

Журнал неорганической химии, 2021, T. 66, № 12, стр. 1693-1699
Синтез наноразмерного WO3 методом химического осаждения с использованием щавелевой кислоты
Ф. Ю. Горобцов a, *, Т. Л. Симоненко a, Н. П. Симоненко a, Е. П. Симоненко a, В. Г. Севастьянов a, Н. Т. Кузнецов a
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
* E-mail: phigoros@gmail.com
Поступила в редакцию 01.07.2021
После доработки 12.07.2021
Принята к публикации 17.07.2021
Аннотация
Изучен процесс синтеза наноразмерного WO3 методом химического осаждения с использованием щавелевой кислоты. Полученный порошок исследован методами ИК-спектроскопии и рентгенофазового анализа, а его термическое поведение изучено с помощью синхронного ТГА/ДСК-анализа. Установлено, что осадок до высокотемпературной обработки представляет собой смесь WO3 ⋅ 2H2O и оксалата вольфрама. Термообработка при 400°С приводит к разложению оксалата, а при 500°С формируется однофазный WO3 с моноклинной кристаллической структурой (средний размер ОКР составляет 36 ± 4 нм, длина частиц – 50 ± 5 нм, ширина – 40 ± 4 нм). С помощью Кельвин-зондовой силовой микроскопии оценена работа выхода электрона с поверхности сформированных частиц WO3 на воздухе. Показано, что химическое осаждение оксида вольфрама(VI) с использованием щавелевой кислоты является перспективным методом получения нанопорошка соответствующего состава.
ВВЕДЕНИЕ
Оксид вольфрама(VI) на сегодняшний день является широко востребованным материалом в самых разных областях применения. Он относится к наиболее часто исследуемым и используемым катодным электрохромным материалам [1–3], чьи электрооптические свойства были открыты почти 50 лет назад [4]. Как катодный электрохромный материал WO3 изменяет свою окраску с прозрачной бесцветной или бледно-желтой на малопрозрачную синюю в результате частичного восстановления W6+ до W5+ и интеркаляции катионов щелочных металлов. Кроме того, WO3 является перспективным фотокатализатором – моноклинная фаза γ-WO3 проявляет высокую каталитическую активность при расщеплении воды [5–8] и обладает меньшей оптической шириной запрещенной зоны (2.7–3.0 эВ) и большей проводимостью, чем TiO2. Помимо электрохромных и фотокаталитических свойств триоксид вольфрама обладает ярко выраженными фотохромными свойствами [9–12], перспективен в газовой сенсорике [13–16] и других областях.
Для получения оксида вольфрама(VI) используются различные методы и подходы, такие как золь-гель технология [17–19], электрохимическое [20–22], химическое газофазное осаждение [23, 24] и др. Гидротермальный метод, являющийся одним из наиболее удобных методов синтеза наноматериалов благодаря возможности получать анизотропные и иерархически организованные микроструктуры, варьировать кристаллическую структуру веществ [25–27], также активно используется для получения WO3 [3, 28–30]. При этом нередко в качестве реагентов, вступающих в реакцию с вольфрамсодержащим соединением в ходе гидротермального синтеза, выступают оксалаты [29] или щавелевая кислота [31–34]. В то же время для синтеза наноразмерного оксида вольфрама(VI) применяют и метод осаждения WO3 из растворов вольфраматов под действием сильных неорганических кислот, таких как соляная [35] или азотная [36]. Щавелевая кислота является сильной органической кислотой и может выступать в качестве хелатного лиганда, образуя комплексные соединения с различными металлами, включая вольфрам [37], но в литературе не удалось найти примеры ее применения при синтезе наноразмерного WO3 методом химического осаждения без гидротермальной обработки. Таким образом, целью настоящей работы является изучение процесса синтеза наноразмерного WO3 методом химического осаждения с использованием щавелевой кислоты.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для синтеза оксида вольфрама(VI) в 43 мл дистиллированной воды при перемешивании растворяли 1.126 г паравольфрамата аммония, 0.582 г щавелевой кислоты и 0.582 г поливинилпирролидона. Затем раствор выдерживали при 25°С в течение 5 сут, после чего наблюдали выпадение осадка светло-желтого цвета. Полученный осадок отделяли от маточного раствора и промывали (два раза дистиллированной водой и один раз этиловым спиртом) путем ступенчатого центрифугирования с последующей сушкой при 50°С в течение 13 ч. Далее, опираясь на результаты термического и рентгенофазового анализа, с целью формирования однофазного оксида WO3 порошок подвергали термообработке при 500°С на протяжении 1 ч. Для более детального изучения процесса кристаллизации оксида вольфрама был также получен порошок при более низкой температуре (400°С, 1 ч).
ИК-спектры пропускания порошка после сушки и высокотемпературной обработки регистрировали в диапазоне волновых чисел 350–4000 см–1 с использованием ИК-Фурье-спектрометра ИнфраЛЮМ ФТ-08 (время накопления сигнала составляло 15 с, разрешение – 1 см–1). Для этого готовили суспензии в вазелиновом масле, которые помещали в виде пленки между стеклами KBr.
Термическое поведение порошка после сушки исследовали с помощью синхронного термоанализатора SDT Q600 (TA Instruments) в диапазоне температур 25–1000°С в токе воздуха 250 мл/мин (скорость нагрева 10 град/мин, масса навески 21.5210 мг).
Рентгенофазовый анализ (РФА) порошка после сушки и высокотемпературной обработки проводили на дифрактометре D8-Advance (Bruker, CuKα-излучение, λ = 1.5418 Å, Ni-фильтр, E = 40 кэВ, I = 40 мА, время накопления сигнала в точке 0.3 с, шаг 0.02°, диапазон углов 5°–80°).
Форму и размер частиц оксида вольфрама(VI) определяли с помощью просвечивающей электронной микроскопии (ПЭМ, JEOL-JEM 1011).
Морфологию и электрофизические характеристики порошка WO3 изучали также с помощью атомно-силовой микроскопии (АСМ). Для этого порошок диспергировали в дистиллированной воде, после чего каплю образовавшейся дисперсии наносили на медную подложку и подвергали сушке при 70°С в течение 2 ч. Нанесенное таким образом покрытие изучали на сканирующем зондовом микроскопе Solver PRO-M (NT-MDT) с использованием зондов с проводящим покрытием HA-HR/W2C+ серии ETALON (резонансная частота ~230 кГц, радиус скругления <35 нм). Измерения проводили в полуконтактном режиме, в котором также было выполнено сканирование в режиме сканирующей емкостной микроскопии и Кельвин-зондовой силовой микроскопии (КЗСМ).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
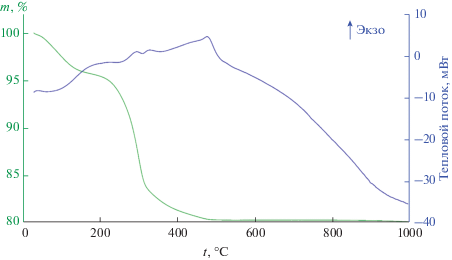
На первом этапе было изучено термическое поведение порошка после сушки. Как следует из термограмм (рис. 1), при нагревании до 1000°С наблюдается несколько ступеней потери массы, которые сопровождаются соответствующими тепловыми эффектами. В интервале температур от 25 до 200°C имеет место заметная потеря массы (4.301%), сопровождаемая эндоэффектом с минимумом около 100°С, что может быть связано с удалением остаточного растворителя и сорбированных газов. Дальнейшему нагреванию в интервале 200–325°С отвечает более интенсивная потеря массы (12.03%) и два экзотермических эффекта с максимумами при 293 и 326°C, относящихся к окислению органических фрагментов оксалата вольфрама. В диапазоне температур 325–475°С наблюдается менее активная потеря массы (3.355%), связанная с окислением остаточного углерода. Соответствующий экзотермический эффект с максимумом при 475°C также может быть связан с кристаллизацией WO3. В ходе последующего повышения температуры от 475 до 1000°С масса вещества практически не изменяется. Таким образом, суммарная потеря массы в исследуемом диапазоне температур составляет 19.91%, а в качестве режима термообработки с целью кристаллизации оксида вольфрама выбран режим с выдержкой при 500°С в течение 1 ч.
С помощью ИК-спектроскопии были изучены порошки после сушки и термообработки при 500°С. В результате анализа полученного спектра (рис. 2а) и сравнения его с литературными данными для различных карбоксилатных комплексов вольфрама [37–40] установлено, что в качестве твердой фазы при осаждении образуется оксалатный комплекс вольфрама. При 1590–1700 см–1 наблюдается набор характеристичных полос, отвечающих асимметричным колебаниям групп COO–, полосу же около 1730 см–1 можно отнести к колебаниям свободной карбоксильной группы. Полоса поглощения с максимумом около 810 см–1 соответствует асимметричным колебаниям связи W–O–W, а три полосы с максимумами при 897, 934, 975 см–1 – симметричным и асимметричным колебаниям связи W=O [41, 42]. На спектре также присутствует полоса с максимумом около 612 см–1, которую можно отнести к симметричным колебаниям группы W–O–W. Колебания связи W–O–W могут относиться как к мостикам в составе оксалата, так и к гидрату WO3 ⋅ 2H2O, имеющему, согласно литературным данным, характеристическую полосу поглощения с максимумом около 610 см–1 [43]. В ходе термообработки при 500°С происходит разложение комплекса и гидрата с образованием WO3, а набор и интенсивность полос поглощения свидетельствуют о формировании моноклинной модификации оксида вольфрама(VI) [43].
Рис. 2.
ИК-спектры полученного порошка после сушки и прокаливания (а); рентгенограммы полученного порошка после сушки и прокаливания (б).
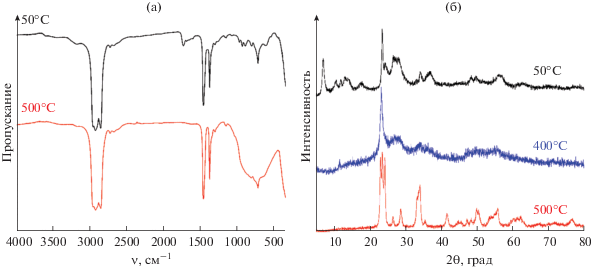
Для определения фазового состава порошка после сушки и высокотемпературной обработки был проведен рентгенофазовый анализ (рис. 2б), который показал, что порошок после сушки представляет собой смесь карбоксилата вольфрама [40, 44] и гидрата состава WO3 ⋅ 2H2O, что подтверждает предположение об отнесении полосы поглощения с максимумом около 612 см–1 на соответствующем ИК-спектре (рис. 2а) к примесной фазе состава WO3 ⋅ 2H2O. Термообработка при 400°С, по данным РФА, приводит к разложению оксалата и образованию однофазного гидрата WO3 ⋅ 2H2O, а при 500°С формируется WO3 с моноклинной структурой (пр. гр. P21/n, PDF 01-072-0677). Каких-либо кристаллических примесей после термообработки при 500°С не обнаружено. С помощью полнопрофильного анализа были рассчитаны параметры кристаллической решетки: a = 7.3010(8), b = 7.5004(9), c = 7.665(1) Å, β = = 90.487(3)°. Средний размер областей когерентного рассеяния (ОКР) при этом составил 36 ± 4 нм.
С помощью ПЭМ были определены форма и размер частиц полученного оксида вольфрама(VI) (рис. 3). Как видно из микрофотографий, порошок в основном состоит из частиц чуть вытянутой формы длиной ~50 ± 5 и шириной ~40 ± ± 4 нм, что достаточно хорошо согласуется с рассчитанными значениями ОКР. Таким образом, можно предположить, что сформировавшиеся частицы WO3 состоят в основном из одного кристаллита.

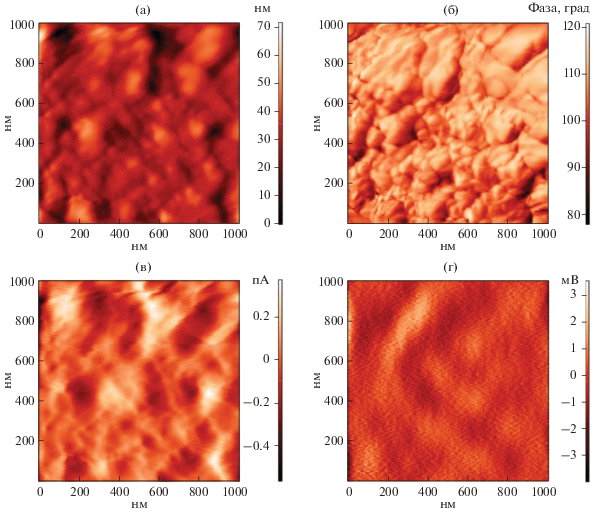
Для изучения морфологии полученного порошка оксида вольфрама(VI) и изучения его локальных электрофизических характеристик применяли также атомно-силовую микроскопию (рис. 4). Так, на полученном топографическом изображении (рис. 4а) и изображении фазового контраста (рис. 4б) прослеживаются та же форма и размер частиц, как и по данным ПЭМ. При этом длина частиц равна 74 ± 9, а ширина – 40 ± ± 4 нм. Больший размер частиц в латеральной плоскости в данном случае объясняется влиянием радиуса скругления (<35 нм) острия зонда. С помощью сканирующей емкостной и Кельвин-зондовой силовой микроскопии были изучены локальные электрофизические свойства сформированных в ходе термообработки при 500°С наночастиц WO3, в частности, распределение носителей заряда на поверхности соответствующей пленки на медной подложке и работа выхода электрона. Так, из карты емкостного контраста (рис. 4в) видно, что на границах между частицами наблюдается повышенная емкость по сравнению с остальной их поверхностью. Это можно объяснить образованием обедненных электронами зон в местах контакта частиц. Распределение поверхностного потенциала (рис. 4г) свидетельствует о том, что носители заряда в большей степени сосредоточены вдали от границ между частицами, хотя в целом поверхностный потенциал распределен достаточно равномерно по всей просканированной площади материала. Из результатов КЗСМ было рассчитано значение работы выхода электрона для полученного WO3, составившее 4.664 ± 0.003 эВ, что близко к нижней границе диапазона значений работы выхода, встречающихся в литературе (4.7–6.4 эВ) [45], свидетельствуя о несколько повышенной электропроводности полученного оксида вольфрама(VI).
ЗАКЛЮЧЕНИЕ
Изучен процесс синтеза наноразмерного WO3 методом химического осаждения с использованием щавелевой кислоты. Полученный в результате осаждения и последующей сушки продукт, по результатам колебательной спектроскопии и рентгенофазового анализа, представляет собой смесь оксалата и гидрата состава WO3 ⋅ 2H2O. При дальнейшей термообработке при 400°С формируется однофазный гидрат WO3 ⋅ 2H2O, а при 500°С образуется нанопорошок WO3 с моноклинной кристаллической структурой. Результаты ПЭМ и АСМ свидетельствуют о формировании оксидных частиц чуть вытянутой формы. Средний размер ОКР для данного нанодисперсного порошка γ-WO3 составляет 36 ± 4 нм, длина частиц – 50 ± 5 нм, ширина – около 40 ± 4 нм. С помощью КЗСМ определено значение работы выхода электрона с поверхности наночастиц оксида вольфрама(VI), которое составило 4.664 ± 0.003 эВ, что близко к нижней границе диапазона значений работы выхода, встречающихся в литературе, свидетельствуя о несколько повышенной электропроводности материала.
Таким образом, в ходе исследования было показано, что с применением щавелевой кислоты методом химического осаждения может быть получен наноразмерный оксид вольфрама(VI).
Список литературы
Granqvist C.G. // Thin Solid Films. 2014. V. 564. P. 1. https://doi.org/10.1016/j.tsf.2014.02.002
Mortimer R.J. // Annu. Rev. Mater. Res. 2011. V. 41. № 1. P. 241. https://doi.org/10.1146/annurev-matsci-062910-100344
Zhang J., Tu J.P., Xia X.H. et al. // J. Mater. Chem. 2011. V. 21. № 14. P. 5492. https://doi.org/10.1039/c0jm04361c
Deb S.K. // Philos. Mag. 1973. V. 27. № 4. P. 801. https://doi.org/10.1080/14786437308227562
Liu Y., Li J., Li W. et al. // J. Phys. Chem. C. 2015. V. 119. № 27. P. 14834. https://doi.org/10.1021/acs.jpcc.5b00966
Zhang J., Chang X., Li C. et al. // J. Mater. Chem. A. 2018. V. 6. № 8. P. 3350. https://doi.org/10.1039/c7ta10056f
Dong P., Hou G., Xi X. et al. // Environ. Sci. Nano. 2017. V. 4. № 3. P. 539. https://doi.org/10.1039/c6en00478d
Liu X., Wang F., Wang Q. // Phys. Chem. Chem. Phys. 2012. V. 14. № 22. P. 7894. https://doi.org/10.1039/c2cp40976c
He T., Yao J. // J. Mater. Chem. 2007. V. 17. № 43. P. 4547. https://doi.org/10.1039/b709380b
Huang R., Shen Y., Zhao L. et al. // Adv. Powder Technol. 2012. V. 23. № 2. P. 211. https://doi.org/10.1016/j.apt.2011.02.009
Songara S., Gupta V., Kumar Patra M. et al. // J. Phys. Chem. Solids. 2012. V. 73. № 7. P. 851. https://doi.org/10.1016/j.jpcs.2012.02.020
Kozlov D.A., Shcherbakov A.B., Kozlova T.O. et al. // Molecules. 2020. V. 25. № 1. P. 154. https://doi.org/10.3390/molecules25010154
Ponzoni A., Comini E., Sberveglieri G. et al. // Appl. Phys. Lett. 2006. V. 88. № 20. P. 28. https://doi.org/10.1063/1.2203932
Ponzoni A., Russo V., Bailini A. et al. // Sensors Actuators, B: Chem. 2011. V. 153. № 2. P. 340. https://doi.org/10.1016/j.snb.2010.10.045
Penza M., Tagliente M.A., Mirenghi L. et al. // Sensors Actuators, B: Chem. 1998. V. 50. № 1. P. 9. https://doi.org/10.1016/S0925-4005(98)00149-X
Llobet E., Molas G., Molinàs P. et al. // J. Electrochem. Soc. 2000. V. 147. № 2. P. 776. https://doi.org/10.1149/1.1393270
Costa C., Pinheiro C., Henriques I. et al. // ACS Appl. Mater. Interfaces. 2012. V. 4. № 3. P. 1330. https://doi.org/10.1021/am201606m
Breedon M., Spizzirri P., Taylor M. et al. // Cryst. Growth Des. 2010. V. 10. № 1. P. 430. https://doi.org/10.1021/cg9010295
Chatzikyriakou D., Krins N., Gilbert B. et al. // Electrochim. Acta. 2014. V. 137. P. 75. https://doi.org/10.1016/j.electacta.2014.05.139
Kwong W.L., Savvides N., Sorrell C.C. // Electrochim. Acta. 2012. V. 75. P. 371. https://doi.org/10.1016/j.electacta.2012.05.019
Giannouli M., Leftheriotis G. // Sol. Energy Mater. Sol. Cells. 2011. V. 95. № 7. P. 1932. https://doi.org/10.1016/j.solmat.2011.02.024
Poongodi S., Kumar P.S., Masuda Y. et al. // RSC Adv. 2015. V. 5. № 117. P. 96416. https://doi.org/10.1039/c5ra19177g
Annanouch F.E., Haddi Z., Vallejos S. et al. // ACS Appl. Mater. Interfaces. 2015. V. 7. № 12. P. 6842. https://doi.org/10.1021/acsami.5b00411
Wu W., Yu Q., Lian J. et al. // J. Cryst. Growth. 2010. V. 312. № 21. P. 3147. https://doi.org/10.1016/j.jcrysgro.2010.07.057
Simonenko T.L., Bocharova V.A., Simonenko N.P. et al. // Russ. J. Inorg. Chem. 2020. V. 65. № 4. https://doi.org/10.1134/S003602362004018X
Simonenko T.L., Bocharova V.A., Gorobtsov P.Y. et al. // Russ. J. Inorg. Chem. 2020. V. 65. № 9. P. 1292. https://doi.org/10.1134/S0036023620090193
Simonenko T.L., Bocharova V.A., Gorobtsov P.Y. et al. // Russ. J. Inorg. Chem. 2020. V. 65. № 9. P. 1304. https://doi.org/10.1134/S0036023620090181
Phuruangrat A., Ham D.J., Hong S.J. et al. // J. Mater. Chem. 2010. V. 20. № 9. P. 1683. https://doi.org/10.1039/b918783a
Zhang J., Liu Z., Liu Z. // ACS Appl. Mater. Interfaces. 2016. V. 8. № 15. P. 9684. https://doi.org/10.1021/acsami.6b00429
Yao S., Qu F., Wang G. et al. // J. Alloys Compd. 2017. V. 724. P. 695. https://doi.org/10.1016/j.jallcom.2017.07.123
Liu Z., Miyauchi M., Yamazaki T. et al. // Sensors Actuators, B: Chem. 2009. V. 140. № 2. P. 514. https://doi.org/10.1016/j.snb.2009.04.059
Ma D., Wang H., Zhang Q. et al. // J. Mater. Chem. 2012. V. 22. № 32. P. 16633. https://doi.org/10.1039/c2jm32784h
Patil V.B., Adhyapak P.V., Suryavanshi S.S. et al. // J. Alloys Compd. 2014. V. 590. P. 283. https://doi.org/10.1016/j.jallcom.2013.12.102
Ding D., Shen Y., Ouyang Y. et al. // Thin Solid Films. 2012. V. 520. № 24. P. 7164. https://doi.org/10.1016/j.tsf.2012.08.003
Nayak A.K., Lee S., Choi Y.I. et al. // ACS Sustain. Chem. Eng. 2017. V. 5. № 3. P. 2741. https://doi.org/10.1021/acssuschemeng.6b03084
Supothina S., Seeharaj P., Yoriya S. et al. // Ceram. Int. 2007. V. 33. № 6. P. 931. https://doi.org/10.1016/j.ceramint.2006.02.007
Dengel A.C., Griffith W.P., Powell R.D. et al. // Dalton Trans. 1987. № 1. P. 17.
Deepa M., Sharma N., Varshney P. et al. // J. Mater. Sci. 2000. V. 35. № 21. P. 5313. https://doi.org/10.1023/A:1004838627252
Zhang H., Zhao H., Jiang Y.Q. et al. // Inorg. Chim. Acta. 2003. V. 351. № 1. P. 311. https://doi.org/10.1016/S0020-1693(03)00177-4
Li D.M., Cui L.F., Xing Y.H. et al. // J. Mol. Struct. 2007. V. 832. № 1–3. P. 138. https://doi.org/10.1016/j.molstruc.2006.08.014
Young C.G., Gable R.W., Mackayla M.F. // Inorg. Chem. 1990. Т. 29. № 9. P. 1777. https://doi.org/ 10.1021/ic00334a036
Llopis E., Ramírez J.A., Doménech A. et al. // J. Chem. Soc., Dalton Trans. 1993. № 7. P. 1121. https://doi.org/10.1039/DT9930001121
Daniel M.F., Desbat B., Lassegues J.C. et al. // J. Solid State Chem. 1987. V. 67. № 2. P. 235. https://doi.org/10.1016/0022-4596(87)90359-8
Cindri M., Strukan N., Vrdoljak V. et al. // Inorg. Chim. Acta. 2000. V. 309. № 1–2. P. 77. https://doi.org/10.1016/S0020-1693(00)00236-X
Han S., Shin W.S., Seo M. et al. // Org. Electron. 2009. V. 10. № 5. P. 791. https://doi.org/10.1016/j.orgel.2009.03.016
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии