–55.
Rahman Pour R., Ehibhatiomhan A., Huang Y., Ashley B., Rashid G.M., Mendel-Williams S., Bugg T.D.H. // Enzyme Microb.Technol. 2019. V. 123. P. 21–29.
Singh R., Eltis L.D. //Arch. Biochem. Biophys. 2015. V. 574. P. 56–65.
Sturm A., Schierhorn A., Lindenstrauss U., Lilie H., Bruser T. // J. Biol. Chem. 2006. V. 281. № 20. P. 13972–13978.
van Bloois E., Torres D.E. // Appl. Microbiol. Biotechnol. 2010. V. 86. № 5. P. 1419–1430.
Kamimura N., Sakamoto S., Mitsuda N., Masai E., Kajita S. // Curr. Opin. Biotechnol. 2019. V. 56. P. 179–186.
Ahmad M., Roberts J.N., Hardiman E.M., Singh R., Eltis L.D., Bugg T.D. // Biochemistry. 2011. V. 50. № 23. P. 5096–5107.
Ramachandra M., Crawford D.L., Hertel G. // Appl. Environ. Microbiol. 1988. V. 54. № 12. P. 3057–3063.
Rahman Pour R., Bugg T.D.H. // Arch. Biochem. Biophys. 2015. V. 574. P. 93–98.
Brown M.E., Barros T., Chang M.C.Y. // ACS Chem. Biol. 2012. V. 7. № 12. P. 2074–2081.
Lončar N., Colpa D.I., Fraaije M.W. // Tetrahedron. 2016. V. 72. № 46. P. 7276–7281.
Brissos V., Tavares D., Sousa A.C., Robalo M.P., Martins L.O. // ACS Catal. 2017. V. 7. № 5. P. 3454–3465.
Karigar C.S., Rao S.S. // Enzym. Res. 2011. Article ID 805187. P. 11. https://doi.org/10.4061/2011/805187
Moussavi G., Shekoohiyan S., Naddafi K. // Chem. Eng. J. 2017. V. 308. P. 1081–1089.
Pourakbar M., Moussavi G., Yaghmaeian K. // RSC Adv. 2018. V. 8. P. 6293–6305.
Moussavi G., Haddad F.A. // Chemosphere. 2019. V. 222. P. 549–555.
Aghayani E., Moussavi G., Naddafi K. // J. Hazard. Mater. 2019. V. 361. P. 259–266.
Baratpour P., Moussavi G. // Chemosphere. 2018. V. 210. P. 1115–1123.
Bharagava R.N., Mani S., Mulla S.I., Saratale G.D. // Ecotoxicol. Environ. Saf. 2018. V. 156. P. 166–175.
Kumar V., Chandra R. // World J. Microbiol. Biotechnol. 2018. V. 34. № 32. https://doi.org/10.1007/s11274-018-2416-9
Bharagava R.N., Chandra R. // Biodegradation. 2010. V. 21. P. 703–711.
Echavarría A.P., Pagán J., Ibarz A. // Food Sci. Technol. Int. 2014. V. 20. № 2. P. 119–126.
Miyata N., Mori T., Iwahori K., Fujita M. // J. Biosci. Bioeng. 2000. V. 89. № 2. P. 145–150.
González T., Terrón M.C., Yagüe S., Junca H., Carbajo J.M., Zapico E.J., Silva R., Arana-Cuenca A., Téllez A., González A.E. // Res. Microbiol. 2008. V. 159. № 2. P. 103–109.
Arimi M.M., Zhang Y., Götz G., Kiriamiti K., Geißen S.U. // Int. Biodeterior. Biodegrad. 2014. V. 87. P. 34–43.
Restrepo-Florez J.-M., Bassi A., Thompson M.R. // Int. Biodeterior. Biodegrad. 2014. V. 88. P. 83–90.
Krueger M.C., Harms H., Schlosser D. // Appl. Microbiol. Biotechnol. 2015. V. 99. № 21. P. 8857–8874.
Sowmya H.V., Ramalingappa M.K., Thippeswamy B. // Adv. Polym. Sci. Technol. Int. J. 2014. V. 4. № 2. P. 28–32.
Margesin R., Zimmerbauer A., Schinner F. // Biotechnol. Tech. 1999. V. 13. № 12. P. 859–863.
Majtan V., Hostacka A., Majtanova L., Trupl J. // Folia Microbiol. 2002. V. 47. № 4. P. 445–449.
van der Maarel M.J.E.C., van der Veen B., Uitdeha-ag J.C.M., Leemhuis H., Dijkhuizen L. // J. Biotechnol. 2002. V. 94. № 2. P. 137–155.
Margesin R., Zimmerbauer A., Schinner F. // Chemosphere. 2000. V. 40. № 4. P. 339–346.
Yerushalmi L., Manuel M.F., Guiot S.R. // Biodegradation. 1999. V. 10. P. 341–352.
Sutton N.B., Grotenhuis J.T.C., Langenhoff A.A.M., Rijnaarts H.H.M. // J. Soils Sediments. 2011. V. 11. P. 129–140.
Гоголева О.А., Немцева Н.В., Бухарин О.В. // Прикл. биохимия и микробиология. 2012. Т. 48. № 6. С. 612–617.
Sazykin I., Sazykina M., Khmelevtsova L., Khammami M., Karchava S., Kudeevskaya E. // Ann. Microbiol. 2016. https://doi.org/10.1007/s13213-015-1188-9
Sazykin I.S., Sazykina M.A., Khmelevtsova L.E., Seliverstova E.Yu., Karchava Sh. K., Zhuravleva M.V. // Arch. Microbiol. 2018. V. 200. № 7. P. 1057–1065.
Achuba F.I., Peretiemo-Clarke B.O. // Int. Agrophys. 2008. V. 22. № 1. P. 1–4.
Leilei Z., Mingxin H., Suiyi Z. // Afr. J. Microbiol. Res. 2012. V. 6. № 6. P. 1213–1220.
Achuba F.I., Okoh P.N. // Open J. Soil Sci. 2014. V. 4. P. 399–406.
Ogbolosingha A.J., Essien E.B., Ohiri R.C. // J. Environ. Earth Sci. 2015. V. 5. № 14. P. 128–142.
Petigara B.R., Blough N.V., Mignerey A.C. // Environ. Sci. Technol. 2002. V. 36. № 4. P. 639–645.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология