

Журнал неорганической химии, 2021, T. 66, № 2, стр. 194-203
Иммобилизация гетероцикл-аннелированных порфиринов на металлорганических координационных полимерах UiO-66, 67
К. П. Бирин a, *, И. А. Абдулаева a, Д. А. Поливановская a, А. А. Синельщикова a, Л. И. Демина a, b, А. Е. Баранчиков b, Ю. Г. Горбунова a, b, А. Ю. Цивадзе a, b
a Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Ленинский пр-т, 31, корп. 4, Россия
b Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
* E-mail: kirill.birin@gmail.com
Поступила в редакцию 21.07.2020
После доработки 28.07.2020
Принята к публикации 30.07.2020
Аннотация
Разработан подход к иммобилизации гетероцикл-аннелированных порфиринов, содержащих одну или две якорные группы на поверхности металлорганических каркасных полимеров типа UiO-66 и UiO-67. С этой целью впервые получены карбоксизамещенные производные Ni-4 и Ni-5. Показано, что наличие одной якорной карбоксильной группы имидазопорфирина недостаточно для прочного удерживания молекулы в составе материала. В то же время пиразинопорфирин, содержащий две якорные группы, прочно удерживается в составе МОКП в соотношении 1 : 16 по отношению к узловым шестиядерным кластерам Zr(IV). Установлено, что постсинтетическая модификация предварительно полученных МОКП является наиболее эффективным методом иммобилизации. Полученные материалы исследованы с помощью рентгенофазового анализа, растровой электронной микроскопии, электронной спектроскопии диффузного отражения и ИК-спектроскопии.
ВВЕДЕНИЕ
Высокопористые Zr(IV)-содержащие металлорганические координационные полимеры (МОКП) привлекают все большее внимание исследователей в связи с их выдающейся химической, термической и механической стабильностью, что позволяет использовать их в различных областях современной науки [1–4]. Принципиальной особенностью ZrIV-МОКП, таких как UiO-66 и UiO-67, построенных из шестиядерных узловых Zr(IV)-содержащих кластеров и линкерных фрагментов ароматических дикарбоновых кислот (рис. 1), является возможность управляемого варьирования количества дефектов в структуре при изменении условий синтеза [5–8]. В свою очередь, это позволяет направленно изменять размер пор в структуре материала, что нашло применение в гетерогенном катализе [9–11].
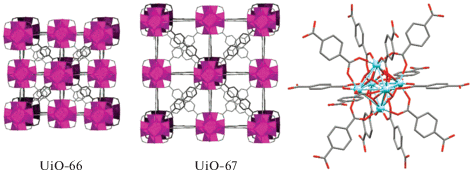
Принципиальной особенностью Zr(IV)-содержащих МОКП UiO-66,67 является способность содержать большое количество дефектов без существенной потери стабильности материала. Дефекты в таких материалах могут представлять собой как отсутствие линкера между двумя узловыми кластерами, так и отсутствие целого кластера и двенадцати связанных с ним молекул-линкеров. В первом случае возникают две координационные вакансии на атомах циркония, принадлежащих соседним узловым кластерам, которые заняты лабильными лигандами. Появление дефектов второго типа приводит к образованию крупных полостей в объеме материала, каждой из которых отвечают двенадцать координационных вакансий на соседних узловых кластерах. Оба типа указанных дефектов могут быть использованы для иммобилизации функциональных молекул в структуру материала.
Возможность модификации получаемых материалов путем иммобилизации в их структуру функциональных молекул расширяет области применения получаемых гибридных материалов. Для получения таких материалов требуется сочетание двух факторов – наличия дефектных кластеров в структуре ZrIV-МОКП и присутствия необходимого количества якорных групп в функциональной молекуле. Так, использование в качестве модификаторов тетракарбоксизамещенных порфиринов позволило получить гибридные материалы для сенсорных [12] и каталитических приложений [13, 14].
Важно отметить, что большинство описанных примеров получения гибридных материалов на основе Zr(IV)-содержащих МОКП и замещенных порфиринов выполнено с использованием симметричного тетракис-(4-карбоксифенил)порфирина, содержащего четыре якорные группы. Такое строение порфирина приводит к его включению в полости материала, равномерному распределению порфирина в объеме и, соответственно, к низкой его концентрации на поверхности частиц. Указанное строение гибридного материала не эффективно для использования в гетерогенных каталитических приложениях из-за недоступности для взаимодействия молекул порфиринов, находящихся в объеме материала. Тем не менее такие материалы были использованы для фотокаталитических приложений [15], а также для внутриклеточного контроля pH и концентрации кислорода [16]. Нам удалось обнаружить единственный пример иммобилизации монофункционализированного порфирина на поверхности частиц UiO-66 с использованием тиольных якорных групп для применения в качестве фотосенсибилизаторов в фотодинамической терапии [17].
Снижение симметрии полиароматического макроцикла порфирина позволяет настраивать электронное и орбитальное строение образующихся производных, что, в свою очередь, определяет их оптические, фотофизические и электрохимические характеристики. Один из таких методов – введение гетероциклических фрагментов на периферийную часть молекулы порфирина [18]. В целом эта стратегия представляет несомненный интерес для тонкой настройки свойств порфиринов и их металлокомплексов с целью использования в катализе.
Ранее нами были разработаны подходы, позволяющие направленно осуществлять модификацию периферийной части макроцикла порфирина путем введения аннелированных пяти- или шестичленных гетероциклических фрагментов [19–24]. Важной особенностью этих подходов является возможность введения одного или двух функциональных фрагментов с сохранением аксиальной симметрии молекулы порфирина. Введение якорных групп с помощью этого метода позволяет обеспечить заданную ориентацию молекулы относительно поверхности неорганической подложки и препятствует кофациальному расположению макроцикла на ней. На данный момент информация о получении гибридных материалов на основе UiO-66,67 и функционализированных гетероцикл-аннелированных порфиринов отсутствует.
Цель настоящей работы – поиск подходов к получению гибридных материалов на основе металлокаркасных координационных полимеров UiO-66,67 и гетероцикл-аннелированных порфиринов, содержащих одну или две якорные карбоксильные группы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все использованные в работе реагенты были получены из коммерческих источников, кроме указанных отдельно. Подготовку растворителей осуществляли по стандартным методикам [25]. Исходный 2-нитро-тетракис(4-бутоксифенил)порфиринат никеля(II) Ni-1 был получен по ранее описанным методикам [20, 21, 26]. Хроматографическую очистку полученных соединений проводили на стеклянных цилиндрических колонках с использованием силикагеля Macherey-Nagel Silica 60, фракция 0.063–0.2 мм. Для тонкослойной хроматографии (ТСХ) использовали алюминиевые пластины Merck TLC Silica 60 F254, элюенты CH2Cl2 и CH2Cl2 : MeOH = 95 : 5.
Масс-спектры MALDI-TOF регистрировали на приборе Bruker Daltonics Ultraflex в режиме положительных ионов без использования матрицы. Электронные спектры поглощения (ЭСП) растворов записывали на приборе Thermo Scientific Evolution 220 в квадратных кварцевых кюветах с длиной оптического пути 1 см. Спектры диффузного отражения (СДО) регистрировали с использованием интегрирующей сферы, спектры ЯМР – с помощью спектрометра Bruker Avance III с рабочей частотой 600.13 МГц в CDCl3 или в смеси CDCl3 : MeOD (80 : 20) при 303 K с использованием остаточного сигнала CHCl3 в качестве внутреннего стандарта.
Рентгенофазовый анализ (РФА) проводили при комнатной температуре в диапазоне углов 2θ 3°–80° на лабораторном порошковом дифрактометре Empyrean (PANalytical) (CuKα-излучение, β-Ni-фильтр, геометрия Брэгга–Брентано). Предварительно перетертые порошки наносили на кремниевые держатели из раствора или в сухом виде. Анализ данных РФА выполняли с использованием программного пакета XPowderX 2019. Средний размер кристаллитов оценивали по величинам полуширины наиболее интенсивных пиков дифрактограммы в интервале углов 2θ = 10°–40°.
Анализ методами растровой электронной микроскопии (РЭМ) и рентгеноспектрального микроанализа (РСМА) проводили с использованием микроскопа Carl Zeiss NVision 40, снабженного энергодисперсионным анализатором Oxford Instruments X-Max. Микрофотографии были получены при ускоряющем напряжении 1 кВ, РСМА проводили при ускоряющем напряжении 20 кВ.
ИК-спектры регистрировали на ИК-фурье-спектрометре Nexus фирмы Nicolet с использованием приставки Pike с алмазным кристаллом методом НПВО в диапазоне 4000–550 см–1.
Синтез Ni-2. Ni-1 (1.00 г, 1 ммоль) растворяли в толуоле (174 мл) и добавляли раствор 4-амино-4H-1,2,4-триазола (2.52 г, 30 ммоль) в этаноле (11.7 мл). Реакционную массу нагревали до 90°C, добавляли твердый KOH (5.60 г, 100 ммоль) и далее полученную смесь нагревали до 110°C. Нагревание реакционной массы продолжали до окончания реакции, контролируя ее протекание методом ТСХ с элюентом CH2Cl2 : гексан = 2 : 1 (~10 мин), затем смесь охлаждали, промывали водой (100 мл), концентрировали и выделяли полученный продукт методом флеш-хроматографии на силикагеле с элюентом CH2Cl2 : гексан (1 : 1). Фракции, содержащие Ni-2, упаривали и получили 960 мг (94%) целевого соединения.
1H ЯМР (CDCl3; δ, м.д.; J, Гц): 8.65 (д, 1H, 3J = = 4.9, Hβ), 8.63 (д, 1H, 3J = 5.0, Hβ), 8.52 (д, 1H, 3J = 4.9, Hβ), 8.50 (с, 2H, Hβ), 8.45 (д, 1H, 3J = 5.0, Hβ), 7.93 (д, 2H, 3J = 8.3, Ho), 7.88 (д, 2H, 3J = 8.3, Ho), 7.87 (д, 2H, 3J = 8.3, Ho), 7.84 (д, 2H, 3J = 8.3, Ho), 7.28 (д, 2H, 3J = 8.3, Hm), 7.20 (д, 2H, 3J = 8.4, Hm), 7.19 (д, 2H, 3J = 8.4, Hm), 7.16 (д, 2H, 3J = 8.4, Hm), 6.57 (с, 2H, NH2), 4.19 (уш.т, 6H, 3J = 6.6, CH2O), 4.15 (т, 2H, 3J = 6.4, CH2O), 1.85–1.97 (м, 8H, CH2), 1.57–1.68 (м, 8H, CH2), 1.04–1.11 (м, 12H, CH3).
13C ЯМР (CDCl3; δ, м.д.): 160.12, 159.23, 159.19, 158.87, 151.54, 147.12, 143.15, 142.85, 142.70, 137.37, 135.04, 134.79, 134.30, 133.60, 133.56, 133.06, 132.72, 132.32, 132.28, 132.22, 131.45, 131.19, 130.99, 129.81, 121.45, 119.67, 117.03, 115.02, 114.69, 113.42, 113.23, 113.20, 68.16, 68.01, 67.76, 31.59, 31.51, 31.40, 19.43, 19.41, 19.37, 14.01, 13.96, 13.94.
MALDI-ТОF MS: m/z вычислено для C60H60N6NiO6 [M]+ 1018.39, найдено 1018.47.
ЭСП (CHCl3; λmax, нм (lg ε)): 448 (4.05), 560 (3.13), 600 (2.98).
Синтез Ni-3. Ni-2 (51 мг, 0.05 ммоль) растворяли в смеси CH2Cl2 (16 мл) и MeOH (0.6 мл) и добавляли в атмосфере аргона 10% Pd/C (25 мг). Далее при интенсивном перемешивании добавляли NaBH4 (19 мг, 0.75 ммоль) и выдерживали полученную смесь при комнатной температуре и перемешивании в течение 15 мин. Окончание реакции контролировали методом ТСХ (элюент CH2Cl2 : гексан = 2 : 1). Далее реакционную массу фильтровали в инертной атмосфере через сорбент Celite 545 и упаривали. Полученный Ni-3 без дополнительной очистки и характеризации сразу использовали в следующей стадии.
Синтез Ni-4 и Ni-5. Полученный на предыдущей стадии Ni-3 (0.05 ммоль) растворяли в 1,2-дихлорбензоле (8 мл) в атмосфере аргона и добавляли 4-карбоксибензальдегид (7.5 мг, 0.05 ммоль). Далее реакционную смесь нагревали при 120°C при перемешивании в инертной атмосфере в течение 18 ч, затем остужали и упаривали в вакууме. Твердый остаток наносили на колонку, заполненную силикагелем в смеси CH2Cl2 : гексан = 1 : 1 и элюировали CH2Cl2 и смесями CHCl3/MeOH (0 → 6% MeOH). Фракции целевых соединений упаривали, получили 8 мг Ni-4 (14%) и 25 мг Ni-5 (80%).
Ni-4. 1H ЯМР (20% MeOD в CDCl3; δ, м.д.; J, Гц): 8.71–8.66 (уш.с, 4H, Hβ), 8.64 (с, 2H, Hβ), 8.04 (д, 2Н, 3J = 8.0, CHm-Ar), 7.86 (д, 4H, 3J = 8.1, ms-Cho-Ar), 7.79 (д, 4H, 3J = 8.2, ms-Cho-Ar), 7.75 (д, 2Н, 3J = 8.0, CHo-Ar), 7.27 (уш.с, 2H, ms-CHm-Ar), 7.18 (уш.с, 2H, ms-CHm-Ar), 7.09 (д, 4H, 3J = 8.2, ms-CHm-Ar), 4.18 (уш.т, 4H, 3J = 6.5, CH2O), 4.10 (т, 4H, 3J = 6.5, CH2O), 1.93–1.85 (уш.м, 4H, CH2), 1.82 (квинт, 4H, 3J = 6.5, CH2), 1.57 (секст, 4H, 3J = 7.6, CH2), 1.52 (секст, 4H, 3J = 7.5, CH2), 1.01 (т, 6H, 3J = 7.4, CH3), 0.97 (т, 6H, 3J = 7.4, CH3).
13C ЯМР (20% MeOD в CDCl3; δ, м.д.): 168.28, 158.93, 150.54, 142.23, 141.54, 134.59, 134.52, 133.66, 132.97, 132.67, 131.64, 130.86, 130.44, 125.54, 119.49, 114.21, 113.19, 112.91, 68.00, 31.42, 19.31, 19.29, 13.76.
MALDI-ТОF MS: m/z вычислено для C68H64N6NiO6 [M]+ 1118.4, найдено 1117.9.
ЭСП (CHCl3; λmax, нм (lgε)): 329 (4.32), 425 (5.13), 534 (4.15), 569 (3.90).
ИК-спектр (νmax, cм–1): 3435 (ср, ш, N–H), 3037 (сл, ш), 2956 (с, ш), 2924(с, ш), 2869(с, ш), 2655 (ш), 2526 (ш), 1724 (ср, С=О), 1690 (с, С=О), 1607 (с), 1571 (ср), 1503 (с), 1464 (ср), 1442 (ср), 1411 (ср), 1365 (ср), 1348 (ср), 1284 (с), 1239(с), 1173 (с), 1107 (ср), 1067 (ср), 1001 (с), 971 (ср), 863 (ср), 816 (ср), 797 (с), 771 (ср), 739 (ср), 664 (ср), 643 (ср).
Ni-5. 1H ЯМР (20% MeOD в CDCl3; δ, м.д.; J, Гц): 8.75 (д, 2H, 3J = 4.9, Hβ), 8.66 (д, 2H, 3J = 4.9, Hβ), 8.61 (с, 2H, Hβ), 7.84 (д, 4H, 3J = 8.1, CHm-Ar), 7.78 (д, 4H, 3J = 8.1, ms-CHo-Ar), 7.71 (д, 4H, 3J = 8.1, ms-CHo-Ar), 7.38 (д, 4H, 3J = 8.0, CHo-Ar), 7.10 (д, 8H, 3J = 8.5, ms-CHm-Ar), 4.14–4.08 (м, 8H, CH2O), 1.91 (квинт, 4H, 3J = 6.4, CH2), 1.83 (квинт, 4H, 3J = 6.3, CH2), 1.60 (секст, 4H, 3J = 7.6, CH2), 1.53 (секст, 4H, 3J = 7.6, CH2), 1.01 (т, 6H, 3J = 7.4, CH3), 0.97 (т, 6H, 3J = 7.4, CH3).
13C ЯМР (20% MeOD в CDCl3; δ, м.д.): 159.19, 159.01, 147.88, 147.40, 143.99, 143.56, 142.60, 141.84, 134.49, 134.05, 132.77, 132.65, 132.40, 132.33, 131.88, 131.47, 130.43, 129.27, 120.31, 116.44, 113.03, 112.97, 68.03, 67.97, 31.53, 31.36, 19.38, 19.23, 13.74, 13.70.
MALDI-ТОF MS: m/z вычислено для C76H68N6NiO8 [M]+ 1250.4, найдено 1249.9.
ЭСП (CHCl3; λmax, нм (lg ε)): 345 (4.51), 448 (5.22), 551 (4.27), 581 пл (3.86).
ИК-спектр (νmax, cм–1): 3033 (сл, ш), 2957 (ср, ш), 2931(ср, ш), 2869 (ср, ш), 2655 (ш), 2526 (ш), 1729 (ср, С=О), 1682 (с, С=О), 1606 (с), 1572 (ср), 1505 (с), 1464 (ср), 1419 (ср), 1386 (ср), 1349 (с), 1283 (с), 1240(с), 1219 (с), 1172 (с), 1106 (ср), 1066 (ср), 1001 (с), 964 (ср), 862 (ср), 847 (ср), 796 (с), 710 (с), 666 (ср), 639 (ср), 617 (ср).
Синтез МОКП UiO-66. ZrCl4 (20 мг, 0.087 ммоль) при перемешивании растворяли в ДМФА (5 мл). К полученному раствору добавляли терефталевую кислоту (15 мг, 0.087 ммоль, 1 экв) и перемешивали смесь до получения однородного раствора. Далее в раствор добавляли PhCOOH (318 мг, 2.61 ммоль, 30 экв), перемешивали до полного растворения и нагревали реакционную смесь при 120°С в течение 24 ч. Затем реакционную массу остужали, полученный осадок отделяли центрифугированием и промывали ДМФА (3 × 10 мл) и EtOH (3 × 10 мл) с промежуточным выдерживанием на каждом этапе в течение 2 ч и центрифугированием после каждой промывки. Полученный материал высушивали на воздухе при 120°С в течение 24 ч и далее в вакууме масляного насоса (~10–2 мм рт. ст.) в течение 8 ч.
Синтез МОКП UiO-67. ZrCl4 (20 мг, 0.087 ммоль) при перемешивании растворяли в ДМФА (3.3 мл). К полученному раствору добавляли бифенил-4,4'-дикарбоновую кислоту (21 мг, 0.087 ммоль, 1 экв) и перемешивали смесь до получения однородного раствора. Далее в раствор добавляли PhCOOH (318 мг, 2.61 ммоль, 30 экв), добивались полного растворения и нагревали реакционную смесь при 120°С в течение 24 ч. Затем реакционную массу остужали, полученный осадок отделяли центрифугированием и промывали ДМФА (3 × 10 мл) и EtOH (3 × 10 мл) с промежуточным выдерживанием на каждом этапе в течение 2 ч и центрифугированием после каждой промывки. Полученный материал высушивали на воздухе при 120°С в течение 24 ч и далее в вакууме масляного насоса (~10–2 мм рт. ст.) в течение 8 ч.
Синтез МОКП UiO-66, 67 в присутствии Ni-4 или Ni-5. Методы синтеза и выделения в целом аналогичны методам синтеза МОКП с тем отличием, что перед нагреванием в реакционную массу добавляли Ni-4 (4 мг) или Ni-5 (4.5 мг), что соответствовало отношению Zr : Ni = 25 : 1. Промывка полученных материалов ДМФА с промежуточным выдерживанием на каждом этапе в течение 2 ч и центрифугированием после каждой промывки продолжалась до тех пор, пока в ЭСП фильтрата не пропадали полосы поглощения Ni-4 или Ni-5. После этого материал промывали EtOH (3 × 10 мл) с промежуточным выдерживанием на каждом этапе в течение 2 ч и центрифугированием после каждой промывки. Полученный материал высушивали на воздухе при 120°С в течение 24 ч, а затем в вакууме масляного насоса (~10–2 мм рт. ст.) в течение 8 ч.
Иммобилизация Ni-4 и Ni-5 на МОКП UiO-66, 67. UiO-66 (35 мг) или UiO-67 (40 мг) нагревали в течение 24 ч в ДМФА (4 мл) в присутствии Ni-4 (4 мг) или Ni-5 (4.5 мг), что соответствовало соотношению Zr : Ni = 25 : 1. Промывка полученных материалов ДМФА с промежуточным выдерживанием на каждом этапе в течение 2 ч и центрифугированием после каждой промывки продолжалась до тех пор, пока в ЭСП фильтрата не пропадали полосы поглощения Ni-4 или Ni-5. Далее материал промывали EtOH (3 × 10 мл) с промежуточным выдерживанием на каждом этапе в течение 2 ч и центрифугированием после каждой промывки. Полученный материал высушивали на воздухе при 120°С в течение 24 ч и далее в вакууме масляного насоса (~10–2 мм рт. ст.) в течение 8 ч.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Для получения целевых карбоксизамещенных гетероцикл-аннелированных порфиринов был использован подход, разработанный нами ранее [19, 20] (Схема 1 ). Нуклеофильное аминирование 2-нитропорфирина Ni-1 позволило получить 2-нитро-3-аминопроизводное Ni-2 с выходом, близким к количественному. В свою очередь, восстановление Ni-2 с использованием NaBH4 в смеси CHCl3/MeOH в присутствии 10% Pd/C приводит к диамину Ni-3. В то время как металлопорфирины Ni-1 и Ni-2 стабильны при длительном хранении, наличие двух электронодонорных аминогрупп приводит к склонности Ni-3 к окислению кислородом воздуха. Такое окисление проходит неселективно, однако можно подобрать условия для получения этим способом 2,3-диоксохлоринов [19] или димерных порфиринов [20]. Высокая чувствительность Ni-3 к кислороду воздуха определяет необходимость соблюдения инертных условий при его выделении, далее полученное соединение вводится в последующую реакцию без какой-либо дополнительной очистки и характеризации. Следует отметить, что, по данным ТСХ реакционной массы, наблюдается полная конверсия Ni-2 в Ni-3, а в реакционной массе после восстановления отсутствуют другие окрашенные продукты реакции.
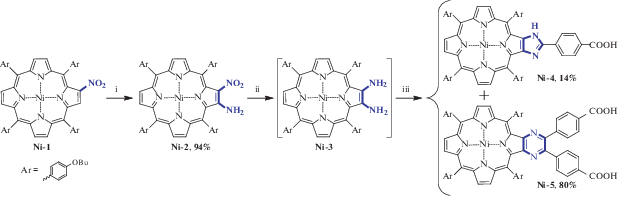
Схема 1 . Синтез карбоксизамещенных порфиринов Ni-4 и Ni-5. i: 4-амино-4Н-1,2,4-триазол, КОН, толуол, 100°С, ii: NaBH4, 10% Pd/C, MeOH, CH2Cl2; iii: 4-карбоксибензальдегид (1 экв.), 1,2-дихлорбензол, 120°С.
Конденсация Ni-3 и 4-карбоксибензальдегида в эквимолярном соотношении неожиданно привела к получению преимущественно пиразинопорфирина Ni-5, в то время как имидазопорфирин Ni-4 был получен с выходом всего 14%. В то время как в [19, 21] было показано, что в случае 4-бромбензальдегида при эквимолярном соотношении реагентов такая конденсация приводит к имидазопорфиринам с высоким выходом и селективностью. Вероятно, в данном случае участвующий в реакции 4-карбоксибензальдегид дополнительно выполняет функцию кислотного катализатора, в присутствии которого направление реакции изменяется и приводит к пиразинопорфирину [19].
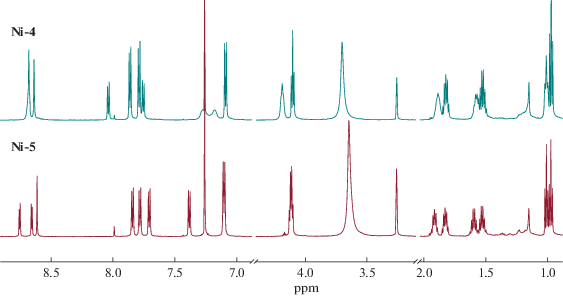
Полученные гетероцикл-аннелированные порфирины были выделены в индивидуальном виде и исследованы различными методами физико-химического анализа. Следует отметить, что в обоих соединениях в одной молекуле сочетаются кислотный и основный центры, кроме того, в имидазопорфирине Ni-4 присутствует лабильный имидазольный NH-протон. Все это приводит к реализации различных динамических обменных процессов. Так, в спектрах ЯМР как Ni-4, так и Ni-5 (рис. 2) не наблюдаются сигналы кислотных протонов COOH-групп, это связано с их обменом с присутствующей в растворителе HDO, а также гидроксильной группой CD3OD, добавление которого необходимо для повышения растворимости изучаемых порфиринов. Кроме того, в спектре Ni-4 отмечается существенное уширение половины наблюдаемых сигналов, в то время как оставшиеся сигналы резонанса характеризуются обычной шириной линий. Уширенные сигналы следует отнести к протонам порфирина, находящимся рядом с аннелированным фрагментом. При этом уширение можно объяснить как миграцией протона между атомами азота имидазольного фрагмента, так и обратимым протонированием этого фрагмента. Протоны карбоксифенильного заместителя проявляются в спектре в виде двух узких хорошо разрешенных дублетов при 8.04 и 7.75 м.д., что объясняется отсутствием влияния симметрии имидазольного цикла на магнитное окружение этих протонов. В случае пиразинопорфирина Ni-5 аналогичное уширение отсутствует, что связано с существенно более низкой основностью пиразинового фрагмента.
В литературе описан ряд методов получения металлорганических каркасных полимеров типа UiO. При этом условия синтеза – природа растворителя, природа и соотношение реагентов, температура реакции, присутствие и природа кислотного модулятора – оказывают существенное влияние на кристалличность и морфологию получаемых материалов. Так, варьирование условий синтеза позволяет получать как аморфные металлорганические каркасные полимеры [27], так и кристаллические материалы [28, 29].
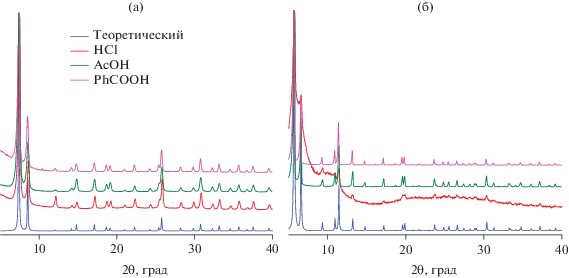
Для получения UiO-66 и UiO-67 нами были воспроизведены описанные подходы к их синтезу, которые заключаются в продолжительном нагревании ZrCl4 и соответствующих дикарбоновых кислот в ДМФА в присутствии кислотного модулятора. Обнаружено, что в отсутствие модулятора взаимодействие приводит к образованию аморфных продуктов реакции, что подтверждается методом рентгенофазового анализа. Использование HCl в качестве модулятора приводит к образованию кристаллического UiO-66 [11], характеризующегося средним размером кристаллитов 32–41 нм (по данным РФА), в то время как попытка синтеза UiO-67 [30] в этих условиях приводит к образованию аморфного продукта реакции. При этом образование осадка координационного полимера в обоих случаях происходит в течение 1 ч.
Изменение природы модулятора с HCl на AcOH приводит к замедлению роста кристаллов UiO-66, однако средний размер кристаллов при этом несколько уменьшается и составляет 31–35 нм при сохранении кристалличности материала. В случае UiO-67 при использовании AcOH в качестве модулятора также удалось получить кристаллический материал, причем средний размер кристаллических частиц равен 62–72 нм.
Переход к PhCOOH в качестве модулятора позволил повысить размер кристаллитов получаемых материалов. Так, в этом случае, по данным РФА, размер кристаллов UiO-66 составил 39–51 нм, а для UiO-67 размер частиц равен 109–122 нм. Полученные этим методом материалы были использованы для дальнейшей модификации. Однофазность образцов и их соответствие описанным в литературе материалам подтверждены методом РФА (рис. 3).
Рис. 3.
Данные РФА для образцов МОКП, полученных с использованием различных модуляторов: а – UiO-66, б – UiO-67.
Данные РЭМ позволяют оценить морфологию полученных кристаллических материалов (рис. 4 ). Так, в случае МОКП UiO-66 размеры образующихся частиц близки и по величине согласуются с данными РФА. В случае UiO-67 наблюдается широкое распределение частиц по размерам и в состав материала входят также кристаллиты, размер которых существенно превосходит вычисленный по данным РФА.
Рис. 4.
РЭМ-изображениия образцов UiO-66 (а) и UiO-67 (б), полученных при использовании PhCOOH в качестве модулятора.

Для модификации UiO-66 и UiO-67 были использованы способы синтеза материала в присутствии карбоксизамещенного порфирина и обмена лигандов полученных материалов (так называемая постсинтетическая модификация). В первом случае синтез материала проводили при добавлении в реакционную массу карбоксизамещенного порфирина Ni-4 или Ni-5 в соотношении Ni : Zr = 1 : 25. Второй подход заключался в продолжительном нагревании предварительно полученных МОКП UiO-66, 67 в ДМФА в присутствии Ni-4 или Ni-5 также в соотношении Ni : Zr = 1 : 25. При этом в обоих случаях можно было ожидать введения карбоксилатных фрагментов порфиринов в координационную сферу атомов ZrIV и, соответственно, привязки молекул порфиринов к узловым кластерам Zr6O4(OH)4.
При реализации обоих подходов к получению гибридных материалов с использованием Ni-4 не наблюдается заметного связывания карбоксизамещенного порфирина с поверхностью частиц материалов. Как при синтезе UiO-66, 67 в присутствии Ni-4, так и при нагревании предварительно полученных материалов в присутствии Ni-4 при последующем выделении и обработке полученных МОКП наблюдается полное удаление порфирина из материала. Присутствие Ni-4 не обнаруживается в материале ни методом рентгеноспектрального микроанализа, ни методом электронной спектроскопии диффузного отражения.
Напротив, в случае Ni-5 обнаружено, что этот порфирин можно успешно использовать для получения гибридных материалов с МОКП UiO-66, 67. И хотя метод синтеза материала в присутствии порфирина не эффективен, постсинтетическая модификация позволила получить целевой продукт по данным РСМА и СДО (рис. 5).
Рис. 5.
Спектры диффузного отражения твердых образцов: a – Ni-5, б – UiO-66 после синтеза в присутствии Ni-5, в – UiO-66, полученный методом постсинтетической модификации.
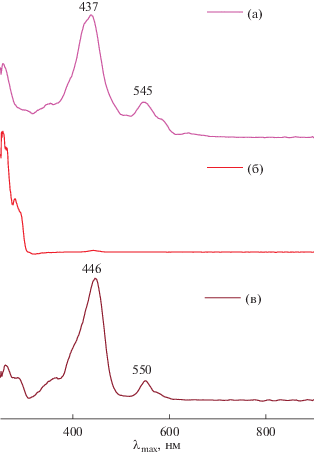
Следует отметить, что полосы порфирина в СДО при образовании гибридного материала смещаются батохромно, что может быть связано со смещением электронной плотности с карбоксилатных групп к атомам ZrIV. Так, в спектре Ni-5 присутствуют полосы с максимумами поглощения при 437 и 545 нм, в то время как в спектре полученного гибридного материала их положение составляет 446 и 550 нм соответственно.
Соотношение Ni/Zr, определяемое по данным РСМА после взаимодействия Ni-5 и UiO-66, составило 1 : 98, что соответствует соотношению порфирина с узловыми кластерами Zr6O4(OH)4 ~ ~ 1 : 16. Вероятно, при проведении синтеза МОКП в присутствии порфирина модификация материала происходит равномерно в объеме, а в случае обмена лигандов процесс протекает преимущественно на поверхности. Кроме того, учитывая, что синтез проводится в присутствии 30-кратного избытка модификатора PhCOOH, разумно предположить пропорциональное вытеснение карбоксильных фрагментов Ni-5 бензоатными фрагментами из координационной сферы атомов циркония. Интересно отметить, что размер кристаллических частиц материала существенно не меняется после взаимодействия с Ni-5, что подтверждается данными РФА.
С целью подтверждения иммобилизации порфирина Ni-5 на поверхность МОКП UiO-66, 67 был использован метод ИК-спектроскопии. В спектрах исходных порфиринов Ni-4 и Ni-5 присутствуют полосы, характерные как для колебаний самого порфиринового кольца, так и для функциональных групп, входящих в их состав (см. Экспериментальную часть). Так, в спектрах в первую очередь необходимо выделить диапазоны, имеющие характеристичные колебания карбоновых кислот. Это прежде всего широкая область поглощения с рядом полос в интервале 3500–2500 см–1, где наблюдаются валентные колебания ОН-групп, участвующих в образовании межмолекулярных водородных связей, которые частично перекрываются с валентными колебаниями связей С–Н. Образование водородных связей приводит к смещению интенсивной полосы валентных колебаний карбонильной группы ν(С=О) в низкочастотную область до 1690 см–1 (Ni-4) и 1682 см–1 (Ni-5). Наряду с этой полосой в спектрах обоих порфиринов присутствует полоса средней интенсивности при 1724 см–1 (Ni-4), 1729 см–1 (Ni-5), характеризующая колебания ν(С=О) свободной карбонильной группы. Следует также отметить, что в спектре Ni-4 дополнительно присутствует характеристичная полоса ν(NH) имидазольного кольца при 3435 см–1.
В ИК-спектрах МОКП UiO-66 и UiO-67 присутствуют полосы колебаний карбоксилатов и фенильных фрагментов, при этом колебания карбонильных групп в спектрах отсутствуют. Интересно, что в спектрах материалов, модифицированных порфирином Ni-5, колебания свободной карбонильной группы, равно как и водородно-связанной ν(С=О) при 1724 и 1682 см–1, не проявляются, что свидетельствует о связывании порфирина с материалом с участием карбоксильных групп (рис. 6 ). Перераспределение интенсивностей в диапазоне 1600–1400 см–1 также косвенно свидетельствует об иммобилизации порфирина в материал с образованием карбоксилата. Известно, что карбонильная полоса ν(С=О) при образовании карбоксилата трансформируется в две полосы: νas(COO–) и νs(COO–), при этом полоса νas(COO–) обычно более интенсивная и расположена в интервале 1500–1600 см–1, а менее интенсивная полоса νs(COO–) расположена около 1400 см–1 [31]. Следует отметить, что в спектрах модифицированных порфиринами МОКП относительная интенсивность полос 1574 см–1 (Ni-5@UiO-66) и 1592 см–1 (Ni-5@UiO-67) увеличивается по сравнению с интенсивностями этих полос в спектрах исходных UiO-66 и UiO-67. При этом полоса при 1592 см–1 в спектре Ni-5@UiO-67 расщепляется на две (1602 и 1582 см–1) и уширяется, также увеличивается по интенсивности полоса при 1512 см–1. Именно в этом диапазоне проявляются интенсивные полосы νas(COO–) карбоксилатных групп и ν(СС) ароматических колец порфирина.
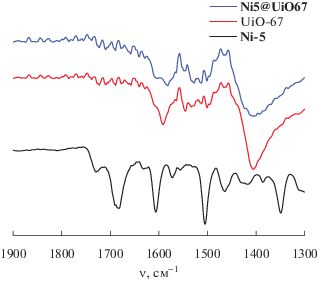
Таким образом, сравнительный анализ ИК-спектров порфирина Ni-5 и МОКП со спектрами гибридных материалов наряду с данными спектров диффузного отражения позволяет сделать вывод, что порфирин связан с материалом обеими якорными карбоксилатными группами.
Описанные структурные данные также согласуются с наблюдаемым эффективным связыванием Ni-5 с полученными МОКП. Так, расстояние между атомами углерода карбоксильных групп 1,2-бис(4-карбоксифенил)-бензола, который может выступать структурным аналогом координационного центра порфирина Ni-5, составляет 7.12 Å [32], что сопоставимо с аналогичным расстоянием в молекулах линкеров, формирующих UiO-66 (5.807 Å) [33, 34] и UiO-67 (10.103 Å) [30]. Расстояния между атомами углерода соседних карбоксилатных групп узлового кластера Zr6O4(OH)4(COO)12 составляют 4.497 и 4.458 Å для UiO-66 и UiO-67 соответственно. В целом эти данные также не противоречат возможной иммобилизации Ni-5 на поверхность МОКП c использованием обеих якорных групп.
ЗАКЛЮЧЕНИЕ
Показана возможность постсинтетической модификации Zr(IV)-содержащих металлорганических координационных полимеров с использованием гетероцикл-аннелированных карбоксизамещенных порфиринов. Выявлено влияние количества якорных карбоксильных групп и метода модификации на соотношение узловых кластеров Zr6O4(OH)4 в МОКП и введенных молекул порфирина в образующемся гибридном материале. Показано, что иммобилизация дикарбоксизамещенного пиразинопорфирина происходит с участием обеих карбоксильных групп при их координации к атомам Zr(IV). Разработанный подход в дальнейшем будет использован для получения гибридных материалов для гетерогенных каталитических приложений.
Список литературы
Zou D., Liu D. // Mater. Today Chem. 2019. V. 12. P. 139. https://doi.org/10.1016/j.mtchem.2018.12.004
Wang B., Xie L.-H., Wang X. et al. // Green Energy Environ. 2018. V. 3. № 3. P. 191. https://doi.org/10.1016/j.gee.2018.03.001
Li H., Li L., Lin R.-B. et al. // EnergyChem. 2019. V. 1. № 1. P. 100006. https://doi.org/10.1016/j.enchem.2019.100006
Liu X. // Front. Chem. Sci. Eng. 2020. V. 14. № 2. P. 216. https://doi.org/10.1007/s11705-019-1857-5
Taddei M. // Coord. Chem. Rev. 2017. V. 343. P. 1. https://doi.org/10.1016/j.ccr.2017.04.010
Ding M., Cai X., Jiang H.-L. // Chem. Sci. 2019. V. 10. № 44. P. 10209. https://doi.org/10.1039/C9SC03916C
Trickett C.A., Gagnon K.J., Lee S. et al. // Angew. Chem. Int. Ed. 2015. V. 54. № 38. P. 11162. https://doi.org/10.1002/anie.201505461
Sapianik A.A., Fedin V.P. // Russ. J. Coord. Chem. 2020. V. 46. № 7. P. 443. https://doi.org/10.1134/S1070328420060093
Dhakshinamoorthy A., Santiago-Portillo A., Asiri A.M. et al. // ChemCatChem. 2019. V. 11. № 3. P. 899. https://doi.org/10.1002/cctc.201801452
Caratelli C., Hajek J., Cirujano F.G. et al. // J. Catal. 2017. V. 352. P. 401. https://doi.org/10.1016/j.jcat.2017.06.014
Vermoortele F., Bueken B., Le Bars G. и дp. // J. Am. Chem. Soc. 2013. V. 135. № 31. P. 11465. https://doi.org/10.1021/ja405078u
Ye Y., Liu H., Li Y. и дp. // Talanta. 2019. V. 200. P. 472. https://doi.org/10.1016/j.talanta.2019.01.086
Wang L., Jin P., Duan S. et al. // Environ. Sci. Nano. 2019. V. 6. № 8. P. 2652. https://doi.org/10.1039/C9EN00460B
Liang J., Xie Y.-Q., Wu Q. et al. // Inorg. Chem. 2018. V. 57. № 5. P. 2584. https://doi.org/10.1021/acs.inorgchem.7b02983
Gao Y., Lu J., Xia J. et al. // ACS Appl. Mater. Interfaces. 2020. V. 12. № 11. P. 12706. https://doi.org/10.1021/acsami.9b21122
Zhao L., Yang J., Gong M. и дp. // J. Mater. Chem. C. 2020. V. 8. № 11. P. 3904. https://doi.org/10.1039/C9TC06624A
Kan J.-L., Jiang Y., Xue A. et al. // Inorg. Chem. 2018. V. 57. № 9. P. 5420. https://doi.org/10.1021/acs.inorgchem.8b00384
Abdulaeva I.A., Birin K.P., Bessmertnykh-Lemeune A. et al. // Coord. Chem. Rev. 2020. V. 407. P. 213108. https://doi.org/10.1016/j.ccr.2019.213108
Birin K.P., Poddubnaya A.I., Abdulaeva I.A. et al. // Dye. Pigment. 2018. V. 156. № March. P. 243. https://doi.org/10.1016/j.dyepig.2018.04.009
Abdulaeva I.A., Polivanovskaia D.A., Birin K.P. et al. // Mendeleev Commun. 2020. V. 30. № 2. P. 162. https://doi.org/10.1016/j.mencom.2020.03.010
Birin K.P., Abdulaeva I.A., Poddubnaya A.I. et al. // Dye. Pigment. 2020. V. 181. P. 108550. https://doi.org/10.1016/j.dyepig.2020.108550
Abdulaeva I.A., Birin K.P., Gorbunova Y.G. et al. // J. Porphyr. Phthalocyanines. 2018. V. 22. № 8. P. 619. https://doi.org/10.1142/S1088424618500475
Abdulaeva I.A., Birin K.P., Sinelshchikova A.A. et al. // CrystEngComm. 2019. V. 21. № 9. P. 1488. https://doi.org/10.1039/C8CE01992D
Abdulaeva I.A., Birin K.P., Michalak J. et al. // New J. Chem. 2016. V. 40. № 7. P. 5758. https://doi.org/10.1039/C5NJ03247D
Armarego W.L.F., Chai C.L.L. // Purification of laboratory chemicals. Elsevier/Butterworth-Heinemann, 2009.
Lo M., Lefebvre J.-F., Leclercq D. et al. // Org. Lett. 2011. V. 13. № 12. P. 3110. https://doi.org/10.1021/ol2010215
Yang F., Li W., Tang B. // J. Alloys Compd. 2018. V. 733. P. 8. https://doi.org/10.1016/j.jallcom.2017.10.129
Katz M.J., Brown Z.J., Colón Y.J. et al. // Chem. Commun. 2013. V. 49. № 82. P. 9449. https://doi.org/10.1039/c3cc46105j
Schaate A., Roy P., Godt A. et al. // Chem. - A Eur. J. 2011. V. 17. № 24. P. 6643. https://doi.org/10.1002/chem.201003211
Nickerl G., Leistner M., Helten S. et al.// Inorg. Chem. Front. 2014. V. 1. № 4. P. 325. https://doi.org/10.1039/C3QI00093A
Deacon G. // Coord. Chem. Rev. 1980. V. 33. № 3. P. 227. https://doi.org/10.1016/S0010-8545(00)80455-5
Hisaki I., Nakagawa S., Tohnai N. et al. // Angew. Chem. Int. Ed. 2015. V. 54. № 10. P. 3008. https://doi.org/10.1002/anie.201411438
Ermer M., Mehler J., Kriesten M. et al. // Dalton Trans. 2018. V. 47. № 41. P. 14426. https://doi.org/10.1039/C8DT02999G
Cavka J.H., Jakobsen S., Olsbye U. et al. // J. Am. Chem. Soc. 2008. V. 130. № 42. P. 13850. https://doi.org/10.1021/ja8057953
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии