

Журнал неорганической химии, 2021, T. 66, № 2, стр. 265-273
Кинетика формирования и растворения анодного оксида алюминия в электролитах на основе серной и селеновой кислот
А. И. Садыков a, А. П. Леонтьев b, С. Е. Кушнир a, b, А. В. Лукашин a, b, c, К. С. Напольский a, b, *
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
119991 Москва, Россия
b Московский государственный университет имени М.В. Ломоносова, Факультет наук о материалах
119991 Москва, Россия
c Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Россия
* E-mail: kirill@inorg.chem.msu.ru
Поступила в редакцию 30.04.2020
После доработки 30.05.2020
Принята к публикации 01.06.2020
Аннотация
Измерена кинетика формирования и растворения пористого анодного оксида алюминия в электролитах на основе серной и селеновой кислот при температуре от 0 до 30°С. Показана существенная разница в кинетике растворения анодного оксида алюминия у основания пор в процессе его формирования и по всей поверхности стенок пористой структуры после прекращения анодирования. При анодировании алюминия в 2.0 М H2SO4 в кинетическом режиме значение кажущейся энергии активации растворения анодного оксида алюминия у основания пор составляет 53.2 ± 1.8 кДж/моль, в то время как для процесса химического растворения стенок пористой структуры соответствующая величина равна 83.3 ± 2.1 кДж/моль. Наблюдаемая разница в кинетике растворения свидетельствует о том, что с понижением температуры электролита увеличивается максимально достижимая толщина пористой пленки анодного оксида алюминия.
ВВЕДЕНИЕ
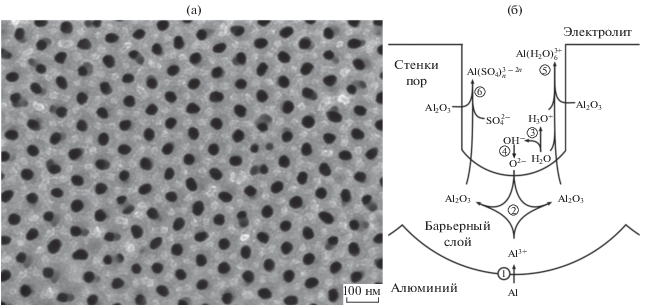
Пленки анодного оксида алюминия (АОА), образующиеся при электрохимическом окислении (анодировании) алюминия или его сплавов в кислых электролитах, находят широкое применение в различных областях, включая получение защитных и декоративных покрытий [1–3], конденсаторов [4], сенсоров [5], мембран для газоразделения [6, 7] и фотонных кристаллов [8–10]. Настолько широкий спектр практического применения АОА обусловлен его высокой термической и химической стабильностью, а также уникальной пористой структурой, параметры которой легко поддаются контролю с помощью варьирования условий анодирования. В структуре АОА присутствует система цилиндрических пор, расположенных перпендикулярно поверхности оксидной пленки (рис. 1а). Диаметр пор можно контролируемо изменять от единиц до нескольких сотен нанометров [6, 11, 12].
Рис. 1.
РЭМ-изображение поверхности пористой пленки АОА, полученной в 0.3 М H2SO4 при 0°С и напряжении 25 В (а). Схема процессов, протекающих при анодном окислении алюминия в растворе серной кислоты (б). Цифрами обозначены номера элементарных реакций, приведенных в тексте.
Основными параметрами, с помощью которых удается контролировать размер пор и расстояние между ними, являются напряжение и плотность тока анодирования. В работах [6, 11–15] изучали влияние напряжения анодирования на пористую структуру АОА, в то время как температуре электролита уделялось гораздо меньше внимания. Тем не менее температура при прочих равных условиях влияет как на геометрические параметры структуры АОА, так и на скорость формирования оксидной пленки [12, 13, 16].
На производстве при получении защитных и декоративных покрытий на поверхности алюминиевых сплавов температуру сернокислого электролита редко опускают ниже 15°С [3]. Это связано с большими затратами электроэнергии на охлаждение при росте разницы температуры электролита и окружающей среды. Часто анодирование проводят даже при температуре окружающей среды без использования системы термостабилизации. Напротив, в лабораторных условиях с того момента, когда была впервые показана возможность получения пленок АОА с высокоупорядоченной структурой [17], при поиске новых условий анодирования, приводящих к самоупорядочению пористой структуры АОА, температуру электролита поддерживают около 0°С [12, 13, 15, 18]. Из литературы также известно, что максимальная толщина формируемой оксидной пленки [19] и ее твердость [20] уменьшаются с ростом температуры электролита. Однако эти закономерности базируются в основном на эмпирических наблюдениях.
Физико-химические процессы, отвечающие росту пор, происходят в сплошном, так называемом барьерном слое оксида, разделяющем металл и электролит у основания пор. В упрощенном виде эти процессы можно представить в виде следующих элементарных реакций (рис. 1б) [21]:
на границе алюминий/оксид
в толще барьерного слоя анодного оксида алюминия
(2)
$2{\text{A}}{{{\text{l}}}^{{3 + }}} + 3{{{\text{O}}}^{{2 - }}} \to {\text{A}}{{{\text{l}}}_{2}}{{{\text{O}}}_{3}},$на границе оксид/электролит у основания пор
(3)
$2{{{\text{H}}}_{2}}{\text{O}} \to {\text{O}}{{{\text{H}}}^{ - }} + {{{\text{H}}}_{3}}{{{\text{O}}}^{ + }},$(4)
${\text{O}}{{{\text{H}}}^{ - }} + {{{\text{H}}}_{2}}{\text{O\;}} \to {\text{O}}_{{\left( {{\text{окс}}} \right)}}^{{2 - }} + {{{\text{H}}}_{3}}{{{\text{O}}}^{ + }}.$Катионы Al3+ образуются в результате электрохимического окисления алюминия на границе металл/оксид (реакция (1)). На интерфейсе оксид/электролит формируются анионы кислорода (О2–) (реакции (3) и (4)) вследствие депротонирования молекул воды под действием электрического поля. Затем катионы и анионы мигрируют [22, 23] навстречу друг другу через слой анодного оксида алюминия, формирование которого происходит в соответствии с реакцией (2) [21].
В кислых электролитах наряду с образованием оксида алюминия (реакции (1)–(4)) происходит его растворение за счет гидратации и/или комплексообразования ионов Al3+ с молекулами воды и анионами кислоты, например сульфат-анионами [24]:
(5)
${\text{A}}{{{\text{l}}}_{2}}{{{\text{O}}}_{3}} + 3{{{\text{H}}}_{2}}{\text{O}} + 6{{{\text{H}}}_{3}}{{{\text{O}}}^{ + }} \to 2{\text{Al}}\left( {{{{\text{H}}}_{2}}{\text{O}}} \right)_{6}^{{3 + }},$(6)
$\begin{gathered} {\text{A}}{{{\text{l}}}_{2}}{{{\text{O}}}_{3}} + 2n{\text{SO}}_{4}^{{2 - }} + 6{{{\text{H}}}_{3}}{{{\text{O}}}^{ + }} \to \\ \to 2{\text{Al}}\left( {{\text{S}}{{{\text{O}}}_{4}}} \right)_{n}^{{3 - 2n}} + 9{{{\text{H}}}_{2}}{\text{O}}{\text{.}} \\ \end{gathered} $По мнению авторов работы [24], энергия активации реакций растворения АОА на дне пор может существенно уменьшаться во внешнем электрическом поле, возникающем при анодной поляризации алюминиевого электрода. При поляризации связей Al–O происходит увеличение их длины, следовательно, связь между алюминием и кислородом становится слабее. Следует отметить, что данные выводы авторы делают исходя из общих соображений, не приводя значений каких-либо физико-химических величин.
Учитывая вышесказанное, фундаментальные исследования кинетики формирования и растворения анодного оксида алюминия в широком диапазоне температур, безусловно, являются актуальными. Знание энергии активации и константы скорости реакции позволит точно прогнозировать результат без необходимости проведения многочисленных экспериментов. Данные о кинетике растворения АОА необходимы для получения высококачественных фотонно-кристаллических материалов [10, 25]. В настоящей работе изучена кинетика формирования и растворения анодного оксида алюминия в электролитах на основе серной и селеновой кислот. Сернокислые электролиты на сегодняшний день широко распространены в промышленности, а также активно используются в лабораторных исследованиях. Растворы селеновой кислоты являются новыми электролитами, которые демонстрируют большой потенциал для получения анодного оксида алюминия для разнообразного оптического применения [26–28].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Формирование АОА проводили на алюминиевой фольге марки А99Т (содержание алюминия не менее 99.99%, ГОСТ 25905-83) толщиной 0.1 мм. Перед анодным окислением фольгу подвергали электрохимической полировке в растворе 1.85 М CrO3 и 13.0 М H3PO4 при анодной плотности тока ~400 мА/см2. Электрополировку проводили в импульсном режиме: 40 импульсов длительностью 3 с с периодом 40 с.
Анодное окисление алюминия осуществляли в двухэлектродной электрохимической ячейке объемом 1 л с донным прижимным электродом. Катодом служила пластина из алюминия марки А5Т. Область металла, подвергаемую анодированию, ограничивали резиновым кольцом с внутренним диаметром 27 мм. Температуру электролита поддерживали постоянной с точностью 0.1°С с помощью термостата Huber Petit Fleur. Анодирование проводили в разных электролитах при нескольких значениях напряжения и температуры (табл. 1). При регистрации стационарных поляризационных кривых напряжение анодирования изменяли только после того, как при данных условиях значение тока выходило на квазистационарное значение (изменение менее 1% за 1 мин).
Таблица 1.
Условия анодирования
| Электролит | t, °С | Напряжение, В |
|---|---|---|
| 2.0 М H2SO4 | 0, 5, 10, 15, 20 | 5–15 с шагом 1 |
| 4.0 М H2SO4 | 0, 5, 10, 15 | 5–15 с шагом 2.5 |
| 1.0 М H2SeO4 | 0, 5, 10, 15, 20, 25, 30 | 10–35 c шагом 5 |
Для изучения кинетики химического растворения пленки АОА получали в 2.0 М H2SO4 при температуре 10°С и напряжении 10 В. Анодирование проводили до достижения заряда 9.96 Кл/см2, что в условиях эксперимента соответствует толщине пористых оксидных пленок ∼5 мкм. Для более точного определения толщины, эффективного показателя преломления и оптической длины пути пленок использовали анализ положения осцилляций Фабри–Перо в спектрах зеркального отражения [29], зарегистрированных на спектрофотометре Perkin Elmer Lambda 950 при углах падения света (относительно нормали) от 8° до 65° в диапазоне длин волн от 650 до 890 нм с шагом 2 нм. Размер светового пучка составлял 1 × 5 мм2. Далее образцы помещали в 2.0 М раствор H2SO4 и наблюдали эволюцию спектров отражения в области длин волн, где проявляются осцилляции Фабри–Перо. Эксперименты проводили при температурах раствора 0, 5.3, 10, 15 и 19.3°С. Спектры отражения регистрировали с помощью спектрометра Ocean Optics QE65000 в интервале от 250 до 1040 нм. Для определения пористости полученных пленок регистрировали спектры отражения в трех средах: на воздухе, в воде и в изопропаноле.
Расстояние между центрами пор в пленках АОА измеряли с помощью растровой электронной микроскопии (РЭМ) алюминиевых реплик, полученных после селективного растворения оксидной пленки в растворе, содержащем 0.2 М CrO3 и 0.7 М H3PO4, при температуре 70°С в течение 30 мин. В работе использовали электронный микроскоп Carl Zeiss NVision 40.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Кинетика химического растворения анодного оксида алюминия
Кинетику химического растворения АОА в 2.0 М H2SO4 в отсутствие внешнего электрического поля определяли с помощью анализа спектров зеркального отражения АОА [29]. Данный метод не является разрушающим и позволяет следить за характеристиками пористой пленки in situ в процессе утончения стенок пор из-за их постепенного растворения в кислом растворе электролита.
Из-за малого диаметра пор в изучаемых оксидных пленках по сравнению с длиной волны падающего света оптические свойства АОА можно рассматривать в терминах эффективной среды. В этом случае величина эффективного показателя преломления neff вычисляется по формуле [30]:
(7)
${{n}_{{{\text{eff}}}}} = \sqrt {n_{{{\text{A}}{{{\text{l}}}_{2}}{{{\text{O}}}_{3}}}}^{2}\left( {1 - P} \right) + n_{p}^{2}P} ,$Толщина пленок АОА, полученных в 2.0 М H2SO4 при температуре 10°С и напряжении 10 В, составляет h0 = 5.04 ± 0.04 мкм согласно результатам анализа спектров зеркального отражения на воздухе при разных углах падения света. Исходная пористость оксидных пленок определена из анализа спектров отражения на воздухе, в воде и изопропаноле при 20°С. Показатель преломления увеличивается в ряду: воздух, вода, изопропанол, поэтому, согласно формуле (7), neff пленок возрастает при заполнении пор этими веществами, и, следовательно, в спектрах отражения наблюдается сдвиг осцилляций Фабри–Перо в область более длинных волн (рис. 2а). Значения оптической толщины L пленки АОА, помещенной в каждую из указанных выше сред, рассчитаны из положения осцилляций Фабри–Перо в спектрах отражения по методике [29]. Поскольку толщина пленок не зависит от вещества, заполняющего поры, neff получается делением L на известную толщину h0. Исходя из формулы (7) квадрат neff линейно зависит от квадрата np:
(8)
$n_{{{\text{eff}}}}^{2} = n_{{{\text{A}}{{{\text{l}}}_{2}}{{{\text{O}}}_{3}}}}^{2}\left( {1 - P} \right) + n_{p}^{2}P.$Рис. 2.
Спектры отражения пористой пленки АОА в трех средах. Стрелками показано смещение осцилляций Фабри–Перо при переходе в среду с бóльшим показателем преломления (а). Данные линейной аппроксимации экспериментальной зависимости квадрата эффективного показателя преломления пленки АОА от квадрата показателя преломления вещества, заполняющего поры, необходимые для определения исходной пористости P0 и показателя преломления оксида алюминия, образующего стенки пористой структуры (б).
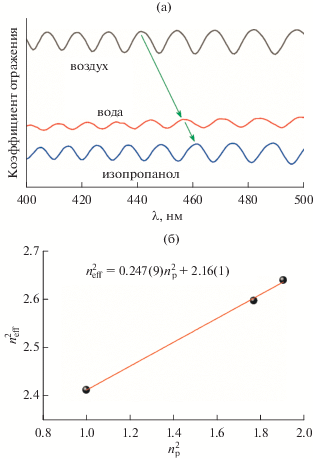
На рис. 2б показана экспериментальная зависимость (neff)2 от (np)2. Линейная аппроксимация этой зависимости дает значения углового коэффициента a = P и свободного члена b. Тогда показатель преломления стенок пористой структуры можно рассчитать следующим образом:
Значение исходной пористости составило P0 = = 0.247 ± 0.009. Величина показателя преломления стенок ${{n}_{{{\text{A}}{{{\text{l}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{3}}}}}}}$ = 1.70 ± 0.02 немного меньше справочного значения показателя преломления для сапфира (1.77 [31]), что согласуется с данными работ [28] (1.64 ± 0.01) и [32] (1.671) для пленок, полученных анодированием алюминия в 1.0 М Н2SeО4 и 0.3 М Н2С2О4 соответственно. Уменьшение показателя преломления оксида алюминия, образующего стенки пористой структуры, по сравнению с показателем преломления сапфира связано с наличием примесей (вода, анионы используемой в качестве электролита кислоты) в структуре АОА.
Для определения скорости химического растворения АОА в растворе электролита регистрировали спектры отражения с интервалом 1 мин (рис. 3а). Эксперименты прекращали, когда осцилляции Фабри–Перо становились плохо различимы в спектре. Из положения осцилляций Фабри–Перо и значения h0 определяли показатель преломления neff.
Рис. 3.
Изменение спектров отражения во времени при выдерживании пленки АОА в 2.0 М растворе H2SO4 при t = 5.3°C (а). Условия получения пленки: 10 В при 10°С в 2.0 М H2SO4. Зависимость эффективного показателя преломления (усредненного по области 450–650 нм) от времени выдержки АОА в 2.0 М растворе H2SO4 при различных температурах (б). Линеаризация кинетических кривых в координатах $\sqrt P $ от t (в).
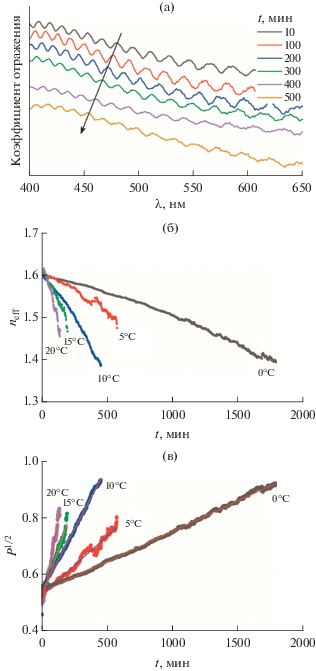
Полученные зависимости neff от продолжительности растворения (t) представлены на рис. 3б. Из графиков видно, что при большей температуре neff уменьшается быстрее. Это говорит о том, что скорость расширения пор в кислом электролите с ростом температуры возрастает.
Поскольку толщина использованной пленки (h0 = 5.04 ± 0.04 мкм) намного больше диаметра пор (~0.02 мкм), изменением толщины за счет химического растворения пленки во время эксперимента можно пренебречь. Отсюда изменение L в ходе эксперимента напрямую дает информацию об изменении neff, который, в свою очередь, зависит от диаметра пор Dp. Для вычисления скорости растворения стенок пор использовали предположение, что скорость (v) увеличения радиуса пор (R) не зависит от кривизны поверхности и остается постоянной в течение всего эксперимента по растворению АОА [10, 33]:
Тогда диаметр пор выражается через диаметр пор до начала растворения (Dp0) следующей формулой:
Пористость пленки с гексагональным массивом вертикальных пор равна:
(12)
$P\left( t \right) = \frac{\pi }{{2\sqrt 3 }}\frac{{D_{p}^{2}\left( t \right)}}{{D_{{{\text{int}}}}^{2}}},$Выражая Dp(t) и Dp0 через P(t) и P0 соответственно и используя формулу (12), получаем:
(13)
$\sqrt {P\left( t \right)} = \sqrt {{{P}_{0}}} + {{\left( {2\pi /\sqrt 3 } \right)}^{{1/2}}}\frac{{v}}{{{{D}_{{{\text{int}}}}}}}t.$С другой стороны, из формулы (8) получаем выражение для P(t) через параметр neff(t), который определяется в ходе эксперимента:
(14)
$P\left( t \right) = \frac{{n_{{{\text{A}}{{{\text{l}}}_{2}}{{{\text{O}}}_{3}}}}^{2} - n_{{{\text{eff}}}}^{2}\left( t \right)}}{{n_{{{\text{A}}{{{\text{l}}}_{2}}{{{\text{O}}}_{3}}}}^{2} - n_{{\text{w}}}^{2}}}~,$(15)
$\begin{gathered} \sqrt {P\left( t \right)} = {{\left( {\frac{{n_{{{\text{A}}{{{\text{l}}}_{2}}{{{\text{O}}}_{3}}}}^{2} - n_{{{\text{eff}}}}^{2}\left( t \right)}}{{n_{{{\text{A}}{{{\text{l}}}_{2}}{{{\text{O}}}_{3}}}}^{2} - n_{{\text{w}}}^{2}}}} \right)}^{{1/2{\text{\;}}}}} = \\ = \sqrt {{{P}_{0}}} + {{\left( {2\pi /\sqrt 3 } \right)}^{{1/2}}}\frac{{v}}{{{{D}_{{{\text{int}}}}}}}t. \\ \end{gathered} $Таким образом, величина v легко вычисляется из углового коэффициента линейной аппроксимации зависимости $\sqrt P $ от t (рис. 3в). Результаты определения v приведены в табл. 2. Полученные значения согласуются со скоростью 0.236 нм/ч, найденной ранее для 2.0 М H2SO4 при температуре 2 ± 1°С [10]. Далее, используя зависимость ln v от (1/T), определяли кажущуюся энергию активации химического растворения АОА $E_{{\text{a}}}^{{{\text{хим}}}},$ которая составила 83.3 ± 2.1 кДж/моль. Полученное значение близко к $E_{{\text{a}}}^{{{\text{хим}}}}$ = 78.6 кДж/моль, найденному ранее для электролита на основе 1.685 M H2SO4 [19].
Таблица 2.
Константы скорости растворения анодного оксида алюминия в 2.0 М H2SO4 при разных температурах
| t, °С | v, нм/ч | vS × 1011, моль/(см2 мин) |
|---|---|---|
| 0.0 | 0.1914 ± 0.0002 | 0.943 ± 0.002 |
| 5.3 | 0.372 ± 0.002 | 1.88 ± 0.08 |
| 10.0 | 0.783 ± 0.002 | 3.9 ± 0.1 |
| 15.0 | 1.23 ± 0.01 | 6.0 ± 0.3 |
| 19.3 | 2.21 ± 0.03 | 11.2 ± 0.6 |
Удельная скорость растворения АОА на единицу площади vS (табл. 2) была рассчитана следующим образом:
(16)
$\begin{gathered} {{{v}}_{{\text{S}}}} = - \frac{1}{S}\frac{{d{{x}_{{{\text{AOА}}}}}}}{{dt}} = - \frac{1}{S}\frac{{{{\rho }_{{{\text{AОA}}}}}}}{{{{M}_{{{\text{A}}{{{\text{l}}}_{2}}{{{\text{O}}}_{3}}}}}}}\frac{{dV}}{{dt}} = \\ = \frac{1}{S}\frac{{{{\rho }_{{{\text{AОA}}}}}S}}{{{{M}_{{{\text{A}}{{{\text{l}}}_{2}}{{{\text{O}}}_{3}}}}}}}\frac{{dR}}{{dt}} = \frac{{{{\rho }_{{{\text{AOA}}}}}}}{{{{M}_{{{\text{A}}{{{\text{l}}}_{2}}{{{\text{O}}}_{3}}}}}}}{v}, \\ \end{gathered} $Кинетика формирования анодного оксида алюминия
Кинетику формирования АОА в электролитах на основе серной и селеновой кислот изучали с помощью регистрации стационарных поляризационных кривых при различной температуре электролита (рис. 4а, 4в, 4д). Использованные условия анодирования относятся к кинетическому режиму, так как при фиксированном напряжении плотность тока после непродолжительного начального участка выходит на постоянное значение и не изменяется с ростом толщины оксидной пленки. Отметим, что с ростом температуры электролита при высоком напряжении плотность тока постоянно уменьшалась со временем, это свидетельствует о переходе в смешанный режим анодирования, при котором диффузия ионов в электролите начинает оказывать влияние на скорость протекающих процессов, что приводит к замедлению скорости реакции с увеличением толщины пленки АОА. В связи с этим при напряжении 40 В в 1.0 М растворе H2SeO4 данные получены лишь для температуры не выше 10°С, а в случае 2.0 и 4.0 М растворов H2SO4 мы ограничили напряжение 20 и 15 В соответственно.
Рис. 4.
Стационарные поляризационные кривые, зарегистрированные при анодировании алюминиевой фольги А99Т для различной температуры электролита в 2.0 М H2SO4 (а), 4.0 М H2SO4 (в), 1.0 М H2SeO4 (д). Рассчитанные значения кажущейся энергии активации формирования анодного оксида алюминия в 2.0 М H2SO4 (б), 4.0 М H2SO4 (г), 1.0 М H2SeO4 (е).
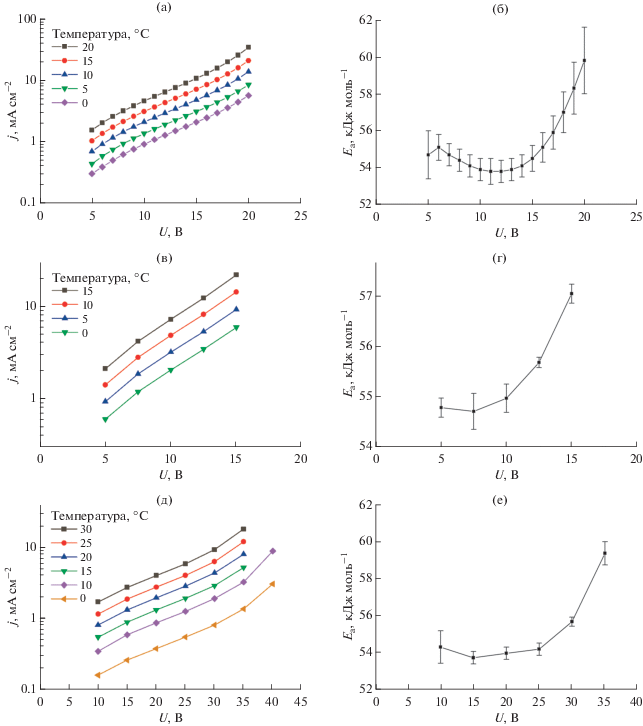
В связи с тем, что в кинетическом режиме анодирования лимитирующей стадией является процесс растворения АОА у основания пор, полученные данные могут быть использованы для определения кажущейся энергии активации растворения АОА на дне пор под действием электрического поля (рис. 4б, 4г, 4е):
(18)
$\ln j\left( {U,\,T} \right) = {\text{const}} - \frac{{{{E}_{{\text{a}}}}\left( U \right)}}{{RT}}.$Полученные значения энергии активации процесса растворения АОА на дне пор в присутствии электрического поля (53–60 кДж/моль, рис. 4б, 4г, 4е) существенно меньше соответствующей величины для процесса химического растворения стенок пор в растворе электролита. Это можно объяснить тем, что под воздействием электрического поля прочность связей Al–O уменьшается.
При всех использованных условиях анодирования наблюдается схожая тенденция. Кажущаяся энергия активации постоянна в среднем диапазоне напряжений и начинает существенно расти при увеличении плотности тока анодирования. Также систематически наблюдается небольшой рост Ea при минимальном использованном напряжении. Рост энергии активации на правой границе используемых диапазонов напряжения мы связываем с ростом температуры на дне пор в связи с протеканием тока более высокой плотности, приводящего к разогреву АОА. К сожалению, точно определить температуру интерфейса АОА/электролит на дне пор не представляется возможным, в связи с этим значения Ea при высоких напряжениях стоит считать менее надежными.
При малом напряжении анодирования плотность тока мала и не может приводить к существенному разогреву. Поэтому рост Ea на нижней границе используемого диапазона напряжений анодирования, очевидно, не связан с отличием температуры интерфейса АОА/электролит от температуры электролита. Известно, что толщина барьерного слоя (db) линейно увеличивается с напряжением анодирования [24]. Следует отметить, что в уравнении пропорциональности присутствует свободный член, величина которого составляет 2–3 нм [35], данное значение близко к толщине естественного оксида на поверхности алюминия. В связи с этим напряженность электрического поля E = U/db при малом напряжении оказывается ниже, что и приводит к росту энергии активации процесса растворения анодного оксида алюминия на дне пор.
Согласно литературным данным, при приложении анодной поляризации кажущаяся энергия активации растворения анодного оксида алюминия на дне пор составляет: 42.5 ± 1.0 кДж/моль [36] и 47.3 ± 1.0 кДж/моль [16] в 0.3 M H2C2O4 при 40 В, что существенно меньше энергии активации химического растворения стенок пор в электролите анодирования. Таким образом, найденные в настоящей работе величины Ea согласуются c ранее опубликованными данными и дополняют их.
Полученные данные позволяют оценить влияние температуры на максимально возможную толщину пленки АОА. В качестве критерия растворения верхней части пленки можно принять равенство диаметра пор и расстояния между порами Dp = Dint. Тогда максимальная толщина пленки АОА (hmax) соответствует толщине пленки, полученной в результате анодирования за время, затраченное на увеличение диаметра пор в верхней части до значения Dint:
(19)
$\begin{gathered} {{h}_{{{\text{max}}}}}\left( T \right) = {{h}_{q}}\left( {{{D}_{{{\text{int}}}}} - {{D}_{{{\text{p}}0}}}} \right)\frac{{j\left( T \right)}}{{2{v}\left( T \right)}} = \\ = {{h}_{q}}\left( {{{D}_{{{\text{int}}}}} - {{D}_{{{\text{p}}0}}}} \right)\frac{{j\left( {{{T}_{0}}} \right)}}{{2{v}\left( {{{T}_{0}}} \right)}}{\text{ex}}{{{\text{p}}}^{{\frac{{E_{a}^{{{\text{хим}}}} - {{E}_{{\text{a}}}}}}{R}\left( {\frac{1}{T} - \frac{1}{{{{T}_{0}}}}} \right)}}}, \\ \end{gathered} $Исходя из полученных выше данных, включая Dp0 = 14.1 нм и hq = 506 нм см2/Кл, процесс растворения в пленке АОА, полученной при напряжении 10 В и 10°С, займет 34 ч при температуре 0°C и всего 3 ч при 19.3°C. При этом, согласно формуле (19), максимальная толщина пленки АОА, полученной при напряжении 10 В, будет уменьшаться с 61 до 25 мкм с увеличением температуры электролита от 0 до 20°C.
ЗАКЛЮЧЕНИЕ
В работе изучена кинетика формирования и растворения пористого анодного оксида алюминия в электролитах на основе серной и селеновой кислот при температуре от 0 до 30°С. На примере электролита 2.0 М H2SO4 показано, что кажущаяся энергия активации растворения анодного оксида алюминия у основания пор в процессе анодирования (53.2 ± 1.8 кДж/моль) существенно ниже, чем соответствующая величина для химического растворения стенок пористой структуры в том же растворе (83.3 ± 2.1 кДж/моль). Это объясняется ускоренным растворением АОА на дне пор за счет наличия в процессе анодирования электрического поля высокой напряженности в барьерном слое. Для всех рассмотренных электролитов (1.0 М H2SeO4, 2.0 М H2SO4, 4.0 М H2SO4) наблюдается небольшое увеличение энергии активации усиленного электрическим полем растворения АОА на дне пор при минимальном использованном напряжении. Эту особенность мы связываем с уменьшением напряженности электрического поля при малом напряжении за счет наличия на поверхности алюминия естественного оксида. В связи с тем, что $E_{{\text{a}}}^{{{\text{хим}}}}$ > Ea, с понижением температуры электролита максимально достижимая толщина пористой пленки анодного оксида алюминия увеличивается. В частности, максимальная толщина пленки АОА, полученной при напряжении 10 В в 2.0 М H2SO4, будет уменьшаться с 61 до 25 мкм с увеличением температуры электролита от 0 до 20°C.
Список литературы
Bengough G.D., Stuart J.M. A process of producing a coloured surface on aluminium or aluminium alloys. GB Patent 223995. 1923.
Bengough G.D., Stuart J.M. Improved process of protecting surfaces of aluminium of aluminium alloys. GB Patent 223994. 1923.
ГОСТ 9.305-84 Единая система защиты от коррозии и старения. Покрытия металлические и неметаллические неорганические. 1984.
Banerjee P., Perez I., Henn-Lecordier L. et al. // Nat. Nanotechnol. 2009. V. 4. № 5. P. 292. https://doi.org/10.1038/nnano.2009.37
Karpov E.E., Karpov E.F., Suchkov A. et al. // Sens. Actuators Phys. 2013. V. 194. P. 176. https://doi.org/10.1016/j.sna.2013.01.057
Inada T., Uno N., Kato T. et al. // J. Mater. Res. 2005. V. 20. № 1. P. 114. https://doi.org/10.1557/JMR.2005.0016
Петухов Д.И., Елисеев А.А., Булдаков Д.А. и др. // Мембраны. 2009. Т. 3. № 43. С. 16.
Wang B., Fei G.T., Wang M. et al. // Nanotechnology. 2007. V. 18. № 36. P. 365601. https://doi.org/10.1088/0957-4484/18/36/365601
Yayoi K., Jinno M., Fujikawa R. et al. // IEEJ Trans. Electr. Electron. Eng. 2007. V. 2. № 4. P. 463. https://doi.org/10.1002/tee.20191
Kushnir S.E., Pchelyakova T.Yu., Napolskii K.S. // J. Mater. Chem. C. 2018. V. 6. № 45. P. 12192. https://doi.org/10.1039/C8TC04246B
Li A.P., Müller F., Birner A. et al. // J. Appl. Phys. 1998. V. 84. № 11. P. 6023. https://doi.org/10.1063/1.368911
Kikuchi T., Nishinaga O., Natsui S. et al. // Electrochim. Acta. 2015. V. 156. № Supplement C. P. 235. https://doi.org/10.1016/j.electacta.2014.12.171
Akiya S., Kikuchi T., Natsui S. et al. // Electrochim.Acta. 2016. V. 190. № Supplement C. P. 471. https://doi.org/10.1016/j.electacta.2015.12.162
Roslyakov I.V., Gordeeva E.O., Napolskii K.S. // Electrochim. Acta. 2017. V. 241. P. 362. https://doi.org/10.1016/j.electacta.2017.04.140
Gordeeva E.O., Roslyakov I.V., Napolskii K.S. // Electrochim. Acta. 2019. V. 307. P. 13. https://doi.org/10.1016/j.electacta.2019.03.098
Leontiev A.P., Roslyakov I.V., Napolskii K.S. // Electrochim. Acta. 2019. V. 319. P. 88. https://doi.org/10.1016/j.electacta.2019.06.111
Masuda H., Fukuda K. // Science. 1995. V. 268. № 5216. P. 1466. https://doi.org/10.1126/science.268.5216.1466
Zhao S., Chan K., Yelon A. et al. // Adv. Mater. 2007. V. 19. № 19. P. 3004. https://doi.org/10.1002/adma.200701284
Patermarakis G., Lenas P., Karavassilis Ch. et al. // Electrochim. Acta. 1991. V. 36. № 3. P. 709. https://doi.org/10.1016/0013-4686(91)85162-Z
Aerts T., Dimogerontakis Th., De Graeve I. et al. // Surf. Coat. Technol. 2007. V. 201. № 16. P. 7310. https://doi.org/10.1016/j.surfcoat.2007.01.044
Davies J.A., Domeij B., Pringle J.P.S. et al. // J. Electrochem. Soc. 1965. V. 112. № 7. P. 675. https://doi.org/10.1149/1.2423662
Mirzoev R.A., Davydov A.D., Zarubenko E.S. et al. // Electrochim. Acta. 2016. V. 218. P. 74. https://doi.org/10.1016/j.electacta.2016.09.115
Mirzoev R.A., Davydov A.D., Vystupov S.I. et al. // Electrochim. Acta. 2017. V. 243. P. 270. https://doi.org/10.1016/j.electacta.2017.05.025
O’Sullivan J.P., Wood G.C. // Proc. R. Soc. Lond. Ser. Math. Phys. Sci. 1970. V. 317. № 1531. P. 511.
Kushnir S.E., Komarova T.Yu., Napolskii K.S. // J. Mater. Chem. C. 2020. V. 8. P. 3991.https://doi.org/10.1039/C9TC07079F
Nazarkina Y., Gavrilov S., Terryn H. et al. // J. Electrochem. Soc. 2015. V. 162. № 9. P. E166. https://doi.org/10.1149/2.0571509jes
Nazarkina Y., Kamnev K., Dronov A. et al. // Electrochim. Acta. 2017. V. 231. P. 327. https://doi.org/10.1016/j.electacta.2017.02.049
Sadykov A.I., Kushnir S.E., Roslyakov I.V. et al. // Electrochem. Commun. 2019. V. 100. P. 104. https://doi.org/10.1016/j.elecom.2019.01.027
Kushnir S.E., Napolskii K.S. // Mater. Des. 2018. V. 144. P. 140. https://doi.org/10.1016/j.matdes.2018.02.012
Choy T.C. Effective medium theory: principles and applications. 2nd ed. Oxford: Oxford University Press, 2016. 241 p.
Malitson I.H. // J. Opt. Soc. Am. 1962. V. 52. № 12. P. 1377. https://doi.org/10.1364/JOSA.52.001377
Santos A., Balderrama V.S., Alba M. et al. // Adv. Mater. 2012. V. 24. № 8. P. 1050. https://doi.org/10.1002/adma.201104490
Rahman M.M., Garcia-Caurel E., Santos A. et al. // Nanoscale Res. Lett. 2012. V. 7. № 1. P. 474. https://doi.org/10.1186/1556-276X-7-474
Beyer K.D., Ravishankara A.R., Lovejoy E.R. // J. Geophys. Res. Atmospheres. 1996. V. 101. № D9. P. 14519. https://doi.org/10.1029/96JD00937
Xing J., Lu S., Zhang C. et al. // Phys. Chem. Chem. Phys. 2017. V. 19. № 32. P. 21696. https://doi.org/10.1039/C7CP03321D
Kashi M.A., Ramazani A. // J. Phys. Appl. Phys. 2005. V. 38. № 14. P. 2396. https://doi.org/10.1088/0022-3727/38/14/015
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии