

Журнал неорганической химии, 2022, T. 67, № 6, стр. 782-793
Формирование соединения BiAl3(PO4)2(OH)6 со структурой вейлендита в гидротермальных условиях
Д. П. Еловиков a, b, *, М. В. Томкович a, А. А. Левин a, О. В. Проскурина a, b
a Физико-технический институт им. А.Ф. Иоффе
194021 Санкт-Петербург, ул. Политехническая, 26, Россия
b Санкт-Петербургский государственный технологический институт (технический университет)
190013 Санкт-Петербург, Московский пр-т, 26, Россия
* E-mail: syncdima@mail.ru
Поступила в редакцию 01.11.2021
После доработки 25.11.2021
Принята к публикации 29.11.2021
- EDN: BBQUWQ
- DOI: 10.31857/S0044457X2206006X
Аннотация
В гидротермальных условиях при температуре 200°С и давлении 7 МПа впервые синтезировано соединение BiAl3(PO4)2(OH)6 со структурой вейлендита. Показано, что формирование соединения происходит через образование промежуточной кристаллической фазы – тригональной модификации BiPO4 и аморфной фазы состава 0.5[3Al2O3 · 2P2O5 · 6H2O]. Согласно результатам растровой электронной микроскопии (РЭМ), частицы, по морфологии напоминающие частицы фазы со структурой вейлендита, обнаруживаются после 6 ч синтеза. Образование BiAl3(PO4)2(OH)6 в количествах, различимых с помощью рентгеновской дифракции, фиксируется через 12 ч и практически полностью заканчивается через 48 ч гидротермальной обработки суспензии прекурсоров. Формирование вейлендита подтверждается методами РЭМ и ИК-спектроскопии. Методом Ритвельда уточнена структура BiAl3(PO4)2(OH)6 по порошковым рентгенодифракционным данным (пр. гр. $R\bar {3}m$ (166) в гексагональных осях, параметры элементарной ячейки: а = 6.99608(5), с = 16.1495(4) Å). Коэффициенты заполнения позиций более тяжелых, чем кислород, атомов в результате уточнения равны единице в пределах стандартного отклонения. Измеренная пикнометрическая плотность образца (4.01(8) г/см3) несколько меньше плотности (4.235(2) г/см3), рассчитанной по результатам уточнения структуры.
ВВЕДЕНИЕ
Анализ имеющейся к настоящему времени литературы показал отсутствие сведений по синтезу соединения BiAl3(PO4)2(OH)6 со структурой вейлендита. Вместе с тем известны данные о природных минералах со структурой вейлендита, которые в зависимости от их происхождения значительно варьируются по своему составу и структурным параметрам (табл. 1). Причиной широкого диапазона составов этого минерала является высокая способность к ионному замещению.
Таблица 1.
Состав и параметры ячейки минералов со структурой вейлендита
| Состав минерала | а, Å | c, Å | Источник |
|---|---|---|---|
| (Bi, Ca)Al3(PO4, SiO4)2(OH)6 | 6.9649(8) | 16.256(1) | [1] |
| (Bi, Ca)Al3(PO4, SiO4)2(OH)6 (примеси Fe, Mn, Cu, Ba) | 6.9834(3) | 16.175(1) | [2] |
| (Bi, Ca)Al3(PO4, SiO4)2(OH)6 | 6.9744(13) | 16.293(4) | [3] |
| (Bi0.71Ca0.26K0.04)Σ1.01(Al1.99Fe0.87)Σ2.86 [(PO4)1.07(AsO4)0.48(PO3OH)0.29(SiO4)0.16]Σ2.00 (OH)5.42 | 6.984(2) | 16.304(6) | [4] |
| (Bi0.54Sr0.31Ca0.25K0.01Ba0.01)Σ1.12H0.18(Al2.96Cu0.02)Σ2.98 [(P0.97S0.03Si0.01)Σ1.00O4]2 (OH)6 | 7.0059(7) | 16.3431(12) | [5] |
| (Bi0.54Ca0.24Na0.02Pb0.02)Σ0.82(Al2.87Fe0.20)Σ3.07 [(PO4)2.07(SiO4)0.02(WO4)0.01]Σ2.10(OH)4.88F0.21 | – | – | [6] |
| (Bi0.92Ca0.11K0.01)Σ1.04(Al2.65Fe0.38)Σ3.04 [(PO4)1.87(SiO4)0.08(SO4)0.01]Σ1.96(OH)6.23 | – | – | [6] |
| (Bi0.93Ca0.14Ba0.01)1.08(Al3.16Fe0.04)3.20 (P0.99Si0.01O4)(PO4)(O0.69OH5.31)6 | 6.995(9) | 16.15(3) | [7] |
| (Bi, Ca)Al3(PO4, SiO4)2(OH)6 | 6.9744–6.9834 | 16.175–16.293 | [8] |
| (Bi, Ca, Sr)Al3(PO4, SiO4)2(OH)6 | 7.031(4) | 16.422(5) | [9] |
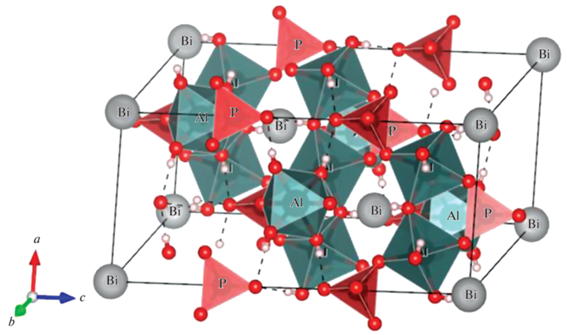
Структуру минерала вейлендита, входящего в группу крандаллита (пр. гр. $R\bar {3}m$ (166)) с общей формулой AAl3(BO4)2(OH, H2O)6 [10–12], можно представить состоящей из слоев искаженных октаэдров [AlO6]. Осевые атомы кислорода октаэдров [AlO6] из меньших тригональных колец направлены друг к другу и представляют собой основу для тетраэдров [BO4]. В случае вейлендита это тетраэдры [PO4] (рис. 1). Катион А окружен шестью антипризматически расположенными октаэдрическими плоскостями, образующими вполне правильный координационный многогранник, состоящий из 20 равносторонних и равнобедренных треугольников. В структуре идеального вейлендита катион А – это Bi (рис. 1). Структура перпендикулярно оси с представляет собой решетку кагомэ. В структуре имеются большие по размеру 12-, средние 6- и маленькие 4-кратно координированные кислородом пустоты, в которых располагаются положительно заряженные ионы [8]. Благодаря своей гибкой кристаллохимии и высокой термодинамической стабильности соединения со структурой крандаллита могут найти применение в качестве матриц для токсичных ионов таких элементов, как As, Pb, Hg, Tl, Sb, Cr, Se, радиоактивных изотопов K, Sr, Th, U и редкоземельных элементов [12].
Соединения с подобной структурой можно получить методом осаждения из растворов солей с продолжительной выдержкой осадка в маточном растворе, однако большим недостатком такого метода является его длительность (в случае синтеза соединений плюмбогуммита и крандаллита – до нескольких недель [13]). Существенного повышения скорости образования гидроксидных и оксидных фаз можно достичь гидротермальной, гидротермально-микроволновой и гидротермально-ультразвуковой обработкой осажденных прекурсоров [14–16]. Так, методом гидротермального синтеза были получены многие искусственные минералы [17–21], отличающиеся в положительную сторону от природных минералов строго определенным составом. Использование в качестве объектов исследования соединений с заданным составом позволяет более точно определить количественные соотношения между составом, структурными параметрами и свойствами веществ, чем в случае природных минералов [22–27].
В связи с этим настоящая работа направлена на изучение процессов формирования соединения BiAl3(PO4)2(OH)6 со структурой минерала вейлендита, определение условий получения и его структурное исследование.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исходных реагентов использовали Bi(NO3)3 ⋅ 5H2O (ч. д. а.), Al(NO3)3 ⋅ 9H2O (х. ч.), (NH4)2HPO4 (ч. д. а.), HNO3 (ос. ч.) и NaOH (х. ч.). Навески нитратов висмута и алюминия, рассчитанные для получения 1.5 г BiAl3(PO4)2(OH)6, растворяли в 2 мл 6 М водного раствора HNO3 при нагревании и перемешивании до полного растворения. К полученному раствору при перемешивании по каплям приливали раствор (NH4)2HPO4, приготовленный растворением навески, рассчитанной согласно стехиометрии, в 10 мл дистиллированной воды. Далее к полученной суспензии по каплям и при перемешивании приливали 4 М водный раствор NaOH для достижения pH 7. Выбранные выше концентрации водных растворов были подобраны экспериментально.
Гидротермальную обработку суспензий проводили в стальных автоклавах с тефлоновыми ампулами при температуре 200°С и давлении 7 МПа с различной продолжительностью изотермической выдержки. Полученный осадок отделяли на центрифуге, промывали дистиллированной водой до нейтральной среды и сушили при 80°С в течение 6 ч. Выход чистого соединения со структурой вейлендита составил ~90 мас. % от возможного.
Определение морфологии и размеров частиц, элементного состава образцов осуществляли методом растровой электронной микроскопии (РЭМ) и рентгеноспектрального микроанализа на электронном микроскопе Tescan Vega 3 SBH (Tescanbrno, Чешская Республика) с приставкой для элементного анализа Inca X-Act (Oxford Instruments plc, Великобритания).
Инфракрасные спектры в режиме пропускания снимали на ИК-фурье-спектрометре ФСМ 1202 (Мониторинг, Россия). Образцы готовили прессованием таблеток с KBr.
Пикнометрическую плотность определяли на гелиевом пикнометре Quantachrome Ultrapycnometer 1000 (Quantachrome Instruments, США).
Рентгенодифракционные (РД) измерения порошкового образца проводили на дифрактометре SmartLab 3 (Rigaku, Япония), оснащенном рентгеновской трубкой с медным анодом, в геометрии Брэгга–Брентано. Монохроматизацию излучения (CuKα-дублетная рентгеновская линия) проводили с помощью никелевого Kβ-фильтра. РД-рефлексы регистрировали посредством полупроводникового линейного детектора D/teX Ultra 250 (Rigaku, Япония). РД-измерения осуществляли в симметричном 2θ–θ-режиме (с углом дифракции 2θ и углом θ отражения рентгеновских лучей от поверхности образца) при комнатной температуре окружающего воздуха (298 K). Для определения угловых поправок дифрактограммы (на ноль детектора Δ2θzero и смещение Displ из-за несовпадения поверхности образца с фокальной плоскостью дифрактометра) выполняли дополнительные РД-измерения синтезированного порошка соединения BiAl3(PO4)2(OH)6 со структурой вейлендита в смеси с порошковым РД-стандартом Si640f (NIST, США) со скоростью съемки дифрактограммы 0.1 град/мин.
Количественный рентгенофазовый анализ (РФА) всех образцов проводили с использованием порошка α-Al2O3 в качестве внутреннего эталона.
Для образца, полученного в результате изотермической выдержки в течение 14 сут, на котором проводили исследования структуры, РФА выполняли с помощью программ EVA [28]. При идентификации возможных кристаллических фаз использовали порошковую базу данных PDF-2 (Powder Diffraction File-2) [29]. При помощи этой же программы EVA определяли параметры наблюдаемых РД-рефлексов (углы Брэгга 2θ и максимальные (Imax) и интегральные (Iint) интенсивности) этого образца для дальнейшего анализа. По найденным углам Брэгга 2θ проиндексированных наблюдаемых рефлексов вейлендита, исправленных на угловые поправки Δ2θzero и Δ2θdispl = = 2Displ/Rgoncosθ, где Rgon – известный радиус гониометра дифрактометра, θ – половина наблюдаемого угла Брэгга рефлекса, методом наименьших квадратов по программе Celsiz [30] были найдены параметры элементарной ячейки кристаллической фазы вейлендита.
Подгонку дифрактограмм образца после изотермической выдержки в течение 14 сут осуществляли методами Ле Бойла (Le Bail, ЛБ) [31] и Ритвельда [32]. В методе ЛБ необходимо задание только пространственной группы соединения и примерных значений параметров его элементарной ячейки, которые уточняются в процессе подгонки. Интенсивности рефлексов в методе ЛБ не рассчитываются из параметров структуры (координат атомов, их температурных факторов и заселенности позиций), как в методе Ритвельда, а берутся прямо из наблюдаемой интенсивности рефлексов на дифрактограмме, что позволяет избежать моделирования влияния преимущественной ориентации. В отличие от метода ЛБ, метод Ритвельда позволяет уточнить не только параметры элементарной ячейки, но и все остальные параметры структуры. Для подгонок ЛБ и Ритвельда использовали ритвельдовскую программу TOPAS [33]. Дублет CuKα описывали спектром из пяти линий [34], как рекомендуется в [33] для излучения, монохроматизированного Ni-фильтром. Фон описывали полиномом Чебышева 5-го порядка [33] с вкладом гиперболической функции фона для малых углов (2θ < 10°). Профили рефлексов описывали в рамках модели псевдо-Войта в модификации Томсона–Кокса–Гастингса (pseudo-Voigt Thomson–Cox–Hastings, pV-TCHZ в TOPAS) [33, 35]. В уточнении использовали весовую схему wi = 1/yi, где yi – интенсивность (в импульсах) на шаге i РД-картины.
Исследование температурных границ стабильности соединения BiAl3(PO4)2(OH)6 проводили с использованием дифрактометра XRD-7000 (Shimadzu, Япония) с высокотемпературной приставкой HTK 1200 N (Anton Paar, Австрия). Температуру повышали пошагово от 100 до 500°С с шагом 100°С и от 500 до 540°С с шагом 10°С. При каждой температуре осуществляли изотермическую выдержку образца в течение 10 мин. Съемку дифрактограмм проводили в следующем режиме: шаг сканирования 0.04°, время выдержки в точке 0.6 с.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
По данным элементного анализа, соотношение висмута, алюминия и фосфора во всех образцах соответствует атомному соотношению элементов в соединении BiAl3(PO4)2(OH)6 с погрешностью до 5 ат. %.
По данным рентгеновской дифрактометрии (рис. 2), до гидротермальной обработки в образце присутствует кристаллический фосфат висмута BiPO4 · 0.67H2O (тригональная сингония, карта PDF-2 01-080-0208) и аморфная фаза. С использованием внутреннего стандарта (α-Al2O3) было определено соотношение кристаллических фаз и аморфного вещества в образцах на разных этапах синтеза (рис. 3). Рефлексы фазы со структурой вейлендита обнаружены после изотермической выдержки в течение 12 ч в гидротермальных условиях при t = 200°С, p = 7 МПа в гидротермальном растворе с pH 7, а формирование соединения BiAl3(PO4)2(OH)6 практически заканчивается после гидротермальной обработки в течение 48 ч (рис. 2).
Рис. 2.
Рентгеновские дифрактограммы образцов с разной продолжительностью гидротермальной обработки: 1 – 0, 2 – 6, 3 – 12, 4 – 24, 5 – 30, 6 – 48 ч, 7 – 14 сут. Квадраты – кристаллическая фаза BiPO4 · 0.67H2O (карта PDF-2 01-080-0208), кружки – кристаллическая фаза BiAl3(PO4)2(OH)6 со структурой вейлендита.
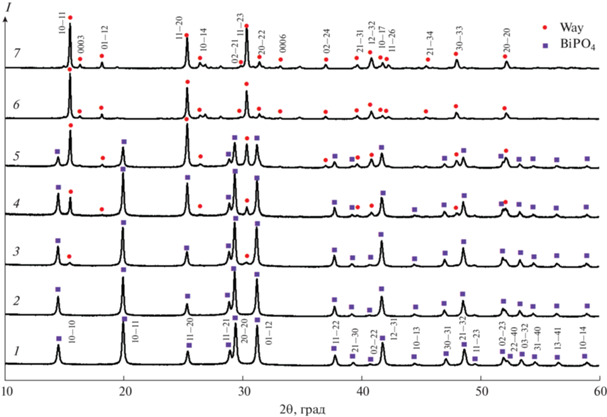
Рис. 3.
Зависимость массового содержания фаз (α) от продолжительности изотермической выдержки (τ) в гидротермальных условиях. 1 – BiPO4, 2 – аморфная фаза, 3 – фаза соединения BiAl3(PO4)2(OH)6 со структурой вейлендита.
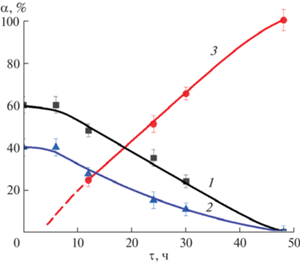
Соотношение массовых долей аморфной фазы и кристаллического BiPO4 до гидротермальной обработки осадка и на протяжении всего процесса фазовых превращений приблизительно соответствует 3 : 2 (рис. 3) с погрешностью определения соотношения фаз 5 мас. %, что хорошо соответствует стехиометрии реакции образования BiAl3(PO4)2(OH)6 из BiPO4 и аморфной фазы состава 0.5[3Al2O3 · 2P2O5 · nH2O] (n ~ 6; количество воды может варьироваться по сравнению со стехиометрическим ее содержанием в BiAl3(PO4)2(OH)6 вследствие адсорбции).
Результаты анализа методом РЭМ показывают, что некоторое количество тонких стержнеобразных частиц, которые по морфологии напоминают частицы фазы со структурой вейлендита, обнаруживается уже после 6 ч синтеза (рис. 4а). Однако доля таких частиц настолько мала, что на рентгеновской дифрактограмме данная фаза, по-видимому, не может быть зафиксирована (рис. 2).
Рис. 4.
Микрофотографии и распределение частиц по длине образцов после изотермической выдержки (L – длина частиц): а – 6; б – 12; в – 48 ч; г – 6; д – 14 сут.
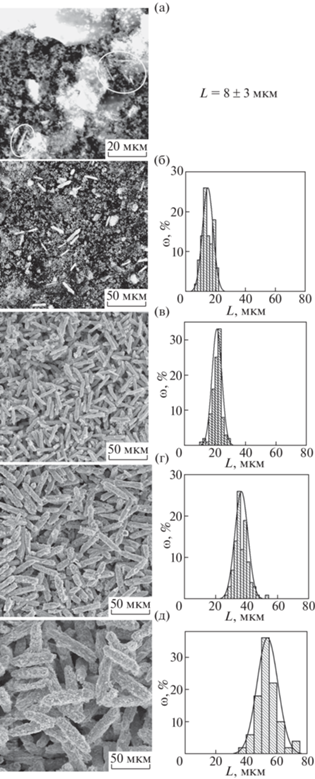
Приведенная на рис. 4в микрофотография может служить подтверждением данных рентгеновской дифракции, показавших, что после 48 ч изотермической выдержки исходных фаз синтез соединения BiAl3(PO4)2(OH)6 практически завершился (рис. 3). Дальнейшая эволюция образцов при их гидротермальной обработке состоит в росте частиц фазы BiAl3(PO4)2(OH)6 (рис. 4г, 4д), являющихся агрегатами, составленными из более мелких частиц размером до ~1–3 мкм.
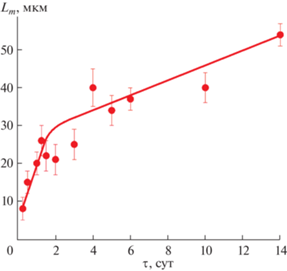
Зависимость средней длины частиц (Lm) фазы со структурой вейлендита от продолжительности гидротермальной обработки можно представить состоящей из двух близких к прямолинейным участков (рис. 5). Изотермической выдержке в гидротермальных условиях продолжительностью до 48 ч (2 сут) соответствует участок более быстрого роста частиц (~10 мкм/сут), а продолжительностью от 2 до 14 сут – участок более медленного роста частиц (~2 мкм/сут). Такое резкое изменение в скорости роста, по-видимому, связано с завершением процесса фазообразования после гидротермальной обработки в течение 2 сут.
Рис. 5.
Зависимость длины частиц (Lm) соединения со структурой вейлендита от продолжительности (τ) изотермической выдержки.
На ИК-спектрах образцов (рис. 6) процессы фазообразования проявляются в появлении и исчезновении определенных полос поглощения в зависимости от продолжительности изотермической выдержки в гидротермальных условиях. В образце 1, в котором, по данным рентгеновской дифрактометрии, присутствуют аморфная фаза и фаза фосфата висмута BiPO4, наблюдается широкая интенсивная полоса при 1045 см–1, которая соответствует симметричным колебательным модам [PO4]3–. В образце 6, состоящем в основном из соединения BiAl3(PO4)2(OH)6 со структурой вейлендита, эта полоса расщепляется на несколько полос, которые соответствуют симметричным (956, 1100 см–1) и асимметричным (1228 см–1) валентным колебаниям связи P–O в тетраэдрическом звене [PO4]3–. Похожие полосы были отмечены и для минерала крандаллита CaAl3(PO4)2(OH)5 · ⋅ (H2O) (967, 1109 и 1223 см–1) [36]. Вокруг широкого пика (605 см–1), по-видимому, наблюдаются асимметричные колебания связи P–O [37–39].
Рис. 6.
ИК-спектры образцов с разной продолжительностью изотермической выдержки. 1 – 0, 2 – 6, 3 – 12, 4 – 24, 5 – 30, 6 – 48 ч, 7 – 14 сут.
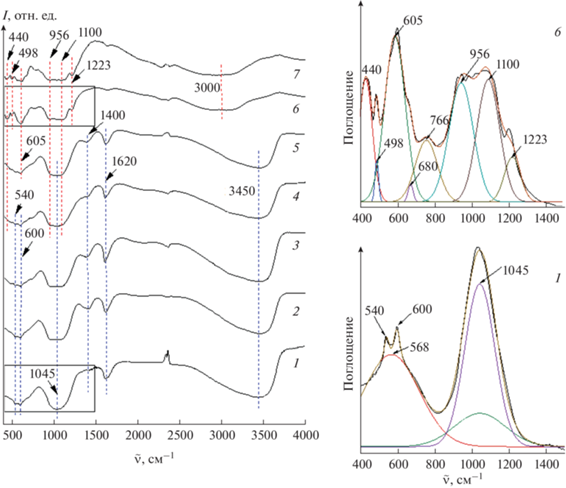
Широкая полоса при 3450 см–1 и полоса при 1620 см–1 указывают на присутствие молекул H2O в образцах 1–5, содержащих фосфат висмута и аморфную фазу.
Малоинтенсивная полоса при 1400 см–1, присутствующая в образцах, содержащих аморфную фазу и фазу фосфата висмута, отвечает валентным колебаниям связи P=O [40], которые также были отмечены в работах [37, 41, 42] для фосфатов алюминия и висмута. Почти полное отсутствие этих пиков в спектрах образцов 6 и 7 соединения BiAl3(PO4)2(OH)6 со структурой вейлендита свидетельствует о том, что колебания связей P=O присущи аморфной фазе и BiPO4.
Широкая полоса около 3000 см–1 в образцах 6, 7 фазы со структурой вейлендита соответствует валентным колебаниям изолированных O–H-групп [43]. Полосы при 440 и 498 см–1 в этих образцах отвечают колебаниям связи Al–O октаэдрического звена [AlO6] в структуре вейлендита.
РФА образца 7, полученного после изотермической выдержки в течение 14 сут, показал, что большинство наблюдаемых рефлексов, в том числе все рефлексы с большой интенсивностью, по наблюдаемым углам Брэгга могут быть отнесены к фазе со структурой вейлендита (тригональная сингония, карты PDF-2 00-013-0267 и 00-061-0545) [44, 45]. Кроме рефлексов, относимых к фазе со структурой вейлендита, на дифрактограмме наблюдаются очень слабые по интенсивности рефлексы, которые проиндицировать не удалось. Большинство из этих непроиндицированных рефлексов характеризуются наблюдаемой максимальной интенсивностью Imax < 1.75·Ibkgr, где Ibkgr – интенсивность фона, что соответствует относительной интенсивности <2.00% от максимальной интенсивности I10-11Wayl рефлекса 10-11 вейлендита. Только девять непроиндицированных рефлексов с углами Брэгга 2θ = 15.098°, 19.576°, 26.634°, 26.971°, 28.261°, 31.812°, 34.899°, 39.491° и 41.591° характеризуются интенсивностью в пределах 1.75 < Imax/Ibkgr < 3.00 и, соответственно, 2.00 < < ${{{{I}_{{{\text{max}}}}}} \mathord{\left/ {\vphantom {{{{I}_{{{\text{max}}}}}} {I_{{10 - 11}}^{{{\text{Wayl}}}}}}} \right. \kern-0em} {I_{{10 - 11}}^{{{\text{Wayl}}}}}}$ < 6.00.
Для образца 7 (после изотермической выдержки в течение 14 сут) методом наименьших квадратов по углам Брэгга наблюдаемых рефлексов с помощью программы Celsiz были рассчитаны параметры гексагональной элементарной ячейки кристаллической фазы со структурой вейлендита. После введения угловых поправок достигнутая точность значений параметров a = 6.9953(8), c = = 16.1467(29) Å достаточно высокая и хорошо согласуется с наблюдаемыми в литературе значениями параметров ячейки фаз со структурой вейлендита (табл. 1), что подтверждает отнесение наблюдаемых рефлексов к этой кристаллической фазе.
Чтобы окончательно убедиться, что наблюдаемые рефлексы могут быть отнесены к фазе со структурой вейлендита, была осуществлена подгонка дифрактограммы образца 7 методом ЛБ. Для подгонки методом ЛБ была использована пр. гр. $R\bar {3}m$ (166) в гексагональной установке, подтвержденная в [5] на основе РД-исследования структуры вейлендита на монокристалле. В качестве исходных для уточнения были использованы параметры элементарной ячейки, определенные на предыдущем этапе программой Celsiz, и угловые поправки из измерений в смеси образца с порошковым стандартом. В ЛБ-подгонке последовательно уточняли фактор сканирования, параметры фона и профиля рефлексов, угловые поправки и параметры элементарной ячейки. Циклы уточнения повторяли до остановки изменений параметров. Был достигнут весовой профильный фактор Rwp = 11.17% (см. определение факторов согласия, приводимых здесь и далее, в [46]), профили всех рефлексов фазы со структурой вейлендита с достаточно большой интенсивностью были подогнаны удовлетворительно.
Однако визуальная инспекция графических результатов подгонки показала, что для уменьшения Rwp программа подняла уровень фона выше многих непроиндицированных рефлексов со слабой интенсивностью. Во избежание этого эффекта и повышения качества подгонки РД-картины была предпринята следующая процедура. Фон был описан вручную полиномом нулевого порядка (константой) таким образом, чтобы на всем угловом диапазоне не превышал наблюдаемую интенсивность экспериментального фона. Гиперболическая часть модельного фона также была подобрана вручную, чтобы описывать малоугловую область экспериментального фона. Во всех угловых позициях 2θ, не совпадающих с угловыми позициями рефлексов вейлендита, где наблюдались слабые непроиндицированные рефлексы, были заданы пики и подогнаны их угловые позиции и профили в модели pV-TCHZ попеременно с уточнением методом ЛБ фазы со структурой вейлендита. На заключительном этапе в уточнение были включены параметры фона в виде полинома Чебышева 5-го порядка, как и ранее, которые уточнялись с параметрами профилей в раздельных циклах. В результате профили всех наблюдаемых рефлексов (как фазы со структурой вейлендита, так и слабых неидентифицированных рефлексов) были подогнаны (Rwp = 4.95%, графические результаты подгонки см. на рис. S1 в Дополнительных материалах).
На следующем этапе было проведено уточнение структуры фазы вейлендита и подгонка симулированной дифрактограммы к экспериментальной дифрактограмме образца 7 (после изотермической выдержки в течение 14 сут) методом Ритвельда [32]. В качестве стартовых использовали координаты атомов и их тепловые факторы из [5], приняв равными единице коэффициенты заполнения всех позиций соответствующими атомами (без дефектов замещения или вакансий). Все остальные параметры дифрактограммы были экспортированы из результатов подгонки ЛБ. Параметры pV-TCHZ профилей неиндицированных рефлексов на первых этапах ритвельдовской подгонки не уточнялись. После уточнения параметров фона, угловых поправок и параметров профиля рефлексов фазы со структурой вейлендита весовой профильный фактор составил Rwp = = 18.55%, а брэгговский фактор согласия RB = = 18.71%. Большие значения факторов согласия были обусловлены повышенной интенсивностью некоторых типов рефлексов, очевидно, в результате влияния эффектов преимущественной ориентации.
Учет эффекта преимущественной ориентации вдоль кристаллографического направления [11–20] в рамках модели Марч–Долласа (March–Dollase, МД) [47] привел к падению факторов до Rwp = 9.55% и RB = 7.56%. Программа TOPAS позволяет учитывать два направления преимущественной ориентации в рамках модели МД. Из других возможных направлений преимущественной ориентации наибольшее падение факторов согласия до Rwp = 8.86% и RB = 6.16% дало направление [02-21]. Остальные направления преимущественной ориентации были учтены в рамках модели сферических гармоник 8-го порядка [48], что после уточнения привело к Rwp = 7.75% и RB = = 2.71%. Окончательные значения параметров преимущественной ориентации МД вдоль направлений [11-20] и [02-21], r[11-20] = 0.28(1) и r[02-21] = = 0.38(1) при долях 12(1) и 88(1)% соответственно.
На следующем шаге проводили несколько циклов уточнения структурных параметров (изотропных температурных факторов Biso и координат x, y, z атомов) с промежуточным уточнением параметров элементарной ячейки a и c, неструктурных параметров по той же методике, что описана в подгонке ЛБ, и параметров преимущественной ориентации. Для уменьшения корреляций при уточнении, угловые поправки и параметры элементарной ячейки, а также структурные параметры и параметры преимущественной ориентации уточняли раздельно. Уточняли индивидуальный температурный фактор тяжелого атома Bi и общие температурные изотропные факторы легких атомов Al и P, а также атомов O и H ($B_{{{\text{iso}}}}^{{\text{P}}}$ = $B_{{{\text{iso}}}}^{{{\text{Al}}}},$ $B_{{{\text{iso}}}}^{{\text{H}}}$ = $B_{{{\text{iso}}}}^{{\text{O}}}$). При уточнении координат атомов было наложено условие на расстояния P–O (не менее 1.495 Å и не более 1.575 Å) и O–H (не менее 0.77 Å и не более 1.10 Å). После нескольких циклов уточнения улучшение факторов согласия прекратилось при достижении величин Rwp = 7.31% и RB = 2.48%.
На финальном этапе было проведено уточнение угловых позиций (углов Брэгга 2θ) и профильных параметров непроиндицированных рефлексов, за которым последовало несколько циклов подгонки дифрактограммы (уточнения параметров, не связанных со структурой) и уточнения параметров структуры анализируемого соединения по вышеописанной методике до остановки изменения факторов согласия. Попытка уточнения коэффициентов заполнения p позиций Bi, Al и P привела к величинам p, отличающимся от единицы (полного заполнения) только в 3-ем знаке после десятичной точки, поэтому в окончательном варианте коэффициенты заполнения были оставлены равными единице.
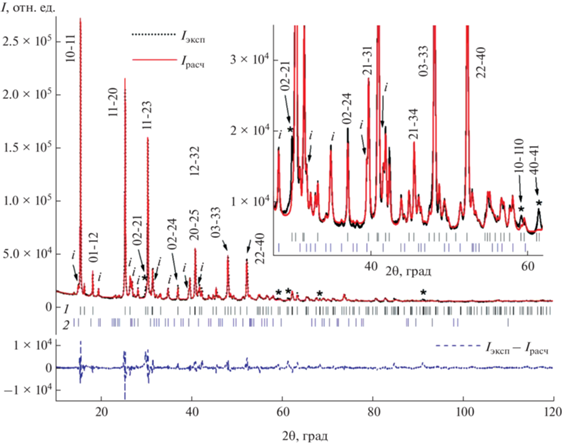
Окончательные достигнутые факторы согласия составили Rwp = 6.74% и RB = 2.18%. Графические результаты ритвельдовской подгонки представлены на рис. 7. Видно, что профили некоторых из рефлексов фазы со структурой вейлендита (hkil = 02-21, 10-110, 40-41 на вкладке к рис. 7, а также групп рефлексов 04-45 и 23-52 с углами Брэгга 2θ ~ 68.4° и 42-65, 15-62 и 20-214 около 2θ ~ 91.1°) были подогнаны методом Ритвельда плохо, хотя в ЛБ-подгонке их подгонка была удовлетворительной. При расчете без преимущественной ориентации все эти рефлексы характеризуются очень низкой относительной интенсивностью Irel = 0.24–1.30% [49] (за исключением рефлекса 10-110 с несколько большей относительной интенсивностью 3.9%). В случае ЛБ-подгонки интенсивность рефлексов экстрагируется из их экспериментального профиля, и если по углу Брэгга рефлексы хорошо соответствуют параметрам элементарной ячейки, то профили этих рефлексов хорошо подгоняются. В случае метода Ритвельда интенсивность рефлексов рассчитывается из параметров структуры и преимущественной ориентации. Вероятно, даже наличие преимущественной ориентации вдоль [02-21] не позволило получить достаточно значимую интенсивность для подгонки наблюдаемого профиля рефлекса 02-21 (хотя профиль рефлекса 04-42, характеризуемого Irel = 5.7%, описан удовлетворительно после ввода преимущественной ориентации вдоль [02-21]). По-видимому, в наблюдаемую высокую интенсивность рефлексов, профили которых не удалось описать в подгонке Ритвельда, дают вклад рефлексы неидентифицированной фазы.
Рис. 7.
Графические результаты подгонки методом Ритвельда синтезированного порошкового образца вейлендита. Указаны индексы Миллера–Браве hkil некоторых избранных рефлексов вейлендита. 1 – рефлексы вейлендита, 2 – непроиндицированные рефлексы. Рефлексы вейлендита с профилями, плохо описываемыми при подгонке Ритвельда, обозначены звездочкой. Только девять непроиндицированных рефлексов имеют интенсивность в пределах 2–6% относительно максимальной интенсивности Imax рефлекса 10-11 вейлендита. Они отмечены на рисунке буквенным символом i.
Количественные результаты уточнения структуры синтезированной фазы со структурой вейлендита методом Ритвельда представлены в табл. 2. Значения параметров элементарной ячейки фазы со структурой вейлендита, полученные уточнением методом Ритвельда, хорошо согласуются с результатами промежуточных этапов анализа (вычисление с помощью программы Celsiz и уточнение методом ЛБ). Избранные межатомные расстояния, рассчитанные на основе данных табл. 2, представлены в табл. S1 . В дополнительных материалах приведен CIF-файл уточненной структуры вейлендита.
Таблица 2.
Параметры структуры BiAl3(PO4)2(OH)6, полученные в результате уточнения методом Ритвельда (R$\bar {3}$m (166) в гексагональных осях; a = 6.99608(5), c = 16.1495(4) Å; Rwp = 6.74%, Rp = 4.43%, RB = 2.18%). Параметры структуры: относительные координаты атомов x/a, y/b, z/c; изотропные температурные факторы Biso; коэффициенты p заполнения позиций атомами
| Атом | Позиция Вайкоффа | x/a | y/b | z/c | p | Biso, Å2 |
|---|---|---|---|---|---|---|
| Bi | 3a | 0 | 0 | 0 | 1.000(1) | 3.41(2) |
| Al | 9d | 1/2 | 1/2 | 1/2 | 1.000(1) | 2.73(6) |
| P | 6c | 0 | 0 | 0.3107(3) | 1.000(5) | BisoAl |
| O1 | 6c | 0 | 0 | 0.4036(5) | 1 | 2.98(7) |
| O2 | 18h | 0.2118(3) | –x/a | 0.9431(4) | 1 | BisoO1 |
| O3 | 18h | 0.1283(2) | –x/a | 0.1356(4) | 1 | BisoO1 |
| H | 18h | 0.1862(24) | –x/a | 0.1149(39) | 1 | BisoO1 |
Рассчитанная рентгеновская плотность образца 7 (после изотермической выдержки в течение 14 сут) составила 4.235(2) г/см3. Экспериментально измеренная пикнометрическая плотность (4.01(8) г/см3) близка, но несколько меньше рентгеновской, вероятно, из-за присутствия небольшого количества неидентифицированной фазы с меньшей плотностью или из-за наличия закрытой пористости в образце.
По данным высокотемпературной рентгеновской дифрактометрии, соединение BiAl3(PO4)2(OH)6 со структурой вейлендита разлагается при температуре ~510°С.
ЗАКЛЮЧЕНИЕ
В гидротермальных условиях было получено соединение BiAl3(PO4)2(OH)6 со структурой вейлендита. Показано, что при температуре 200°С в гидротермальных условиях образование фазы вейлендита происходит путем взаимодействия фосфата висмута и аморфной фазы. Практически однофазный образец соединения со структурой вейлендита образуется через 48 ч изотермической выдержки, после чего происходит увеличение длины его стержнеобразных частиц за счет массопереноса в гидротермальном флюиде. Методом Ритвельда уточнена структура вейлендита. Определены параметры элементарной ячейки соединения BiAl3(PO4)2(OH)6 со структурой вейлендита: а = 6.99608(5), с = 16.1495(4) Å. Установлено, что рассматриваемое соединение устойчиво до температуры ~510°С.
Список литературы
Von Knorring O., Mrose M.E. // Abstr. Am. Mineralogist. 1962. V. 48. № 216. P. 156A. https://doi.org/10.1017/S0885715600012033
Clark A.M., Couper A.G., Embrey P.G., Fejer E.E. // Mineral. Mag. 1986. V. 50. P. 731. https://doi.org/10.1180/minmag.1986.050.358.23
Bayliss P. // Powder Diffraction. 1986. V. 1. № 4. P. 331. https://doi.org/10.1017/S0885715600012033
Novák F., Pauliš P., Sűsser C. // Bull. Mineral.-Petrolog. 2001. V. 9. P. 230.
Mills S.J., Kampf A.R., Raudsepp M. et al. // Mineral. Petrol. 2010. V. 100. № 3. P. 249. https://doi.org/10.1007/s00710-010-0133-7
Losertová L., Buřival Z., Losos Z. // Bull. Mineral.-Petrolog. 2014. V. 22. № 2. P. 269.
Pribavkin S.V., Kozlov P.S., Mazur S.V. et al. // Mineralogy. 2017. V. 3. № 2. P. 3. [Прибавкин С.В., Козлов П.С., Мазур С.В. и др. // Минералогия. 2017. V. 3. № 2. P. 3.]
Database “Mindat.org”. The Hudson Institute of Mineralogy, USA. https://www.mindat.org/min-4252.html
Database “RRUFF Project” (University of Arizona, USA) https://rruff.info/waylandite/
Bayliss. P., Kolitsch U., Nickel E. H. et al. // Mineralogical Magazine. 2010. V. 5. P. 919. https://doi.org/10.1180/minmag.2010.074.5.919
Schwab R.G., Pimpl T., Schukow H. et al. // J. Mineral. Geochem. 2004. V. 9. P. 385. https://doi.org/10.1127/0028-3649/2004/2004-0385
Kolitsch U., Pring A. // J. Mineral. Petrol. Sci. 2001. V. 96. P. 67. https://doi.org/10.2465/jmps.96.67
Hikichi Y., Ohsato H., Miyamoto M. // J. Mineralogy. 1989. V. 19. № 2. P. 67. (in Japanese) https://doi.org/10.2465/gkk1952.19.67
Pozhidaeva O.V., Korytkova E.N., Romanov D.P. et al. // Russ. J. Gen. Chem. 2002. V. 72. № 6. P. 849. [Пожидаева О.В., Корыткова Э.Н., Романов Д.П. и др. // Журн. общ. химии. 2002. Т. 72. № 6. С. 910.]https://doi.org/10.1023/A:1020409702215
Enikeeva M.O., Proskurina O.V., Danilovich. D.P. et al. // Nanosyst.: Phys, Chem., Math. 2020. V. 11. № 6. P. 705. https://doi.org/10.17586/2220-8054-2020-11-6-705-715
Meskin P.E., Gavrilov A.I., Maksimov V.D. et al. // Russ. J. Inorg. Chem. 2007. V. 52. № 11. P. 1648. [Мескин П.Е., Гаврилов А.И., Максимов В.Д. и др. // Журн. неорган. химии. 2007. V. 52. № 11. С. 1755.]https://doi.org/10.1134/S0036023607110022
Nikolaev A.I., Gerasimova L.G., Maslova M.V. et al. // IOP Conf. Ser.: Mater. Sci. Eng. 2019. V. 704. https://doi.org/10.1088/1757-899X/704/1/012003
Enikeeva M.O., Kenges K.M., Proskurina O.V. et al. // Russ. J. Appl. Chem. 2020. V. 93. № 4. P. 540. [Еникеева М.О., Кенес К.М., Проскурина О.В. и др. // Журн. прикл. химии. 2020. Т. 93. № 4. С. 529.]https://doi.org/10.1134/S1070427220040084
Thomas V.G., Demin S.P., Foursenko D.A. et al. // J. Cryst. Growth. 1999. V. 206. № 3. https://doi.org/10.1016/S0022-0248(99)00312-7
Gavryushkin P.N., Thomas V.G., Bolotina N.B. et al. // Crystal Growth Design. 2016. V. 16. № 4. P. 1893. https://doi.org/10.1021/acs.cgd.5b01398
Korytkova E.N., Pivovarova L.N., Drosdova I.A. et al. // Russ. J. Gen. Chem. 2007. V. 77. № 10. P. 1669. [Корыткова Э.Н., Пивоварова Л.Н., Дроздова И.А. и др. // Журн. общ. химии. 2007. Т. 77. № 10. С. 1600.]https://doi.org/10.1134/S1070363207100039
Korytkova E.N., Pivovarova L.N., Semenova O.E. et al. // Russ. J. Inorg. Chem. 2007. V. 52. № 3. P. 338. [Корыткова Э.Н., Пивоварова Л.Н., Семенова О.Е. и др. // Журн. неорган. химии. 2007. Т. 52. № 3. P. 388.]https://doi.org/10.1134/S0036023607030084
Korytkova E.N., Maslov A.V., Pivovarova L.N. et al. // Glass Phys. Chem. 2004. V. 30. № 1. P. 51. [Корыткова Э.Н., Маслов А.В., Пивоварова Л.Н. и др. // Физика и химия стекла. 2004. Т. 30. № 1. С. 72.]
Mashkovtsev R.I., Stoyanov E.S., Thomas V.G. // J. Struct. Chem. 2004. V. 45. № 1. P. 56. https://doi.org/10.1023/B:JORY.0000041501.77617.72
Schmetzer K., Schwarz D., Bernhardt H.-J. et al. // J. Gemm. 2006. V. 30. № 1/2. P. 59. https://doi.org/10.15506/JoG.2006.30.1.59
Korytkova E.N., Maslov A.V., Pivovarova L.N. et al. // Inorg. Mater. 2005. V. 41. № 7. P. 743. [Корыткова Э.Н., Маслов А.В., Пивоварова Л.Н. и др. // Неорган. материалы. 2005. Т. 41. № 7. С. 849.]https://doi.org/10.1007/s10789-005-0202-1
Ogorodova L.P., Kiselev I.A., Korytkova E.N. et al. // Russ. J. Phys. Chem. 2006. V. 80. № 7. P. 1021. [Огородова Л.П., Киселева И.А., Корыткова Э.Н. и др. // Журн. физ. химии. 2006. Т. 80. № 7. С. 1170.]https://doi.org/10.1134/S003602440607003X
Diffrac Suite Eva Version 5.1.0.5; Brucker AXS: Karlsruhe, Germany, 2019.
Fawcett T.G., Kabekkodu S.N., Blanton J.R. et al. // Powder Diffr. 2017. V. 32. № 2. P. 63. https://doi.org/10.1017/S0885715617000288
Maunders C., Etheridge J., Wright N. et al. // Acta Crystallogr. 2005. V. 61. P. 154. https://doi.org/10.1107/S0108768105001667
Le Bail A., Duroy H., Fourquet J.L. // Mater. Res. Bull. 1988. V. 23. № 3. P. 447. https://doi.org/10.1016/0025-5408(88)90019-0
Rietveld H.M. // Acta Crystallogr. 1967. V. 22 P. 151. https://doi.org/10.1107/S0365110X67000234
Coelho A.A. // J. Appl. Crystallogr. 2018. V. 51. P. 210. https://doi.org/10.1107/S1600576718000183
Berger H. // X-ray Spectrom. 1986. V. 15. P. 241. https://doi.org/10.1002/xrs.1300150405
Thompson P., Cox D.E., Hastings J.B. // J. Appl. Crystallogr. 1987. V. 20. P. 79. https://doi.org/10.1107/S0021889887087090
Frost R.L., Xi Y., Palmer S.J. et al. // Mineral. Petrol. 2010. V. 82. № 461. https://doi.org/10.1016/j.saa.2011.07.078
Marimuthu A., Devamani R.H.P. // Int. J. Appl. Sci. Eng. Res. 2012. V. 1. № 6. P. 769. https://doi.org/10.6088/ijaser.0020101078
Rosseeva E.V., Buder J., Simon P. et al. // Chem. Mater. 2008. V. 20. № 19. P. 6003. https://doi.org/10.1021/cm8005748
Frost R.L., Lopez A., Xi Y. et al. // Spectrochim. Acta A: Mol. Biomol. Spectrosc. 2013. V. 114. P. 309. https://doi.org/10.1016/j.saa.2013.05.033
Trivedi M.K., Branton A., Trivedi D. et al. // J. Chromatogr. Sep. Tech. 2015. V. 6. № 5. P. 1.https://doi.org/10.4172/2157-7064.1000282
Shao M., Wang A., Cui H. et al. // J. Mater. Sci. – Mater. Electron. 2020. V. 30 № 15. P. 12056. https://doi.org/10.1007/s10854-020-03711-1
Roming M., Feldmann C. // J. Mater. Sci. 2009. V. 44. № 5. P. 1412. https://doi.org/10.1007/s10853-009-3258-5
Repina S.A., Popova V.I., Churin E.I. et al. // Geology Ore Deposits. 2011. V. 53. № 7. P. 564. https://doi.org/10.1134/S1075701511070191
d`Yvoire F. // C. R. Seances. Acad. Sci. Paris. 1960. V. 251. P. 2182.
Rankin J., Sharpe J., Williams P. // Aust. J. Mineral. 2008. V. 14. № 2. P. 79. http://handle.uws.edu.au: 8081/1959.7/538585
Hill R.J., Fischer R.X. // J. Appl. Crystallogr. 1990. V. 23. P. 462. https://doi.org/10.1107/S0021889890006094
Dollase W.A. // J. Appl. Crystallogr. 1986. V. 19. P. 267. https://doi.org/10.1107/S0021889886089458
Järvinen M. // J. Appl. Crystallogr. 1994. V. 26. P. 525. https://doi.org/10.1107/S0021889893001219
Kraus W., Nolze G. // J. Appl. Crystallogr. 1996. V. 29. P. 301. https://doi.org/10.1107/S0021889895014920
Дополнительные материалы
- скачать EMS_1.docx
- Приложение 1
- скачать EMS_2.cif
- Приложение 2
Инструменты
Журнал неорганической химии