

Журнал неорганической химии, 2021, T. 66, № 2, стр. 204-219
Cпектроскопия ЯМР – универсальный метод исследования структуры и магнитных свойств комплексов парамагнитных лантанидов в растворах (обзор)
Ю. Г. Горбунова a, b, *, А. Г. Мартынов b, К. П. Бирин b, А. Ю. Цивадзе a, b
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
b Институт физической химии и электрохимии РАН им. А.Н. Фрумкина
119071 Москва, Ленинский пр-т, 31, корп. 4, Россия
* E-mail: yulia@igic.ras.ru
Поступила в редакцию 10.08.2020
После доработки 19.08.2020
Принята к публикации 25.08.2020
Аннотация
Координационные соединения лантанидов привлекают все большее внимание исследователей благодаря уникальным физико-химическим (в том числе оптическим и магнитным) свойствам. При этом тонкая настройка свойств функциональных материалов на их основе возможна за счет управления структурой комплексов. В данном обзоре на примере серий соединений парамагнитных лантанидов с тетрапиррольными лигандами рассмотрено применение спектроскопии ЯМР для решения сложной задачи идентификации парамагнитных комплексов лантанидов, а также для установления корреляции между строением координационной сферы ионов лантанидов и магнитными характеристиками комплексов. Показано, как анализ лантанид-индуцированных сдвигов и скоростей лантанид-индуцированной релаксации в сериях изоструктурных соединений способствует выявлению спектрально-структурных корреляций, которые с привлечением рентгеноструктурных и расчетных данных позволяют не только устанавливать структуру соединения в растворе, но и проводить первичную оценку возможности использования комплексов в качестве мономолекулярных магнитов.
ВВЕДЕНИЕ
Уникальные свойства ионов лантанидов с частично заполненными 4f-подуровнями обусловливают широкое применение координационных соединений на их основе в качестве релаксационных агентов для магнитно-резонансной томографии, парамагнитных и люминесцентных меток для изучения биологических объектов [1–5 ]. Среди новых высокотехнологичных областей применения комплексов лантанидов особое внимание привлекает создание мономолекулярных материалов для хранения больших объемов информации и разработки технологий квантовых вычислений [6–9], катализаторов для полимеризации диенов и циклических эфиров [10], а также редокс-активных материалов для элементов записи и хранения информации [11–13]. Кроме того, в последнее время все большее внимание уделяется гибридным органо-неорганическим соединениям, когда комплексы лантанидов вводятся либо на поверхность/в объем неорганической матрицы [14], либо, наоборот, функциональные органические молекулы встраиваются в неорганические лантанидсодержащие матрицы [15]. Особого внимания заслуживает относительно новый класс соединений – слоистые гидроксиды лантанидов, особенностью которых является возможность изменения их состава в широких пределах при изменении основного структурного мотива. Так, за счет анионного обмена с органическими лигандами возможно получение уникальных люминесцентных материалов [16–19], сенсоров и препаратов для биомедицинских приложений, в том числе тераностики [20], а также катализаторов и фотокатализаторов [21–24]. Недавно было показано, что комбинирование наночастиц диоксида церия с фталоцианинами позволяет контролировать процесс генерации синглетного кислорода фотосенсибилизатором [25, 26].
Такой широкий диапазон применения координационных соединений лантанидов обусловлен их специфической 4f-электронной конфигурацией и разнообразием координационных чисел. Все это в совокупности со значительным уменьшением атомных и ионных размеров лантанидов в ряду от La к Lu (так называемое лантанидное сжатие) позволяет тонко варьировать геометрию и электронную структуру комплексов на их основе. В свою очередь, корреляция между строением комплексов лантанидов и функциональными характеристиками материалов на их основе определяет важность разработки подходов к определению их структуры при совместном применении спектральных и структурных методов для создания материалов с заданными свойствами.
Комплементарное применение структурных методов в сочетании со спектроскопией ЯМР для исследования парамагнитных комплексов лантанидов позволяет решать две взаимосвязанные задачи. С одной стороны, знание структуры, полученной на основе рентгеноструктурного анализа (РСА) или квантово-химического моделирования, позволяет интерпретировать спектры ЯМР парамагнитных комплексов и определять тем самым характеристики, важные для дизайна, например, магнитных материалов [27, 28]. С другой стороны, анализ спектров ЯМР позволяет изучать особенности геометрической структуры и электронного строения комплексов в растворе [29–31]. Данные методы применимы также к исследованию внутримолекулярной динамики конформационно-подвижных соединений – макроциклов, полидентатных лигандов, биомолекул и т.п. [32–35].
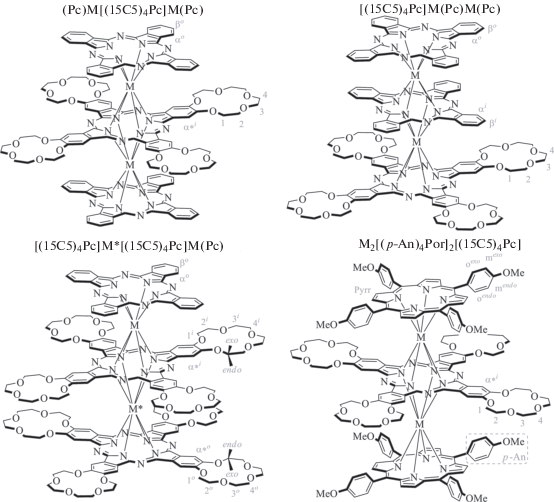
В представленном обзоре подробно описан подход к решению обеих задач с применением спектрально-структурного анализа на примере работ авторов по исследованию сэндвичевых трехпалубных комплексов лантанидов с тетрапиррольными лигандами (рис. 1) – перспективных компонентов новых супрамолекулярных оптоэлектронных и магнитных материалов [36–38].
ПРИМЕНЕНИЕ МЕТОДА ЯМР ДЛЯ ИЗУЧЕНИЯ КОМПЛЕКСОВ ЛАНТАНИДОВ
Характерной особенностью комплексов парамагнитных лантанидов является наличие в их спектрах ЯМР сигналов резонанса, существенно смещенных относительно их положения в спектрах диамагнитных аналогов. Наблюдаемые лантанид-индуцированные сдвиги (ЛИС, $\Delta \delta _{k}^{{{\text{Ln}}}},$ уравнение (1), где M – диамагнитный ион РЗЭ – La3+, Lu3+ или Y3+) могут быть как положительными, так и отрицательными в зависимости от природы лантанида в составе комплекса и взаимного расположения рассматриваемого атома и иона лантанида [39].
Кроме того, наличие парамагнитных ионов в таких комплексах приводит к лантанид-индуцированному увеличению скорости релаксации (ЛИР), что еще больше усложняет задачу анализа спектров ЯМР комплексов лантанидов. Так, ускорение релаксации в комплексах лантанидов приводит к уширению сигналов в спектрах соединений, пропорциональному времени поперечной релаксации ${{T}_{2}}.$ При этом пропадает расщепление сигналов, которое позволяет выделить группы спиново-связанных протонов, а в предельном случае уширение приводит к полному исчезновению сигналов в спектре. Кроме того, применение двумерных методик с переносом поляризации становится практически невозможным в силу низкой эффективности такого переноса в условиях быстрой продольной релаксации ядер ${{T}_{1}}.$
Тем не менее задача исследования комплексов парамагнитных лантанидов методом спектроскопии ЯМР может быть решена на основании функциональных зависимостей ЛИС и ЛИР от геометрических характеристик, определяющих взаимное расположение рассматриваемого атома и иона лантанида.
ЛИС протона Hk определяется как суперпозиция контактной ($\Delta \delta _{k}^{{{\text{con}}}},$ уравнение (2)) и дипольной ($\Delta \delta _{k}^{{{\text{dip}}}},$ уравнение (3)) составляющих, передающихся через систему связей и пространство соответственно:
(1)
$\Delta \delta _{k}^{{{\text{Ln}}}} = \delta _{k}^{{{\text{Ln}}}} - \delta _{k}^{{\text{M}}} = \Delta \delta _{k}^{{{\text{con}}}} + \Delta \delta _{k}^{{{\text{dip}}}},$(2)
$\Delta \delta _{k}^{{{\text{con}}}} = {{F}_{k}}{{\left\langle {{{S}_{Z}}} \right\rangle }^{{{\text{Ln}}}}},$(3)
$\Delta \delta _{k}^{{{\text{dip}}}} = \frac{{\chi _{{ax}}^{{{\text{Ln}}}}}}{{12\pi }}{{G}_{k}},\,\,\,\,{{G}_{k}} = \frac{{3{{{\cos }}^{2}}{{\theta }_{k}} - 1}}{{r_{k}^{3}}}.$Коэффициент ${{F}_{k}}$ в уравнении (2) характеризует величину контактной составляющей ЛИС, возникающей благодаря делокализации спиновой плотности в молекуле комплекса по системе химических связей в результате взаимодействия Ферми. Данное взаимодействие быстро затухает с увеличением количества связей между ионом лантанида и рассматриваемым протоном. Величина ${{\left\langle {{{S}_{Z}}} \right\rangle }^{{{\text{Ln}}}}}$ является расчетной табулированной характеристикой магнитных свойств ионов лантанидов [40, 41].
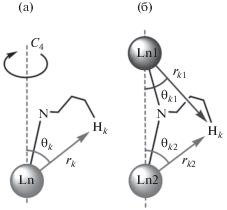
Коэффициент ${{G}_{k}}$ в уравнении (3) характеризует дипольную составляющую ЛИС, связанную с взаимодействием рассматриваемого k-го ядра с парамагнитным центром через пространство: ${{\theta }_{k}}$ – азимут относительно главной оси симметрии молекулы, ${{r}_{k}}$ – расстояние от k-го протона до иона Ln3+ (рис. 2a), $\chi _{{ax}}^{{{\text{Ln}}}}$ – аксиальная анизотропия тензора магнитной восприимчивости. В случае биядерных комплексов, содержащих два иона лантанидов, величина $\Delta \delta _{k}^{{{\text{dip}}}}$ является аддитивной и определяется индивидуальными вкладами от каждого иона (рис. 2б, уравнение (4)). Таким образом, входящая в выражение для дипольного вклада ЛИС величина ${{G}_{k}}$ несет информацию о геометрических характеристиках молекулы комплекса в растворе, в то время как $\chi _{{{\text{ax}}}}^{{{\text{Ln}}}}$ характеризует магнетохимические свойства комплекса.
(4)
$\Delta \delta _{k}^{{{\text{dip}}}} = \frac{{\chi _{{{\text{ax}}}}^{{{\text{Ln1}}}}}}{{12\pi }}\left[ {\frac{{3{{{\cos }}^{2}}{{\theta }_{{k1}}} - 1}}{{r_{{k1}}^{3}}}} \right] + \frac{{\chi _{{{\text{ax}}}}^{{{\text{Ln2}}}}}}{{12\pi }}\left[ {\frac{{3{{{\cos }}^{2}}{{\theta }_{{k2}}} - 1}}{{r_{{k2}}^{3}}}} \right].$Рис. 2.
Определение параметров, входящих в уравнение (3) и определяющих величину дипольного вклада ЛИС в случае моно- (а) и биядерных (б) комплексов.
Разделение ЛИС на контактную и дипольную составляющие основано на использовании приближенного представления дипольного вклада в виде произведения параметра кристаллического поля $A_{2}^{0}$ как характеристики рассматриваемого комплекса и табулированной расчетной константы Блини ${{D}^{{{\text{Ln}}}}}$ как характеристики иона лантанида [42]. Дальнейшей трансформацией уравнения (5) могут быть получены различные линейные формы, применение которых для серий изоструктурных комплексов с различными лантанидами позволяет исследовать поведение коэффициентов ${{F}_{k}},$ $A_{2}^{0}$ и ${{G}_{k}}$ по мере увеличения числа f-электронов и уменьшения ионных радиусов лантанидов [39, 43].
(5)
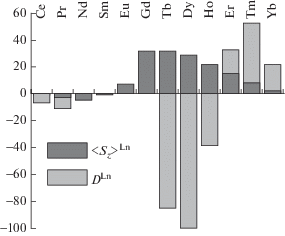
$\Delta \delta _{k}^{{{\text{Ln}}}} = {{F}_{k}}{{\left\langle {{{S}_{z}}} \right\rangle }^{{{\text{Ln}}}}} + A_{2}^{0}{{D}^{{{\text{Ln}}}}}{{G}_{k}}.$Многочисленные примеры применения такого подхода к анализу сэндвичевых комплексов с парамагнитными лантанидами свидетельствуют о пренебрежимо малом вкладе контактной составляющей в величину ЛИС [44–49], поскольку величина $\Delta \delta _{k}^{{{\text{con}}}}$ быстро уменьшается с увеличением числа связей между ионом лантанида и рассматриваемым протоном. Такое поведение ЛИС также связано со спецификой изменения термов ${{\left\langle {{{S}_{Z}}} \right\rangle }^{{{\text{Ln}}}}}$ и ${{D}^{{{\text{Ln}}}}}$ в ряду лантанидов (рис. 3).
Благодаря преимущественно дипольной природе ЛИС становится возможным прямое сопоставление структурных характеристик, найденных с помощью рентгеноструктурного анализа или квантово-химического моделирования с данными, полученными из анализа спектров ЯМР.
Другим подходом спектрально-структурного изучения комплексов лантанидов методом ЯМР является исследование времен релаксации ядер [43]. ЛИР является функцией парамагнитных характеристик ядра и $r_{k}^{6},$ где ${{r}_{k}}$ – расстояние от парамагнитного лантанида до изучаемого ядра k. Таким образом, информация об уменьшении времени релаксации позволяет выполнить отнесение сигналов в спектре по данным о строении соединения, а также уточнить межъядерные расстояния внутри соединения в растворе. Время продольной релаксации ${{T}_{1}}$ может быть измерено с помощью стандартной двухимпульсной последовательности. Время поперечной релаксации ${{T}_{2}}$ определяется из соотношения $\Delta {{\nu }_{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}} = {{\left( {{\pi }{{T}_{2}}} \right)}^{{ - 1}}},$ где $\Delta {{\nu }_{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}$ – полуширина сигнала.
Выявление парамагнитного ускорения релаксации ${{T}_{1}}$ возможно при наличии данных о времени релаксации изоструктурного комплекса диамагнитного лантанида. В этом случае наблюдаемая скорость релаксации определяется как
(6)
$\frac{1}{{T_{1}^{{{\text{exp}}}}}} = \frac{1}{{T_{1}^{{{\text{dia}}}}}} + \frac{1}{{T_{1}^{{{\text{para}}}}}}.$В этом выражении $T_{1}^{{{\text{exp}}}}$ – наблюдаемое время релаксации ядра, $T_{1}^{{{\text{dia}}}}$ – время релаксации ядра в диамагнитном реперном комплексе, ${1 \mathord{\left/ {\vphantom {1 {T_{1}^{{{\text{para}}}}}}} \right. \kern-0em} {T_{1}^{{{\text{para}}}}}}$ – парамагнитный вклад в скорость релаксации. Вычисленные из приведенного соотношения ${1 \mathord{\left/ {\vphantom {1 {T_{1}^{{{\text{para}}}}}}} \right. \kern-0em} {T_{1}^{{{\text{para}}}}}}$ являются прямой характеристикой расстояния лантанид–ядро. Для пары ядер k и l в соединении справедливо соотношение (7), которое позволяет проводить отнесение сигналов резонанса на основе структурных моделей и уточнять геометрические параметры структуры соединения:
(7)
$\frac{{T_{{1k}}^{{{\text{para}}}}}}{{T_{{1l}}^{{{\text{para}}}}}} = {{\left( {\frac{{{{r}_{k}}}}{{{{r}_{l}}}}} \right)}^{6}}.$В случае соединений, содержащих два парамагнитных центра, анализ усложняется в силу их взаимного влияния на каждый рассматриваемый протон. Таким образом, наблюдаемая скорость релаксации определяется тремя вкладами: диамагнитным и двумя парамагнитными:
(8)
$\frac{1}{{T_{1}^{{{\text{exp}}}}}} = \frac{1}{{T_{1}^{{{\text{dia}}}}}} + \frac{1}{{T_{1}^{{{\text{para(Ln1)}}}}}} + \frac{1}{{T_{1}^{{{\text{para(Ln2)}}}}}}.$В связи с тем, что парамагнитное ускорение релаксации быстро спадает с удалением ядра от парамагнитного центра, а также с учетом симметричного строения изучаемых комплексов для наиболее точного определения отклонений межатомных расстояний в расчетной геометрической модели от реальных значений следует использовать данные о парамагнитном ускорении, определяемом ближайшим к ядру лантанидом. Для анализа расстояний Hi ↔ Ln внутри комплекса необходимо разложить суммарное парамагнитное ускорение релаксации на парамагнитные вклады, определяемые влиянием каждого из лантанидов. С учетом соотношения ${1 \mathord{\left/ {\vphantom {1 {T_{1}^{{{\text{para}}}}}}} \right. \kern-0em} {T_{1}^{{{\text{para}}}}}} \sim {{r}^{{ - 6}}}$ для любого протона системы Hk:
(9)
$T_{1}^{{{\text{para(Ln1)}}}} = T_{1}^{{{\text{para}}}} + T_{1}^{{{\text{para}}}}{{\left( {\frac{{{{r}_{1}}}}{{{{r}_{2}}}}} \right)}^{6}}.$Точное определение расстояний по данным времен релаксации требует разложения парамагнитного ускорения на вклады, определяемые различными механизмами релаксации. Кроме того, необходима информация о ряде параметров ядра лантанида, зависящих от его окружения. Эта информация отсутствует в литературе для комплексов лантанидов с тетрапиррольными лигандами, однако возможна верификация расчетной структурной модели на основании отношений расстояний и парамагнитных ускорений релаксации. Для двух протонов k и l комплекса должно выполняться равенство:
(10)
$\frac{{{{r}_{1}}({{{\text{H}}}_{k}})}}{{{{r}_{1}}({{{\text{H}}}_{l}})}} = {{\left[ {\frac{{T_{1}^{{{\text{para(Ln1)}}}}({{{\text{H}}}_{k}})}}{{T_{1}^{{{\text{para(Ln1)}}}}({{{\text{H}}}_{l}})}}} \right]}^{{{1 \mathord{\left/ {\vphantom {1 6}} \right. \kern-0em} 6}}}}.$Приведенное соотношение позволяет экспериментально определить соотношение расстояний Ln ↔ Hk для любой пары протонов комплекса, а также для модельной структуры. Соотношение экспериментальной и рассчитанной величин может служить характеристикой точности модели.
Структурно-спектральные характеристики гетеролептических краун-замещенных трехпалубных фталоцианинатов лантанидов
В данном разделе рассмотрено применение метода ЯМР-спектроскопии для изучения двух серий изомерных комплексов, содержащих два незамещенных фталоцианиновых лиганда (Рс) и один тетра-15-краун-5-замещенный лиганд [(15С5)4Рс] в терминальном или центральном положении – [(15C5)4Pc]M(Pc)M(Pc) [46, 50] и (Pc)M[(15C5)4Pc]M(Pc) [51, 52], а также ряд комплексов с двумя соседними замещенными лигандами – [(15C5)4Pc]M*[(15C5)4Pc]M(Pc) (рис. 1). Для последнего семейства комплексов были получены как гомо- (M* = M [51, 52]), так и гетероядерные (M* ≠ M [49, 53, 54]) производные. В качестве металлов-комплексообразователей использовали лантаниды(III) Nd, Sm, Eu, Tb, Dy, Ho, Er, Tm, Yb, в качестве диамагнитного репера – Y. Интерес к исследованию данных комплексов обусловлен их способностью к супрамолекулярной сборке [37, 38, 55], что позволяет рассматривать их как перспективные компоненты функциональных магнитных материалов [56] и молекулярных переключателей с управляемыми свойствами [57].
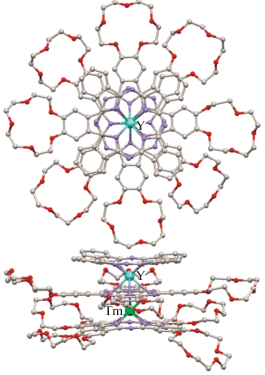
Для различных типов обсуждаемых комплексов были выращены монокристаллы, структура которых расшифрована рентгеноструктурным методом [49, 51], что позволило установить единообразие строения координационных полиэдров ионов лантанидов вне зависимости от количества и расположения краун-эфирных заместителей. Координационные полиэдры ионов металлов представляют собой искаженные тетрагональные антипризмы, образованные развернутыми под углом ~45° тетрапиррольными лигандами. Связи ионов металлов с изоиндольными атомами азота, образованные терминальными лигандами, систематически короче на ~0.3 Å по сравнению со связями от ионов M3+ до атомов Niso мостиковых внутренних лигандов. Расстояния между фталоцианиновыми лигандами составляют 2.9–3.1 Å, расстояния между металлоцентрами – 3.43–3.53 Å. Для примера на рис. 4 показана структура комплекса [(15C5)4Pc]Y[(15C5)4Pc]Tm(Pc) [49].
Рис. 4.
Структура гетероядерного комплекса [(15C5)4Pc]Tm[(15C5)4Pc]Y(Pc), вид сверху и сбоку. Атомы водорода и сольватные молекулы растворителей не показаны [49].
Для всех трех серий соединений были получены спектры 1H ЯМР растворов в CDCl3. Сдвиги для комплекса каждого парамагнитного лантанида индивидуальны и характеризуются его природой. На рис. 5 приведены примеры спектров изоструктурных комплексов [(15C5)4Pc]M(Pc)M(Pc) со спектральными диапазонами, характеристичными для ионов лантанидов с максимальными положительными (Tm3+) и отрицательными (Tb3+) ЛИС в тетрапиррольном окружении. В то же время спектр комплекса иттрия имеет типичный для диамагнитных соединений вид.
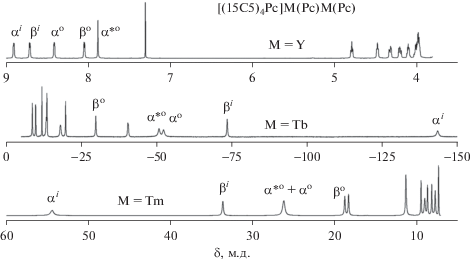
В табл. 1 и 2 приведены химические сдвиги сигналов резонанса протонов в спектрах ЯМР изомерных гомоядерных комплексов (Pc)M[(15C5)4Pc]M(Pc) и [(15C5)4Pc]M(Pc)M(Pc) в CDCl3. Из этих данных видно, что в гомоядерных комплексах сигналы резонанса всех протонов претерпевают смещение либо в слабое (M = = Eu, Er, Tm, Yb), либо в сильное поле (Nd, Sm, Tb, Dy, Ho) относительно их положения в спектре диамагнитного комплекса иттрия. Во всех случаях величина этого смещения уменьшается с увеличением расстояния между протоном и ионами Ln3+.
Таблица 1.
Положение сигналов резонанса (δ, м.д.) в спектрах 1H ЯМР гомоядерных комплексов (Pc)M[(15C5)4Pc]M(Pc) [51, 52] в CDCl3. Обозначения протонов приведены на рис. 1
| H | Nd | Sm | Eu | Tb | Dy | Ho | Er | Tm | Yb | Y |
|---|---|---|---|---|---|---|---|---|---|---|
| αo | 6.46 | 8.02 | 9.72 | –50.47 | –21.51 | –2.13 | 21.77 | 25.1 | 10.38 | 8.58 |
| βo | 6.69 | 7.56 | 8.39 | –30.07 | –11.57 | 0.8 | 15.61 | 17.76 | 8.93 | 7.92 |
| α*i | 1.49 | 6.59 | 11.92 | –154.76 | –74 | –21.96 | 44.23 | 53.58 | 13 | 8.25 |
| 1-CH2 | 2.37 | 4.42 | 6.62 | –67.6 | –31.59 | –8.51 | 20.81 | 23.4 | 7.34 | 5.13 |
| 2-CH2 | 3.07 | 4.29 | 5.68 | –35.27 | –15.37 | –2.58 | 13.58 | 15.99 | 5.9 | 4.67 |
| 3-CH2 | 3.29 | 4.06 | 4.96 | –21.68 | –8.7 | –0.41 | 10.07 | 11.64 | 5.11 | 4.31 |
| 4-CH2 | 3.35 | 4.06 | 4.85 | –19.23 | –7.46 | –0.01 | 9.45 | 10.89 | 4.98 | 4.26 |
Таблица 2.
Положение сигналов резонанса (δ, м.д.) в спектрах 1H ЯМР гомоядерных комплексов [(15C5)4Pc]M(Pc)M(Pc) [46, 50] в CDCl3. Обозначения протонов приведены на рис. 1
| H | Nd | Sm | Eu | Tb | Dy | Er | Tm | Y |
|---|---|---|---|---|---|---|---|---|
| αo | 6.23 | 7.87 | 9.80 | –53.27 | –24.00 | 22.17 | 26.14 | 8.44 |
| βo | 6.76 | 7.72 | 8.70 | –31.67 | –12.78 | 15.41 | 18.70 | 8.09 |
| α*i | 2.48 | 7.39 | 13.13 | –148.51 | –72.05 | 45.10 | 54.40 | 8.97 |
| β*i | 4.99 | 7.85 | 11.37 | –76.35 | –34.37 | 28.86 | 33.56 | 8.71 |
| α*o | 5.51 | 7.30 | 9.27 | –54.81 | –24.00 | 22.64 | 26.14 | 7.86 |
Указанная выше возможность пренебрежения контактной составляющей ЛИС позволяет рассматривать ЛИС как величину, пропорциональную аксиальной анизотропии тензора магнитной восприимчивости $\chi _{{ax}}^{{{\text{Ln}}}}$ иона Ln3+ и геометрическому параметру ${{G}_{k}},$ аддитивному в случае биядерных комплексов (рис. 2б, уравнение (4)). В случае несимметричных гомоядерных трис-фталоцианинатов [(15C5)4Pc]M[(15C5)4Pc]M(Pc) ионы лантанидов оказываются в неэквивалентном координационном окружении – гомо- и гетеролептическом, что приводит к разнице в величинах $\chi _{{ax}}^{{{\text{Ln1}}}}$ и $\chi _{{ax}}^{{{\text{Ln2}}}}$ [49], однако в первом приближении этим различием можно пренебречь из-за близости электронных свойств незамещенных и краун-замещенных лигандов. Аналогичное приближение используется для анализа спектров комплексов [(15C5)4Pc]M(Pc)M(Pc) [46].
Таким образом, с использованием указанного приближения отношения ЛИС для различных пар протонов Hk и Hl можно с достаточной точностью описать отношением соответствующих геометрических факторов – ${{R}_{{kl}}}.$ Подставив в уравнение (11) выражения для ЛИС из уравнения (1), получим уравнение (12), согласно которому можно оценить положение сигналов резонанса в спектрах парамагнитных комплексов $\delta _{k}^{{{\text{Ln}}}}$ исходя из положения соответствующих сигналов в спектре ЯМР диамагнитного комплекса $\delta _{k}^{Y}$ и отношений ${{R}_{{kl}}}$ для соответствующих пар протонов, которые могут быть найдены из рентгеноструктурных данных или данных квантово-химического моделирования [44, 48]:
(11)
$\frac{{\Delta \delta _{k}^{{{\text{Ln}}}}}}{{\Delta \delta _{l}^{{{\text{Ln}}}}}} \approx \frac{{\Delta \delta _{k}^{{{\text{dip}}}}}}{{\Delta \delta _{l}^{{{\text{dip}}}}}} = {{R}_{{kl}}},\,\,\,\,{{R}_{{kl}}} = \frac{{{{G}_{k}}}}{{{{G}_{l}}}},$(12)
$\delta _{k}^{{{\text{Ln}}}} = \delta _{k}^{Y} + \Delta \delta _{l}^{{{\text{Ln}}}}{{R}_{{kl}}}.$Применение уравнения (12) позволяет объяснить особенность спектров ЯМР изомерных гетероядерных комплексов [(15C5)4Pc]M*[(15C5)4Pc]M(Pc) – [M*, M] по сравнению с гомоядерными аналогами. В спектрах комплексов [Ln*, Y] и [Y*, Ln] наблюдаются сигналы резонанса, смещенные как в сильное, так и в слабое поле относительно их положения в спектре комплекса [Y*, Y], тогда как в спектрах гомоядерных комплекcов [Ln*, Ln] величина ЛИС имеет одинаковый знак для всех протонов (табл. 3).
Таблица 3.
Положение сигналов резонанса (δ, м.д.) в спектрах 1H ЯМР гомо- и гетероядерных комплексов [(15C5)4Pc]M*[(15C5)4Pc]M(Pc) – [M*, M] в CDCl3 [49, 51, 53]. Обозначения протонов приведены на рис. 1
| Параметр | [Tb*, Tb] | [Tb*, Y] | [Y*, Tb] | [Y*, Y] |
|---|---|---|---|---|
| αo | –56.34 | 24.82 | –71.45 | 8.58 |
| βo | –34.72 | 9.19 | –34.22 | 8.02 |
| α*i | –157.94 | –70.67 | –71.45 | 8.27 |
| α*o | –56.34 | –71.09 | 23.86 | 7.95 |
| экзо-1i | –36.99 | –19.67 | –42.05 | 5.19 |
| эндо-1i | –36.25 | –29.76 | –28.04 | 5.19 |
| экзо-1o | –33.84 | –22.30 | 9.18 | 4.79 |
| эндо-1o | –17.78 | –33.77 | 0.08 | 4.44 |
Такое поведение гетероядерных комплексов обусловлено нелинейностью функции ${{G}_{k}},$ из-за которой вокруг иона Ln3+ образуются области как с отрицательными, так и с положительными знаками ${{G}_{k}}$ (рис. 6). Так, в случае комплексов [Tb*, Y] в области с ${{\theta }_{k}} > 0$ оказываются ароматические протоны терминального незамещенного фталоцианинового лиганда αo и βo (рис. 6a), тогда как в случае изомерного комплекса [Y*, Tb] в эту область попадают протоны замещенного лиганда – ароматические протоны α*o и экзопротоны первой метиленовой группы краун-эфирных заместителей (рис. 6б).
Рис. 6.
Контурные карты функции $G(\theta ,r)$ для гетероядерных комплексов [Ln*, Y] (a) и [Y*, Ln] (б) и гомоядерных комплексов [Ln*, Ln] (в) [49]. Координаты протонов получены усреднением данных РСА для комплекса [Tm*, Y]. Овалами отмечены протоны, попадающие в область противоположного знака функции $G(\theta ,r)$ по сравнению с большинством протонов молекул. Обозначения протонов даны на рис. 1.
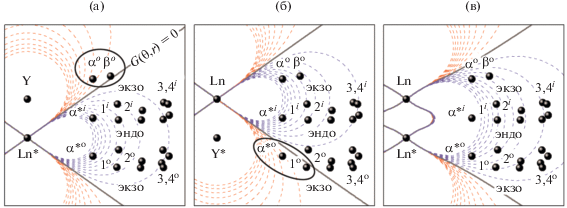
В спектрах гомоядерных комплексов функции ${{G}_{k}}$ суммируются для обоих ионов лантанидов и все протоны находятся в знакопостоянной области с ${{\theta }_{k}} < 0$ (рис. 6в). Наблюдаемое согласие между величинами ${{G}_{k}},$ рассчитанными из данных РСА и 1H ЯМР, свидетельствует об изоструктурности комплексов в кристалле и в растворе, благодаря чему данные РСА являются адекватной моделью для интерпретации спектров ЯМР трис-фталоцианинатов лантанидов.
Наличие данных РСА для исследованных комплексов позволяет определить величину входящей в уравнение (12) важной характеристики магнитных свойств комплексов – аксиальной анизотропии тензора магнитной восприимчивости $\chi _{{ax}}^{{{\text{Ln}}}}.$ Знак данной величины определяет знаки ЛИС протонов, попадающих в области с различными знаками ${{G}_{k}}.$
С помощью метода наименьших квадратов были найдены угловые коэффициенты зависимости величин ЛИС протонов от соответствующих им значений функции ${{G}_{k}},$ рассчитанных исходя из рентгеноструктурных данных (рис. 7). Проведение такого анализа для серий гетероядерных комплексов [(15C5)4Pc]M*[(15C5)4Pc]M(Pc) – [Ln*, Y] и [Y*, Ln] показало, что в комплексах [Y*, Ln] с гетеролептическим окружением ионов Ln3+ $\chi _{{ax}}^{{{\text{Ln}}}}$ выше, чем в изомерных комплексах [Ln*, Y] с гомолептическим окружением иона Ln3+ (табл. 4).
Рис. 7.
Определение аксиальной анизотропии $\chi _{{{\text{ax}}}}^{{{\text{Tb}}}}$ регрессионным методом на основании данных ЯМР и РСА для гетероядерных комплексов [(15C5)4Pc]M*[(15C5)4Pc]M(Pc) – [M*, M] [49].
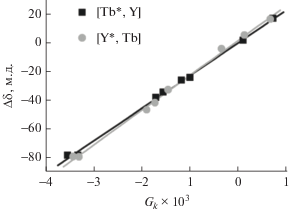
Таблица 4.
Аксиальная анизотропия тензора магнитной восприимчивости в комплексах [(15C5)4Pc]M*[(15C5)4Pc]M(Pc) – [M*, M] по данным спектроскопии 1H ЯМР и РСА [49]
| Ln | [Ln*, Y] $\chi _{{ax}}^{{{\text{Ln}}}}$, м–3 | [Y*, Ln] $\chi _{{ax}}^{{{\text{Ln}}}}$, м–3 |
|---|---|---|
| Nd | 3.42 × 10–32 | 3.55 × 10–32 |
| Eu | –2.06 × 10–32 | –2.22 × 10–32 |
| Tb | 8.41 × 10–31 | 8.98 × 10–31 |
| Dy | 4.30 × 10–31 | 4.92 × 10–31 |
| Er | –1.90 × 10–31 | –2.17 × 10–31 |
| Tm | –2.39 × 10–31 | –2.57 × 10–31 |
| Yb | –2.77 × 10–32 | –2.95 × 10–32 |
Определение величин $\chi _{{ax}}^{{{\text{Ln}}}}$ позволило с помощью уравнения (4) интерпретировать особенности спектрального поведения гетероядерных комплексов, содержащих два различных парамагнитных ядра. Это было сделано на примере пары изомерных комплексов [(15C5)4Pc]M*[(15C5)4Pc]M(Pc), содержащих ионы Tb3+ и Tm3+.
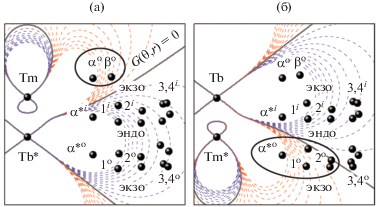
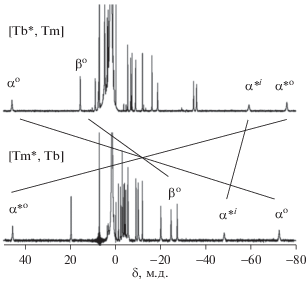
Поскольку величина $\chi _{{ax}}^{{{\text{Tb}}}}$ в 3.5 раза больше $\chi _{{ax}}^{{{\text{Tm}}}},$ именно влияние иона тербия доминирует в проявлении совокупного $\Delta \delta _{k}^{{{\text{dip}}}}.$ Однако из-за того, что эти ионы характеризуются противоположными по знаку величинами $\chi _{{ax}}^{{{\text{Ln}}}},$ границы, в которых величина дипольной составляющей является знакопостоянной, деформируются – области с $\Delta \delta _{k}^{{{\text{dip}}}} < 0$ сокращаются, а области с $\Delta \delta _{k}^{{{\text{dip}}}} > 0$ увеличиваются (рис. 8). В результате спектральный диапазон, в котором наблюдаются сигналы резонанса комплексов, содержащих ионы Tb3+ и Tm3+ расширяется до 130 м.д. (рис. 9) по сравнению с комплексами, содержащими ионы Tb3+ и Y3+ (95 м.д., табл. 3).
Рис. 8.
Контурные карты функции $G(\theta ,r)$ для изомерных гетероядерных комплексов [(15C5)4Pc]M*[(15C5)4Pc]M(Pc) – [Tb*, Tm] и [Tm*, Tb]. Координаты протонов получены усреднением данных РСА для комплекса [Tm*, Y]. Овалами отмечены протоны, попадающие в область положительных значений ЛИС, по сравнению с большинством протонов молекул [49]. Обозначения протонов даны на рис. 1.
Структурно-спектральные характеристики гетеролептических порфиринато-фталоцианинатов лантанидов
Описанный подход применим к другим классам гетеролептических трехпалубных комплексов, например к гетеролептическим порфиринато-фталоцианинатам, для которых пренебрежение разницей в анизотропии $\chi _{{ax}}^{{{\text{Ln}}}}$ для ионов металлов в окружении Por/Pc или Pc/Pc даже в случае гомоядерных комплексов не корректно [58]. Для спектрального анализа таких соединений были разработаны селективные методы получения гетеролептических трехпалубных комплексов, содержащих тетра(15-краун-5)фталоцианин и мезо-арилзамещенные порфирины с заданным расположением металлоцентров и тетрапиррольных лигандов [59–63]. Этот класс соединений был подробно изучен на примере симметричных гетеролептических трехпалубных комплексов, содержащих центральный тетра(15-краун-5)фталоцианиновый и терминальные тетра-мезо(4-метоксифенил)порфириновыe лиганды – Ln2[(p-An)4Por]2[(15C5)4Pc], где Ln = La(III), Ce(III), Pr(III), Nd(III), Sm(III) и Eu(III). Спектр 1H ЯМР диамагнитного комплекса лантана представлен на рис. 10.
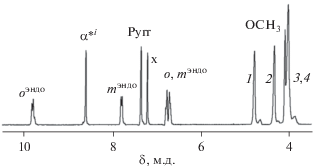
Как описано выше, для надежного отнесения сигналов в спектрах гетеролептических комплексов парамагнитных лантанидов необходима структурная информация о молекуле. Однако для данного класса соединений не удалось вырастить монокристаллы, в связи с этим нами был разработан подход, позволяющий построить расчетную модель их строения. Расчет геометрии сложных макроциклических структур квантово-химическими методами, например DFT, с достаточной точностью требует большой вычислительной мощности и длительного времени, поэтому нами был разработан упрощенный метод, позволяющий получить геометрические параметры протонов, определяемые их расположением относительно парамагнитного центра. Для получения пространственной модели строения молекулы оказались пригодны методы молекулярной механики и молекулярной динамики, такие как MM+ и AMBER98. В качестве модели был выбран комплекс церия с центральным октаметоксифталоцианиновым и терминальными тетра-мезо(4-метоксифенил)порфириновыми лигандами. Для этого соединения был выполнен расчет геометрии и полученные результаты сопоставлены с данными рентгеноструктурного анализа для родственных комплексов [64]. На основе найденной расчетной структуры был проведен анализ ЛИС сигналов резонанса протонов.
В рассматриваемых комплексах ЛИС сигналов ароматических протонов фталоцианинового лиганда и пиррольных протонов порфирина, отдаленных от ионов лантанидов на пять и четыре связи соответственно, могут иметь существенный контактный вклад, в то время как остальные протоны удалены от парамагнитного центра на большее число связей, что позволяет построить первичный анализ спектров серии соединений на допущении о преимущественно дипольной природе ЛИС. Для этого с использованием уравнения (4) были вычислены значения геометрических параметров Gk для каждого из типов протонов молекулы (табл. 5). В последнем столбце приведены значения суммарных геометрических функций Gk, нормированные на геометрический параметр ароматического протона фталоцианиновой палубы.
Таблица 5.
Геометрические параметры ароматических протонов модельного комплекса Ln2[(p-An)4Por]2[(15C5)4Pc] [48]
| Hk | Ln1 | Ln2 | $\begin{gathered} \sum {{{G}_{k}} = } \\ = {{G}_{{k1}}} + {{G}_{{k2}}} \\ \end{gathered} $ | $\frac{{\sum {{{G}_{k}}} }}{{\sum {G(\alpha {\kern 1pt} {{*}^{i}})} }}$ | ||||
|---|---|---|---|---|---|---|---|---|
| θk1, град | rk1, Å | Gk1 × 103 | θk1, град | rk1, Å | Gk1 × 103 | |||
| oэндо | 93.61 | 5.19 | –7.06 | 57.51 | 6.14 | –0.58 | –7.64 | 1.05 |
| oэкзо | 49.22 | 5.86 | 1.39 | 30.77 | 8.68 | 1.86 | 3.25 | –0.45 |
| mэндо | 89.50 | 7.52 | –2.35 | 63.85 | 8.38 | –0.71 | –3.06 | 0.42 |
| mэкзо | 58.19 | 8.02 | –0.32 | 40.95 | 10.40 | 0.63 | 0.31 | –0.04 |
| Pyrr | 70.68 | 5.39 | –4.28 | 43.25 | 7.43 | 1.44 | –2.84 | 0.39 |
| α*i | 108.17 | 5.79 | –3.65 | 71.83 | 5.79 | –3.65 | –7.29 | 1.00 |
Сигнал ароматического протона наиболее удобен для анализа сигналов в спектрах и ЛИС, так как его отнесение выполнено с хорошей точностью для всех трехпалубных гетеролептических комплексов. Из этого рассмотрения становится ясно, что два протона молекулы попадают в область магнитного воздействия лантанида, противоположную по знаку и, как следствие, направлению ЛИС большей части протонов системы. Это внешние орто- и мета-протоны мезо-заместителя порфириновой палубы, причем величины ЛИС этих протонов отличаются на порядок. Абсолютное значение геометрического параметра мета-протона мало по сравнению с параметрами остальных протонов молекулы, и его теоретически ожидаемый ЛИС также мал. Следует отметить, что самым высоким значением ${{G}_{k}}$ характеризуется не протон фталоцианиновой палубы, эффективно взаимодействующий с обоими парамагнитными центрами, а внутренний орто-протон мезо-заместителя, ЛИС которого определяется преимущественно одним лантанидом.
Графическое представление взаимного расположения протонов и парамагнитных металлоцентров в молекуле трехпалубного комплекса приведено на рис. 11. Схема представляет собой сечение молекулы по оси Ln–Ln. В каждой точке пространства вычислено значение $G(\theta ,r)$ и определено геометрическое место точек с $G(\theta ,r) = 0,$ что является границей, при пересечении которой знак $G(\theta ,r)$ изменяется на противоположный.
Рис. 11.
Контурные карты функции $G(\theta ,r)$ для комплексов Ln2[(p-An)4Por]2[(15C5)4Pc], координаты атомов получены на основании расчета методом MM+ [48]. Овалами отмечены протоны, попадающие в область противоположного знака функции $G(\theta ,r)$ по сравнению с большинством протонов молекул. Обозначения протонов даны на рис. 1.
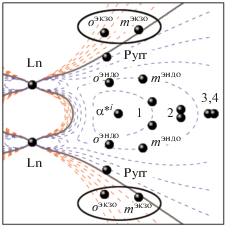
Нулевая огибающая этой системы нелинейна и по мере удаления от системы двух лантанидов стремится к углу наклона 54.7° по отношению к оси Ln–Ln, характерному для одноядерных комплексов [44]. Однако в пределах молекулы этот угол больше, что объясняет размещение протонов комплекса в областях разного знака ЛИС. Бóльшая часть протонов молекулы комплекса находится в зоне ЛИС одного знака, однако внешние орто- и мета-протоны мезо-анизильных заместителей порфириновой палубы попадают в область противоположного знака. Важно отметить, что пиррольные протоны порфириновых палуб находятся близко к нулевой линии, что позволяет предположить их малый дипольный ЛИС несмотря на близкое расположение к металлоцентрам.
Полученные численные значения геометрических параметров и их графическое представление позволили выполнить отнесение сигналов в спектрах комплексов парамагнитных лантанидов. Для этого было найдено такое соответствие сигналов в спектре диамагнитного комплекса лантана и комплексов парамагнитных лантанидов, при котором отношение ${{\Delta \delta ({{{\text{H}}}_{k}})} \mathord{\left/ {\vphantom {{\Delta \delta ({{{\text{H}}}_{k}})} {\Delta \delta ({\alpha }{{{\text{*}}}^{i}})}}} \right. \kern-0em} {\Delta \delta ({\alpha }{{{\text{*}}}^{i}})}}$ близко к вычисленному ${{G({{{\text{H}}}_{k}})} \mathord{\left/ {\vphantom {{G({{{\text{H}}}_{k}})} {G({\alpha }{{{\text{*}}}^{i}})}}} \right. \kern-0em} {G({\alpha }{{{\text{*}}}^{i}})}}.$ На рис. 12 приведены примеры отнесения сигналов для комплексов европия и неодима, сдвигающих сигналы в область слабых и сильных полей соответственно. В табл. 6 приведены численные значения химических сдвигов сигналов резонанса протонов молекулы и относительные ЛИС для полученной серии комплексов парамагнитных лантанидов. Анализ спектров серии комплексов лантанидов позволил выполнить точное отнесение сигналов протонов мезо-арильных заместителей, которое невозможно выполнить другими методами.
Рис. 12.
Отнесение сигналов в спектрах 1H ЯМР трехпалубных комплексов Ln2[(p-An)4Por]2[(15C5)4Pc] в CDCl3, где Ln = La, Ce, Eu [48].
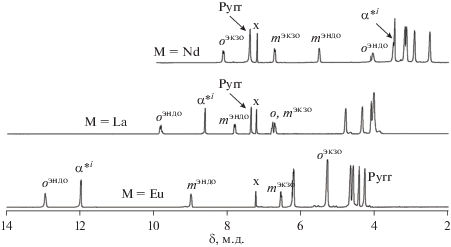
Таблица 6.
Положение сигналов резонанса (δ, м.д.) в спектрах 1H ЯМР трехпалубных комплексов Ln2[(p-An)4Por]2[(15C5)4Pc] [48]. Относительные значения $\Delta \delta _{k}^{{{\text{rel}}}}$ соответствуют отношениям ЛИС рассматриваемых протонов к ЛИС ароматических протонов фталоцианинового лиганда α*
| Параметр | La | Ce | Pr | Nd | Sm | Eu | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| δk | δk | $\Delta \delta _{k}^{{{\text{rel}}}}$ | δk | $\Delta \delta _{k}^{{{\text{rel}}}}$ | δk | $\Delta \delta _{k}^{{{\text{rel}}}}$ | δk | $\Delta \delta _{k}^{{{\text{rel}}}}$ | δk | $\Delta \delta _{k}^{{{\text{rel}}}}$ | |
| 1-CH2 | 4.83 | –0.35 | 0.46 | 1.30 | 0.45 | 2.57 | 0.44 | 4.22 | 0.46 | 6.23 | 0.42 |
| 2-CH2 | 4.38 | 1.32 | 0.27 | 2.27 | 0.27 | 2.99 | 0.27 | 4.01 | 0.28 | 5.32 | 0.28 |
| 3-CH2 | 4.06 | 2.12 | 0.17 | 2.73 | 0.17 | 3.20 | 0.17 | 3.85 | 0.16 | 4.68 | 0.18 |
| 4-CH2 | 4.06 | 2.28 | 0.16 | 2.83 | 0.16 | 3.25 | 0.16 | 3.85 | 0.16 | 4.61 | 0.16 |
| oэндо | 9.84 | –2.64 | 1.12 | 1.13 | 1.12 | 4.12 | 1.12 | 8.16 | 1.26 | 13.00 | 0.94 |
| oэкзо | 6.75 | 10.08 | –0.30 | 9.04 | –0.30 | 8.17 | –0.28 | 7.16 | –0.31 | 5.32 | –0.43 |
| mэндо | 7.85 | 2.75 | 0.46 | 4.33 | 0.45 | 5.57 | 0.45 | 6.94 | 0.68 | 9.01 | 0.35 |
| mэкзо | 6.80 | 6.67 | 0.01 | 6.74 | 0.01 | 6.77 | 0.01 | 6.73 | 0.05 | 6.57 | –0.07 |
| α*i | 8.65 | –2.53 | 1.00 | 0.89 | 1.00 | 3.55 | 1.00 | 7.32 | 1.00 | 12.01 | 1.00 |
| Pyrr | 7.40 | 3.15 | 0.38 | 5.81 | 0.20 | 7.45 | –0.01 | 6.60 | 0.60 | 4.30 | –0.92 |
| OMe | 4.14 | 2.77 | 0.12 | 3.19 | 0.12 | 3.52 | 0.12 | 3.94 | 0.15 | 4.45 | 0.09 |
Величины относительного ЛИС протонов молекулы, нормированные на величину ЛИС ароматического протона фталоцианинового лиганда, совпадают качественно и близки количественно к рассчитанным на основе параметризованной модели комплекса (табл. 6). Однако при детальном рассмотрении полученного массива данных обнаруживаются некоторые отклонения наблюдаемых соотношений от расчетных.
Так, ЛИС внешнего мета-протона мезо-заместителя отличается от рассчитанного как по знаку (для комплексов Ce, Pr, Nd), так и по абсолютной величине. Это отклонение объясняется сравнительно малой величиной ЛИС, сравнимой с уширением сигнала при уменьшении времени поперечной релаксации ${{T}_{2}},$ что снижает точность ее определения. Отклонения относительных ЛИС сигналов протонов комплекса самария определяются уменьшением точности определения ЛИС в связи с малыми величинами ${{\left\langle {{{S}_{Z}}} \right\rangle }^{{{\text{Sm}}}}}$ и ${{D}^{{{\text{Sm}}}}}$ и, как следствие, малыми ЛИС. Выполненное отнесение сигналов в спектрах трехпалубных комплексов согласуется с данными о скорости релаксации протонов (табл. 7).
Таблица 7.
Экспериментальное время продольной релаксации протонов трехпалубных комплексов Ln2[(p-An)4Por]2[(15C5)4Pc] [48]
| Параметр | La | Nd | Eu | ||
|---|---|---|---|---|---|
| $1/T_{1}^{{{\text{exp}}}}$, с–1 | $1/T_{1}^{{{\text{exp}}}}$, с–1 | $1/T_{1}^{{{\text{para}}}}$, с–1 | $1/T_{1}^{{{\text{exp}}}}$, с–1 | $1/T_{1}^{{{\text{para}}}}$, с–1 | |
| 1-CH2 | 5.88 | 9.39 | 3.51 | 7.87 | 1.99 |
| 2-CH2 | 5.88 | 7.85 | 1.97 | 6.45 | 0.57 |
| 3-CH2 | 4.00 | 5.47 | 1.47 | 4.76 | 0.76 |
| 4-CH2 | 4.00 | 5.38 | 1.38 | 4.76 | 0.76 |
| oэндо | 2.22 | 48.78 | 46.56 | 11.11 | 8.89 |
| oэкзо | 1.67 | 13.95 | 12.28 | 6.45 | 4.78 |
| mэндо | 1.67 | 5.21 | 3.55 | 2.86 | 1.19 |
| mэкзо | 1.67 | 4.08 | 2.42 | 2.86 | 1.19 |
| α*i | 3.13 | 16.61 | 13.49 | 11.11 | 7.99 |
| Pyrr | 1.25 | 23.09 | 21.84 | 7.87 | 6.62 |
| OMe | 1.96 | 3.09 | 1.13 | 2.27 | 0.31 |
Имея данные о временах релаксации протонов диамагнитного репера – комплекса лантана, можно выделить диамагнитный и парамагнитный вклады экспериментально наблюдаемой скорости релаксации. Учитывая, что ${1 \mathord{\left/ {\vphantom {1 {T_{1}^{{{\text{para}}}}}}} \right. \kern-0em} {T_{1}^{{{\text{para}}}}}} \sim {{r}^{{ - 6}}}$ (r – расстояние от протона до парамагнитного центра), парамагнитный вклад в скорость релаксации быстро уменьшается с удалением рассматриваемого протона от иона лантанида, и, таким образом, данные о временах релаксации предоставляют дополнительные сведения для верификации расчетной модели структуры комплекса. Однако следует отметить, что даже малая ошибка в определении расстояний в расчетной модели комплекса существенно влияет на результаты анализа за счет шестой степени отношения расстояний. В целом данные парамагнитного ускорения релаксации согласуются с определенными расстояниями и подтверждают выполненное отнесение сигналов.
Сравнение параметров модели и полученных релаксационных данных для трехпалубных гетеролептических комплексов неодима и европия показало, что ошибка расчетных величин для протонов мезо-заместителей порфириновых палуб находится в интервале 1–5%, что подтверждает корректность описания геометрии всего ряда полученных комплексов с помощью полученной оптимизированной структурной модели. Необходимо отметить, что данное уточнение невозможно для краун-эфирных заместителей молекулы из-за большого набора их конформаций без возможности определить наиболее выгодную. Отношения, включающие параметры пиррольного и фталоцианинового протонов молекулы, демонстрируют существенно бóльшую ошибку. Это может определяться их близостью к парамагнитному центру и существенным вкладом контактного механизма ускорения релаксации аналогично ЛИС.
Магнетохимические свойства гетеролептических фталоцианинатов РЗЭ
Для серии комплексов [(15C5)4Pc]M*[(15C5)4Pc]M(Pc) – гомоядерного [Tb*, Tb] и изомерных гетероядерных [Tb*, Y] и [Y*, Tb] – были исследованы магнетохимические свойства с использованием ac-магнитометрии [54].
Для гомоядерного комплекса при нулевом постоянном внешнем магнитном поле в интервале температур 1.9–28 K наблюдались зависимости действительной (χ') и мнимой (χ'') компонент магнитной восприимчивости от частоты осциллирующего магнитного поля, что свидетельствовало о наличии медленной магнитной релаксации. В случае гетероядерных комплексов медленную магнитную релаксацию можно было наблюдать только при наложении постоянного внешнего магнитного поля напряженностью 1.5 кЭ.
Такое различие в поведении гомо- и гетероядерных комплексов обусловлено реализацией механизма квантового туннелирования в случае гетероядерных комплексов в отсутствие внешнего магнитного поля. Реализация этого механизма обусловлена искажением координационных полиэдров ионов тербия от правильной квадратной антипризмы за счет разницы в длинах связей с атомами азота терминальных и внутреннего лигандов (рис. 4). В случае гомоядерного комплекса благодаря близости расположения ионов Tb3+ между ними реализуется ферромагнитное взаимодействие, эффективно подавляющее квантовое туннелирование [65, 66]. Наличие такого взаимодействия в гомоядерном комплексе было подтверждено измерением магнитной восприимчивости комплексов [M*, M].
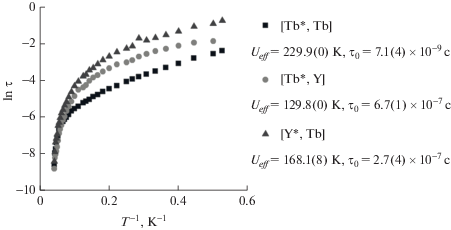
С использованием обобщенной модели Дебая были определены времена релаксации намагниченности τ при разных температурах. С помощью уравнения Аррениуса были найдены высоты энергетического барьера перемагничивания Ueff и времена релаксации τ0 (рис. 13). Полученные данные свидетельствуют о том, что в гетероядерных комплексах более высокий барьер Ueff наблюдается у комплекса c ионом тербия, находящимся между [(15C5)4Pc] и (Pc). Следует отметить, что для этого же комплекса характерно более высокое значение аксиальной анизотропии $\chi _{{ax}}^{{Tb}},$ найденное по данным спектроскопии 1H ЯМР и РСА (табл. 4). Подобное усиление SMM-свойств с ростом анизотропии было отмечено в работах М. Ямашиты [67, 68]. Таким образом, найденные корреляции между величиной барьера и координационным окружением ионов Tb3+ в трехпалубных комплексах могут быть использованы для направленного получения комплексов с улучшенными магнитными характеристиками.
Рис. 13.
Зависимость времени релаксации намагниченности от 1/Т, значения барьера перемагничивания и времени τ0, найденные по уравнению Аррениуса для гомо- и гетероядерных комплексов [(15C5)4Pc]M*[(15C5)4Pc]M(Pc) – [Tb*, Tb], [Tb*, Y] и [Y*, Tb] [54].
В то время как особенности магнитного поведения, в том числе явление молекулярного магнетизма, могут быть полноценно изучены лишь с использованием dc- и ac-магнитометрии, спектроскопия ЯМР как существенно более доступный метод может дать ценную первичную информацию о магнитных свойствах комплексов и способствовать выявлению новых корреляций для получения молекулярных магнетиков с улучшенными характеристиками.
Список литературы
Harnden A.C., Parker D., Rogers N.J. // Coord. Chem. Rev. 2019. V. 383. P. 30. https://doi.org/10.1016/j.ccr.2018.12.012
Staszak K., Wieszczycka K., Marturano V. et al. // Coord. Chem. Rev. 2019. V. 397. P. 76. https://doi.org/10.1016/j.ccr.2019.06.017
Kaczmarek M. // J. Lumin. 2020. V. 222. P. 117174. https://doi.org/10.1016/j.jlumin.2020.117174
Zairov R.R., Yagodin A.V., Khrizanforov M. et al. // J. Nanoparticle Res. 2019. V. 21. № 1. P. 12. https://doi.org/10.1007/s11051-018-4455-4
Ning Y., Zhu M., Zhang J.-L. // Coord. Chem. Rev. 2019. V. 399. P. 213028. https://doi.org/10.1016/j.ccr.2019.213028
Wang H., Wang B.W., Bian Y. et al. // Coord. Chem. Rev. 2016. V. 306. № P1. P. 195. https://doi.org/10.1016/j.ccr.2015.07.004
Cador O., Le Guennic B., Pointillart F. // Inorg. Chem. Front. 2019. V. 6. № 12. P. 3398. https://doi.org/10.1039/c9qi00875f
Lan Y., Klyatskaya S., Ruben M. // Bis(phthalocyaninato) Lanthanide(III) Complexes - from Molecular Magnetism to Spintronic Devices / Eds. Layfield R.A., Murugesu M. Lanthanides Actinides Mol. Magn., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015: pp. 223–292. https://doi.org/10.1002/9783527673476.ch8
Harriman K.L.M., Errulat D., Murugesu M. // Trends Chem. 2019. V. 1. № 4. P. 425. https://doi.org/10.1016/j.trechm.2019.04.005
Lyubov D.M., Tolpygin A.O., Trifonov A.A. // Coord. Chem. Rev. 2019. V. 392. P. 83. https://doi.org/10.1016/j.ccr.2019.04.013
MacDonald M.R., Bates J.E., Ziller J.W. et al. // J. Am. Chem. Soc. 2013. V. 135. № 26. P. 9857. https://doi.org/10.1021/ja403753j
Fedushkin I.L., Maslova O.V., Morozov A.G. et al. // Angew. Chem. Int. Ed. 2012. V. 51. № 42. P. 10584. https://doi.org/10.1002/anie.201204452
Shokurov A.V., Kutsybala D.S., Martynov A.G. et al. // Langmuir. 2020. V. 36. № 6. P. 1423. https://doi.org/10.1021/acs.langmuir.9b03403
Kaczmarek A.M., Van Der Voort P. // Materials. 2020. V. 13. № 3. P. 566. https://doi.org/10.3390/ma13030566
Yapryntsev A.D., Baranchikov A.E., Ivanov V.K. // Russ. Chem. Rev. 2020. V. 89. № 6. P. 629. https://doi.org/10.1070/RCR4920
Yorov K.E., Kottsov S.Y., Baranchikov A.E. et al. // J. Sol-Gel Sci. Technol. 2019. V. 92. № 2. P. 304. https://doi.org/10.1007/s10971-019-04958-9
Yapryntsev A., Abdusatorov B., Yakushev I. et al. // Dalton Trans. 2019. V. 48. № 18. P. 6111. https://doi.org/10.1039/C9DT00390H
Yapryntsev A.D., Baranchikov A.E., Skogareva L.S. et al. // CrystEngComm. 2015. V. 17. № 13. P. 2667. https://doi.org/10.1039/C4CE02303J
Xiang Y., Yu X.-F., He D.-F. et al. // Adv. Funct. Mater. 2011. V. 21. № 22. P. 4388. https://doi.org/10.1002/adfm.201101808
Yapryntsev A.D., Bykov A.Y., Baranchikov A.E. et al. // Inorg. Chem. 2017. V. 56. № 6. P. 3421. https://doi.org/10.1021/acs.inorgchem.6b02948
Gándara F., Puebla E.G., Iglesias M. et al. // Chem. Mater. 2009. V. 21. № 4. P. 655. https://doi.org/10.1021/cm8029517
Demel J., Kubát P., Millange F. et al. // Inorg. Chem. 2013. V. 52. № 5. P. 2779. https://doi.org/10.1021/ic400182u
Wang X., Chen W., Song Y.-F. // Eur. J. Inorg. Chem. 2014. V. 2014. № 17. P. 2779. https://doi.org/10.1002/ejic.201400122
Sokolov M.R., Enakieva Y.Y., Yapryntsev A.D. et al. // Adv. Funct. Mater. 2020. V. 30. № 27. P. 2000681. https://doi.org/10.1002/adfm.202000681
Shekunova T.O., Lapkina L.A., Shcherbakov A.B. et al. // J. Photochem. Photobiol., A. Chem. 2019. V. 382. P. 111925. https://doi.org/10.1016/j.jphotochem.2019.111925
Lapkina L.A., Gorbunova Y.G., Gil D.O. et al. // J. Porphyrins Phthalocyanines. 2013. V. 17. № 06n07. P. 564. https://doi.org/10.1142/S1088424613500648
Hiller M., Krieg S., Ishikawa N. et al. // Inorg. Chem. 2017. V. 56. № 24. P. 15285. https://doi.org/10.1021/acs.inorgchem.7b02704
Santria A., Fuyuhiro A., Fukuda T. et al. // Dalton Trans. 2019. V. 48. № 22. P. 7685. https://doi.org/10.1039/C9DT00915A
Joss D., Häussinger D. // Prog. Nucl. Magn. Reson. Spectrosc. 2019. V. 114–115. P. 284. https://doi.org/10.1016/j.pnmrs.2019.08.002
Babailov S.P. // Prog. Nucl. Magn. Reson. Spectrosc. 2008. V. 52. № 1. P. 1. https://doi.org/10.1016/j.pnmrs.2007.04.002
Geraldes C.F.G.C., Zhang S., Sherry A.D. // Inorg. Chim. Acta 2004. V. 357. № 2. P. 381. https://doi.org/10.1016/j.ica.2003.03.001
Babailov S.P., Coutsolelos A.G., Dikiy A. et al. // Eur. J. Inorg. Chem. 2001. V. 2001. № 1. P. 303. https://doi.org/10.1002/1099-0682(20011)2001:1<303::AID-EJIC303>3.0.CO;2-Y
Babailov S.P. // Inorg. Chem. 2012. V. 51. № 3. P. 1427. https://doi.org/10.1021/ic201662q
Babailov S.P., Stabnikov P.A., Zapolotsky E.N. et al. // Inorg. Chem. 2013. V. 52. № 9. P. 5564. https://doi.org/10.1021/ic400525r
Birin K.P., Gorbunova Y.G., Tsivadze A.Y. // Dalton Trans. 2011. V. 40. № 43. P. 11474. https://doi.org/10.1039/c1dt11231g
Горбунова Ю.Г., Лапкина Л.А., Мартынов А.Г. и др. // Коорд. химия. 2004. V. 30. № 4. P. 263. https://doi.org/10.1023/B:RUCO.0000022799.63314.fc
Gorbunova Y.G., Martynov A.G., Tsivadze A.Y. // Crown-Substituted Phthalocyanines: From Synthesis Towards Materials, in: K.M. Kadish, K.M. Smith, R. Guilard (Eds.), Handbook of Porphyrin Science, World Scientific Publishing, 2012: pp. 271–388 https://doi.org/10.1142/9789814397605_0015
Martynov A.G., Gorbunova Y.G., Tsivadze A.Y. // Russ. J. Inorg. Chem. 2014. V. 59. № 14. P. 1635. https://doi.org/10.1134/S0036023614140046
Piguet C., Geraldes C.F.G.C. // Paramagnetic NMR Lanthanide Induced Shifts for Extracting Solution Structures, in: K.A. Gschneidner, J.-C.G. Bünzli, V.K. Pecharsky (Eds.), Handbook on the Physics and Chemistry of Rare Earths, Elsevier Science B.V., 2003: pp. 353–463 https://doi.org/10.1016/S0168-1273(02)33005-8
Golding R., Halton M. // Aust. J. Chem. 1972. V. 25. № 12. P. 2577. https://doi.org/10.1071/CH9722577
Pinkerton A.A.A., Rossier M., Spiliadis S. et al. // J. Magn. Reson. 1985. V. 64. № 3. P. 420. https://doi.org/10.1016/0022-2364(85)90104-0
Bleaney B. // J. Magn. Reson. 1972. V. 8. № 1. P. 91. https://doi.org/10.1016/0022-2364(72)90027-3
Peters J.A., Huskens J., Raber D.J. // Prog. Nucl. Magn. Reson. Spectrosc. 1996. V. 28. № 3–4. P. 283. https://doi.org/10.1016/0079-6565(95)01026-2
Ishikawa N., Iino T., Kaizu Y. // J. Phys. Chem. A. 2003. V. 107. № 39. P. 7879. https://doi.org/10.1021/jp034971n
Arnold D.P., Jiang J. // J. Phys. Chem. A. 2001. V. 105. № 32. P. 7525. https://doi.org/10.1021/jp0105847
Martynov A.G., Gorbunova Y.G. // Polyhedron. 2010. V. 29. № 1. P. 391. https://doi.org/10.1016/j.poly.2009.06.009
Martynov A.G., Gorbunova Y.G., Tsivadze A.Y. // Dalton Trans. 2011. V. 40. № 27. P. 7165. https://doi.org/10.1039/c1dt10455a
Birin K.P., Gorbunova Y.G., Tsivadze A.Y. // Magn. Reson. Chem. 2010. V. 48. № 7. P. 505. https://doi.org/10.1002/mrc.2612
Polovkova M.A., Martynov A.G., Birin K.P. et al. // Inorg. Chem. 2016. V. 55. № 18. P. 9258. https://doi.org/10.1021/acs.inorgchem.6b01292
Martynov A.G., Zubareva O.V., Gorbunova Y.G. et al. // Inorg. Chim. Acta. 2009. V. 362. № 1. P. 11. https://doi.org/10.1016/j.ica.2008.01.008
Martynov A.G., Zubareva O.V., Gorbunova Y.G. et al. // Eur. J. Inorg. Chem. 2007. № 30. P. 4800. https://doi.org/10.1002/ejic.200700489
Мартынов А.Г., Сафонова Е.А., Горбунова Ю.Г. и др. // Журн. неорган. химии. 2010. Т. 55. № 3. С. 389.
Цивадзе А.Ю., Мартынов А.Г., Половкова М.А. и др. // Изв. АН. Сер. химическая. 2011. № 11. P. 2216.
Holmberg R.J., Polovkova M.A., Martynov A.G. et al. // Dalton Trans. 2016. V. 45. № 22. P. 9320. https://doi.org/10.1039/C6DT00777E
Martynov A.G., Polovkova M.A., Berezhnoy G.S. et al. // Inorg. Chem. 2020. V. 59. № 13. P. 9424. https://doi.org/10.1021/acs.inorgchem.0c01346
Horii Y., Kishiue S., Damjanović M. et al. // Chem. Eur. J. 2018. V. 24. № 17. P. 4320. https://doi.org/10.1002/chem.201705378
Martynov A.G., Safonova E.A., Tsivadze A.Y. et al. // Coord. Chem. Rev. 2019. V. 387. P. 325. https://doi.org/10.1016/j.crr.2019.02.004
Sakaue S., Fuyuhiro A., Fukuda T. et al. // Chem. Commun. 2012. V. 48. № 43. P. 5337. https://doi.org/10.1039/c2cc31125a
Birin K.P., Gorbunova Y.G., Tsivadze A.Y. // Dalton Trans. 2011. V. 40. № 43. P. 11539. https://doi.org/10.1039/c1dt11141h
Birin K.P., Gorbunova Y.G., Tsivadze A.Y. // Dalton Trans. 2012. P. 23. https://doi.org/10.1039/c2dt30841j
Birin K.P., Poddubnaya A.I., Gorbunova Y.G. et al. // Macroheterocycles. 2017. V. 10. № 4–5. P. 514. https://doi.org/10.6060/mhc171258b
Birin K.P., Gorbunova Y.G., Tsivadze A.Y. et al. // J. Porphyrins Phthalocyanines. 2009. V. 13. № 2. P. 283. https://doi.org/10.1142/S1088424609000358
Birin K.P., Kamarova K.A., Gorbunova Y.G. et al. // Prot. Met. 2013. V. 49. № 2. P. 173. https://doi.org/10.1134/S2070205113020032
Sun X., Li R., Wang D. et al. // Eur. J. Inorg. Chem. 2004. № 19. P. 3806. https://doi.org/10.1002/ejic.200400289
Ishikawa N., Iino T., Kaizu Y. // J. Am. Chem. Soc. 2002. V. 124. № 38. P. 11440. https://doi.org/10.1021/ja027119n
Katoh K., Kajiwara T., Nakano M. et al. // Chem. Eur. J. 2011. V. 17. № 1. P. 117. https://doi.org/10.1002/chem.201002026
Katoh K., Breedlove B.K., Yamashita M. // Chem. Sci. 2016. V. 7. P. 4329. https://doi.org/10.1039/C5SC04669F
Morita T., Damjanović M., Katoh K. et al. // J. Am. Chem. Soc. 2018. V. 140. № 8. P. 2995. https://doi.org/10.1021/jacs.7b12667
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии