

Химическая физика, 2023, T. 42, № 4, стр. 20-30
Исследование поверхности потенциальной энергии реакционных систем H + O + C2H4
А. Г. Давтян 1, З. О. Манукян 1, С. Д. Арсентьев 1, *, Л. А. Тавадян 1, В. С. Арутюнов 2
1 Институт химической физики им. А.Б. Налбандяна Национальной академии наук Республики Армения
Ереван, Республика Армения
2 Федеральный исследовательский центр химической физики им. Н.Н. Семёнова Российской академии наук
Москва, Россия
* E-mail: arsentiev53@mail.ru
Поступила в редакцию 10.02.2022
После доработки 25.03.2022
Принята к публикации 20.04.2022
- EDN: MWHERN
- DOI: 10.31857/S0207401X23040052
Аннотация
Поверхность потенциальной энергии (ППЭ) реакционной системы H + O + C2H4, которая играет важную роль в окислительной конверсии углеводородов в целом, была теоретически исследована с использованием различных квантовохимических методов. На ППЭ, соответствующей реакции атома кислорода с этиленом, локализован ряд ранее неизвестных интермедиатов, проанализированы возможные пути их дальнейшего превращения. Получены данные относительно последовательности энергетических уровней образования этоксильного радикала, представлена детальная диаграмма энтальпий мономолекулярных реакций его распада и изомеризации, сделан вывод о вероятности их протекания. Полученные результаты дают возможность оценочно ранжировать отдельные элементарные акты в процессах горения и окисления углеводородов и оценить вероятность различных направлений превращения химических компонентов в исследуемых системах.
ВВЕДЕНИЕ
В основе многих прикладных задач газохимии и горения лежат реакции окислительного превращения углеводородов [1–7], поэтому получение надежной информации о детальном механизме этих процессов является актуальной задачей. При этом важно иметь также данные о строении и химической активности возникающих в ходе превращения интермедиатов, в частности радикалов. В связи с этим достоверное описание реакций между молекулярными реагентами и активными частицами, возникающими в ходе окислительной конверсии углеводородов, является необходимым условием для моделирования этих сложных процессов.
Поскольку получение достоверной информации о реакциях короткоживущих частиц экспериментальным путем не всегда возможно, возникает необходимость использования различных методов вычислительной химии, в частности, квантовохимических расчетов. Например, в исследовании [8], в котором проведено квантовохимическое моделирование углеводородных соединений с высокой энтальпией образования, отмечено, что одним из таких подходов является компьютерный дизайн новых веществ, как правило, еще не синтезированных, но по различным соображениям перспективных для создания компонентов новых топлив.
Существует ряд вычислительных методов, позволяющих определять возможные пути превращения реагентов в многокомпонентных смесях, включая обнаружение ранее неизвестных химически активных частиц и стабильных соединений, а также нахождение геометрической структуры переходных состояний. Наиболее часто для проведения теоретического анализа используются методы B3LYP [9–11] и CBS [12–17]. По сравнению с другими гибридными методами теории функционала плотности (DFT) метод B3LYP предъявляет относительно низкие требования к используемым вычислительным ресурсам, обеспечивая при этом более высокую скорость работы. Методы CBS, использующие основанные на положениях квантовой химии ab initio расчеты более высокого уровня, значительно требовательнее к вычислительным ресурсам, но позволяют получать более точные результаты.
Многочисленными исследованиями показано, что газофазная окислительная конверсия углеводородов протекает с участием короткоживущих частиц с неспаренным электроном, атомов и радикалов, которые вносят существенный вклад в развитие процесса [18–20]. Простейшими реакционноспособными частицами в процессах окислительной конверсии углеводородов можно считать атомы водорода и кислорода. Квантовохимическое построение ППЭ их реакций в единой энергетической шкале позволяет более надежно, с общих позиций, соотносить их энергетические и геометрические характеристики. При этом очень важно установить связь между строением конкретного реагента и его интермедиатов и их реакционной способностью.
Взаимодействие возникающих в ходе окислительной конверсии углеводородов атомов водорода с этиленом представляет собой простейшие элементарные реакции реакционноспособных частиц, имеющих неспаренный электрон, с углеводородами олефинового ряда [21–23]. Наличие в системах окислительной конверсии углеводородов в газовой фазе атомов кислорода делает необходимым выяснение механизма взаимодействия атома кислорода с углеводородами и возникающими при этом промежуточными соединениями. Взаимодействие атома кислорода с двойной связью олефинов, в частности этилена, играет заметную роль как процессах окисления и горения углеводородов, так и в процессах, протекающих в атмосфере, поэтому достоверная информация об этой реакции также имеет большое значение. Теоретический анализ этой реакции проведен в работах [24–31], в которых использовали квантовохимические расчеты. Экспериментально определенное значение энергии активации (∆Hа) реакции присоединения атома кислорода к этилену, которое составляет 2 ккал/моль, приводится в базе данных NIST [32].
В настоящей работе поставлена задача теоретического исследования поверхностей потенциальной энергии, соответствующих реакциям в системе H–O–C2H4, включая обнаружение стабильных и метастабильных интермедиатов. С использованием различных квантовохимических методов также исследованы возможные каналы их дальнейшего превращения.
МЕТОДИКА РАСЧЕТОВ
Профили ППЭ были исследованы с использованием различных квантовохимических методов, в том числе гибридными методами B3LYP теории функционала плотности и ab initio расчетов в рамках композитного метода CBS-QB3. Оба метода входят в состав используемого в работе современного программного пакета Gaussian 16 [33]. Ввод исходных параметров реагентов и просмотр результатов счета проводили с использованием программы визуализации GaussView 6 [34], которая позволяет запускать расчеты из графического интерфейса без необходимости использования командной строки и упрощает интерпретацию выходных данных.
Для предварительного скрининга ППЭ применили метод B3LYP, поскольку он не требует особых вычислительных ресурсов. Этот метод, как правило, дает достаточно хорошие результаты при расчете ППЭ различных химических процессов и определении геометрических структур исходных реагентов и промежуточных соединений. Локализация экстремумов, соответствующих стабильным продуктам и химически активным интермедиатам, проводилась с применением стандартного набора базисных функций 6-31G(d), позволяющего получать надежные данные для систем с открытыми электронными оболочками [35, 36]. Однако в ряде случаев этот метод не учитывает наличие дисперсионных взаимодействий в реакциях с участием ненасыщенных соединений, поэтому он не всегда точно определяет величины барьеров реакций с переносом атомов водорода [37]. Для более точного анализа ППЭ с определением геометрических и энергетических параметров структур, локализованных методом B3LYP/6-31G(d), был выполнен расчет по методу CBS-QB3. Для нахождения переходных состояний на ППЭ исследуемой системы использовали алгоритмы Берни и квадратичного синхронного транзита STQN-QST2 [38, 39], которые являются составной частью программного пакета Gaussian 16. Все расчеты проводили для температуры T = 298.15 K и давления P = 1 атм.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В присутствии углеводородов, содержащих двойную связь, атом водорода может вступать в реакции присоединения и отрыва. Так, при взаимодействии с молекулой этилена атом водорода присоединяется к нему, образуя этильный радикал, или отрывает от него другой атом водорода с образованием молекулы водорода и ненасыщенного винильного радикала:
(1)
${\text{H}} + {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{4}} \leftrightarrow {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{5}},$(1а)
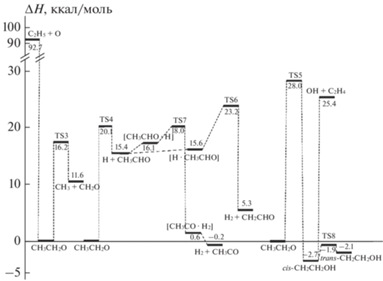
${\text{H}} + {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{4}} \leftrightarrow {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{3}} + {{{\text{H}}}_{2}}.$Как показали расчеты методом B3LYP/6-31G(d), наиболее вероятным с энергетической точки зрения каналом протекания реакции (1) является образование переходного состояния (TS2). Рассчитанная величина энергии активации этой реакции составляет 0.4 ккал/моль при изменении энтальпии реакции, составляющей –39.6 ккал/моль. Для более точного определения параметров был произведен расчет ППЭ этой реакции методом CBS-QB3, который дал значения энергии активации прямой реакции 0.6 ккал/моль, а обратной – 36.1 ккал/моль. При этом анализ полученных данных показал, что реакция присоединения атома водорода к этилену не является одностадийной, а представляет собой результат сложного процесса, протекающего в два этапа, с промежуточным образованием предреакционного ван-дер-ваальсового комплекса [H · C2H4] (рис. 1). Установлено также, что реакция (1а) отрыва атома водорода, как и реакция (1), тоже является многостадийным процессом, протекающим в три этапа с промежуточным образованием предреакционного и постреакционного ван-дер-ваальсовых комплексов ([C2H4 · H] и [C2H3 · H2]), и имеет переходное состояние (TS1), как это показано на рис. 1.
Рис. 1.
Диаграмма, описывающая сечения ППЭ для реакций (1) и (1а) системы H + C2H4, рассчитанные с использованием методов B3LYP/6-31G(d) и CBS-QB3. В скобках приведены значения, полученные методом B3LYP/6-31G(d).

В исследовании [40] геометрические параметры оптимизировались на уровне CCSD(T)/cc-pCVQZ, а величины энергии были вычислены с поправкой на релятивистские эффекты и приближение Борна–Оппенгеймера. Величина энергии активации, найденная для реакции (1а) отщепления атома водорода от молекулы этилена, составила 9.65 ккал/моль, что находится в хорошем согласии с нашими расчетами методами B3LYP (9.6 ккал/моль) и CBS-QB3 (8.9 ккал/моль). На основании существующих данных и результатов настоящего исследования можно утверждать, что реакция (1а) имеет значительно более высокую энергию активации, чем реакция (1) (табл. 1) и, следовательно, должна сильно уступать ей в скорости. Найденные нами относительные величины энергии Гиббса (ΔG) и энтальпии (ΔH) экстремумов ППЭ реакционной системы H + C2H4 приведены в табл. 1.
Таблица 1.
Параметры экстремумов на ППЭ системы H + C2H4, рассчитанные методами B3LYP/6-31G(d) и CBS-QB3
| Система | ΔG а, ккал/моль | ΔH б, ккал/моль | ν$_{1}^{в}$, см–1 | |||
|---|---|---|---|---|---|---|
| B3LYP/6-31G(d) | CBS-QB3 | B3LYP/6-31G(d) | CBS-QB3 | B3LYP/6-31G(d) | CBS-QB3 | |
| H + C2H4 | 0 | 0 | 0 | 0 | ||
| [C2H4 · H] | 2.8 | 1.2 | 0.2 | 0.3 | ||
| TS1 | 16.1 | 15.4 | 9.6 | 8.9 | –984.82 | –934.78 |
| [C2H3 · H2] | 7.5 | 5.7 | 5.4 | 4.5 | ||
| C2H3 + H2 | 3.5 | 2.5 | 4.8 | 3.8 | ||
| [H · C2H4] | –3.6 | 2.5 | 0.08 | 0.06 | ||
| TS2 | 4.9 | 6.2 | –0.4 | 0.6 | –203.88 | –309.2 |
| C2H5 | –34.1 | –30.0 | –39.6 | –35.5 | ||
Рассчитанная методом B3LYP/6-31G(d) величина энергии активации для реакции, обратной реакции (1), составила 39.2 ккал/моль, что находится в хорошем согласии со значением 38 ккал/моль, определенным в работе [41]. Несколько отличающиеся результаты теоретического изучения обратимой реакции (1) получены в работе [42], в которой приведены следующие значения: ΔHа = = 5.1 ккал/моль (ΔH° = –43.4 ккал/моль) и в работе [43] – ΔHа = 3.1 ккал/моль (с интервалом значений энтальпии ΔH° от –37.4 до –40.1 ккал/моль). Различия между расчетными величинами энергии активации и теплового эффекта реакции (1) из работы [41], нашего исследования и данными из работ [42–44], по всей видимости, обусловлены использованием различных методов расчета.
Экспериментально измеренные величины энергетических параметров рассматриваемой реакции (1) приведены также в работах [45, 46]. Получены следующие оценочные значения: ΔHа = = 0.5–7.0 ккал/моль [45] и ΔH° = –39.6 ккал/моль [46].
Приведенные выше результаты экспериментов и данные теоретических работ показывают, что, результатом конкуренции реакций (1) и (1а) должно быть образование преимущественно этильного радикала C2H5. Поскольку в реальных процессах окислительной конверсии углеводородов и их производных всегда образуется атомарный кислород, то естественно ожидать, что он будет вступать во взаимодействие с алкильными радикалами, в частности с этильным радикалом, что может привести к образованию этоксильного радикала по следующей реакции:
(2)
${\text{O}} + {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{5}} \to {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{O}}.$Тепловой эффект реакции присоединения атома кислорода к этильному радикалу с образованием этоксильного радикала, вычисленный на основе существующих данных [47–49], составляет ΔH ° = –91.72 ккал/моль. Это значение хорошо согласуется с полученной нами методом CBS-QB3 величиной, равной –92.70 ккал/моль.
В связи с тем, что этоксильный радикал играет важную роль при окислительной конверсии углеводородов, была поставлена задача исследования поверхности потенциальной энергии этого радикала с целью установления возможных каналов его превращения. Результаты проведенных вычислений, направленных на поиск экстремумов на ППЭ системы CH3CH2O, приведены в табл. 2. Из данных этой таблицы следует, что постреакционный комплексный ассоциат [H2 · CH2CHO] реакции H + CH3CHO → H2 + CH2CHO не удалось локализовать методом CBS-QB3. Для всех остальных реакций комплексные ассоциаты были локализованы на соответствующих ППЭ как методом B3LYP, так и CBS-QB3. С целью выяснения причин обнаруженного несоответствия проведены дополнительные расчеты с использованием других методов теории функционала плотности – M062X/6-31G(d,p) и UwB97XD/6-31G(d,p), входящими в состав программного пакета Gaussian 16 [33]. Оба метода, как и метод B3LYP, показали наличие пост-реакционного комплексного ассоциата [H2 · CH2CHO] реакции H + CH3CHO → → H2 + CH2CHO. По всей видимости, такой результат, полученный для данной конкретной реакции методом CBS-QB3, связан со спецификой применяемого этой программой алгоритма. В проанализированных литературных источниках отсутствуют как теоретические, так и экспериментальные данные, свидетельствующие об обнаружении другими исследователями найденных нами ван-дер-ваальсовых комплексов.
Таблица 2.
Относительные величины энтальпии и свободной энергии Гиббса экстремумов на ППЭ и значения мнимых частот переходных состояний системы C2H4–H–O, рассчитанные методами B3LYP/6-31G(d) и CBS-QB3
| Система | ΔH а, ккал/моль | ΔG б, ккал/моль | ν$_{1}^{в}$, см–1 | |||
|---|---|---|---|---|---|---|
| B3LYP/6-31G(d) | CBS-QB3 | B3LYP/6-31G(d) | CBS-QB3 | B3LYP/6-31G(d) | CBS-QB3 | |
| CH3 + CH2O | 14.70 | 11.58 | 2.99 | 1.14 | ||
| H + CH3CHO | 19.01 | 15.41 | 11.43 | 9.04 | ||
| H2 + CH2CHO | 7.07 | 5.32 | –1.38 | –1.93 | ||
| H2 + CH3CO | 2.58 | –0.19 | –6.58 | –8.14 | ||
| OH + C2H4 | 33.62 | 25.42 | 23.76 | 16.78 | ||
| [CH3CHO · H] | 18.84 | 16.13 | 15.36 | 13.53 | ||
| [CH3CO · H2] | 2.91 | 0.57 | –1.76 | –3.63 | ||
| [H · CH3CHO] | 19.13 | 15.63 | 14.51 | 11.02 | ||
| [H2 · CH2CHO] | 7.90 | * | 1.57 | * | ||
| cis-CH2CH2OH | 4.40 | –2.69 | 3.97 | –1.90 | ||
| trans-CH2CH2OH | 5.7 | –2.1 | 4.9 | –1.7 | ||
| CH3CH2O | 0 | 0 | 0 | 0 | ||
| TS5 (CH3CH2O ↔ CH2CH2OH) | 31.39 | 27.98 | 31.92 | 29.71 | –2019.62 | –1986.17 |
| TS3 (CH3CH2O ↔ CH3 + CH2O) | 17.73 | 16.21 | 16.63 | 16.42 | –301.86 | –340.61 |
| TS6 (H + CH3CHO ↔ ↔ H2 + CH2CHO) | 22.39 | 23.22 | 21.85 | 23.83 | –1388.79 | –1304.39 |
| TS4 (CH3CH2O ↔ H + CH3CHO) | 22.59 | 20.09 | 22.16 | 20.81 | –815.03 | –813.73 |
| TS7 (H + CH3CHO ↔ ↔ H2 + COCH3) | 17.27 | 17.96 | 16.26 | 17.82 | –702.24 | –128.34 |
| TS8 (cis-CH2CH2OH ↔ ↔ trans-CH2CH2OH) | 5.94 | –1.92 | 5.60 | –1.06 | –275.28 | –268.76 |
Образовавшийся в реакции (2) этоксильный радикал CH3CH2O вследствие высокой химической активности может участвовать в реакциях мономолекулярного распада и изомеризации. В качестве вероятных реакций были проанализированы следующие:
(3а)
${\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} \leftrightarrow {\text{C}}{{{\text{H}}}_{{\text{3}}}} + {\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{O}},$(3б)
${\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} \leftrightarrow {\text{H}} + {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CHO}},$(3в)
${\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} \leftrightarrow {\text{OH}} + {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{4}},$(3г)
${\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} \leftrightarrow {\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}},$(3д)
${\text{H}} + {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CHO}} \leftrightarrow {{{\text{H}}}_{{\text{2}}}} + {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CO}},$(3е)
${\text{H}} + {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CHO}} \leftrightarrow {{{\text{H}}}_{2}} + {\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{CHO}}.$На рис. 2 приведена диаграмма энтальпии, соответствующая сечениям ППЭ для реакции C2H5 + O. Как видно из этого рисунка, образование этоксильного радикала по реакции (2) протекает с нулевой энергией активации. Действительно, расчеты показали, что при изменении длины связи в аддукте CH3CH2 · O до значений 3.85 Å происходит монотонное возрастание полной энергии системы, что свидетельствует об отсутствии переходного состояния.
Рис. 2.
Диаграмма энтальпии для реакционной системы C2H5 + O, рассчитанной методом CBS-QB3. Экстремумы ППЭ приведены относительно структуры CH3CH2O.
Радикал C2H5O может также образоваться при взаимодействии этилена с гидроксильным радикалом. На возможность такого канала указывается в работе [50]. Согласно нашим расчетам, такое направление возможно, но при этом реакция проходит через промежуточное образование радикала CH2CH2OH.
Из рис. 2 видно, что гидроксиэтильный радикал, образующийся по реакции (3г) с ΔH° = = ‒2.7 ккал/моль, может иметь две различающиеся пространственные структуры. На рис. 3 представлены структуры cis- и trans-конформеров этого радикала, а на рис. 2 и в табл. 2 приводятся их энергетические параметры.
Рис. 3.
Пространственные структуры конформеров (a, б) радикала CH2CH2OH, рассчитанные методом CBS-QB3.
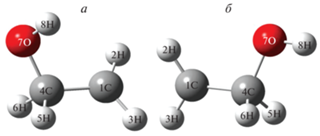
Диаграмма на рис. 2 показывает, что существует также возможность мономолекулярного распада гидроксиэтильного радикала по реакции
(4)
$cis{\text{ - }}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}} \to {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{4}} + {\text{OH}}.$Рассчитанная энтальпия изомеризации cis-формы гидроксиэтильного радикала с последующим распадом на этилен и гидроксильный радикал по реакции (4) составляет ΔH° = 28.1 ккал/моль.
Переходные состояния на ППЭ удалось локализовать для следующих реакций: (3е) с энергией активации 7.8 ккал/моль, (3а) с энергией активации 16.2 ккал/моль, (3б) с энергией активации 20.1 ккал/моль, (3г) с энергией активации 28.0 ккал/моль, (3д) с энергией активации 2.6 ккал/моль.
Как видно из рис. 2, реакция (3в) является сложной и протекает в две стадии через промежуточное образование радикала CH2CH2OH, который распадается затем на этилен и гидроксильный радикал. Реакция (3е) также может идти в две стадии – на первом (промежуточном) этапе в реакции распада (3б) образуются ацетальдегид и атома водорода, которые затем могут взаимодействовать по двум каналам. Как следует из рис. 2, энергия активации реакции (3е) равна 7.8 ккал/моль, а энтальпия – 10.1 ккал/моль.
Мономолекулярные реакции изомеризации этоксильного радикала (3г), распада по связи C–H на ацетальдегид и атом водорода (3б), а также по связи C–C на формальдегид и метильный радикал (3а) являются важными реакциями продолжения цепей при окислении углеводородов [51]. Величина энергии активации этих каналов уменьшается в ряду TS5 > TS4 > TS3. Очевидно, что в таком же порядке должна расти вероятность мономолекулярных превращений этоксильного радикала. В частности, в работе [51] приведены следующие значения активационного барьера: для реакции (3а) – 17.1 ккал/моль, для реакции (3е) – 8.04 ккал/моль, а для реакции (3д) – 1.9 ккал/моль, что согласуется с полученными нами величинами.
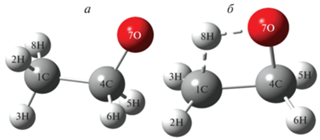
Энергетический барьер образования cis-конформера гидроксиэтильного радикала (28 ккал/моль) в результате изомеризации радикала C2H5O по реакции (3г) высок, что, скорее всего, является следствием возникновения напряжений в четырехчленной циклической структуре переходного состояния реакции внутримолекулярной миграции атома водорода. Для сравнения на рис. 4 приведены структуры этоксильного радикала и переходного состояния реакции (3г).
Рис. 4.
Пространственные структуры этоксильного радикала (а) и переходного состояния (б) реакции CH3CH2O ↔ cis-CH2CH2OH.
В последнее время реакция взаимодействия атомарного кислорода с этиленом привлекает большое внимание исследователей, поскольку она является фундаментальной стадией при окислительной конверсии и может служить прототипом реакций, в которых кислород присоединяется к двойной связи. Довольно подробно реакция (3)O + C2H4 исследована в работах [52–56], в которых проведены исследования поверхности потенциальной энергии методами B3LYP, G3, CBS-QB3, G2M(CC, MP2). Получены данные относительно энтальпии возможных направлений реакции этилена с атомом кислорода.
В настоящей работе на ППЭ системы, описывающей взаимодействие атома кислорода с этиленом, обнаружен ряд ранее не исследованных переходных состояний. Проанализированы ППЭ следующих возможных реакций этилена с атомом кислорода:
(5)
${{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}} + {\text{O}} \to {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{3}}}} + {\text{OH}},$Расчеты методом CBS-QB3, проведенные в данной работе и представленные на рис. 5, показали, что энергия активации реакции (5), имеющей триплетное переходное состояние, составляет 9.0 ккал/моль, что близко к значению 11 ккал/моль, приведенному в работе [52].
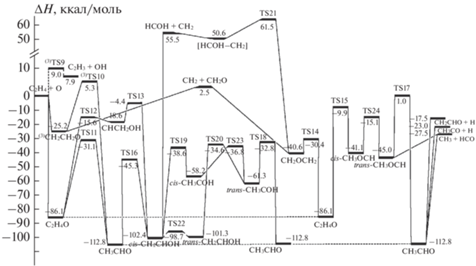
Согласно нашим расчетам реакция присоединения атома кислорода к этилену в синглетном состоянии системы протекает с нулевой энергией активации, а в триплетном, по данным работы [52], она имеет сравнительно небольшую энергию активации, равную 1.3 ккал/моль. В то же время большая энергия связи C–H в молекуле этилена (~110.7 ккал/моль), приводит к высокой энергии активации реакции отрыва. Из вышесказанного следует, что реакция (5) не может конкурировать с реакцией (5а). Поэтому реакции, связанные с образованием и превращениями винильного радикала в дальнейшем не рассматривались. Величины энтальпии и свободной энергии Гиббса для экстремумов на ППЭ, а также значения мнимых частот переходных состояний системы C2H4 + O, приведены в табл. 3.
Таблица 3.
Относительные величины энтальпия (ккал/моль) и свободной энергии Гиббса (ккал/моль) для экстремумов на ППЭ и значения мнимых частот переходных состояний системы C2H4 + O, рассчитанные методами B3LYP/6-31G(d) и CBS-QB3
| Система | B3LYP/6-31G(d) | CBS-QB3 | ν$_{1}^{в}$, см–1 | |||
|---|---|---|---|---|---|---|
| ΔHа | ΔG б | ΔH а | ΔG б | B3LYP/6-31G(d) | CBS-QB3 | |
| C2H4 + O* | 0 | 0 | 0 | 0 | ||
| CH2CH2O* | –31.31 | –23.69 | –25.15 | –17.54 | ||
| C2H3 + OH | 11.44 | 9.37 | 7.88 | 5.82 | ||
| HCOH + CH2 | 65.18 | 62.53 | 55.46 | 52.83 | ||
| CH3 + HCO | –29.61 | –33.30 | –27.49 | –31.13 | ||
| CH3CO + H | –24.43 | –24.47 | –22.99 | –23.03 | ||
| CH2CHO + H | –19.93 | –19.24 | –17.50 | –16.82 | ||
| CH2 + CH2O | 11.71 | 9.08 | 2.53 | –0.05 | ||
| C2H4O | –84.10 | –74.49 | –86.12 | –76.53 | ||
| CHCH2OH | –12.48 | –3.52 | –18.58 | –9.68 | ||
| CH2OCH2 | –39.56 | –30.93 | –40.61 | –31.85 | ||
| CH3CHO | –112.29 | –103.70 | –112.75 | –104.15 | ||
| cis-CH2CHOH | –96.57 | –87.48 | –102.36 | –93.28 | ||
| trans-CH2CHOH | –94.54 | –85.77 | –101.26 | –92.44 | ||
| cis-CH3COH | –54.47 | –45.87 | –58.17 | –49.64 | ||
| trans-CH3COH | –56.02 | –47.60 | –61.25 | –52.93 | ||
| cis-CH3OCH | –40.58 | –31.96 | –41.05 | –32.50 | ||
| trans-CH3OCH | –43.78 | –35.09 | –44.98 | –36.34 | ||
| [HCOH + CH2] | 54.29 | 62.42 | 50.55 | 58.41 | ||
| TS9 (C2H4 + O ↔ C2H3 + OH)* | 6.62 | 13.41 | 8.97 | 15.90 | –842.49 | –1172.74 |
| TS10 (CH2CH2O ↔ CH3CHO)* | 0.63 | 8.82 | 5.34 | 13.47 | –1779.30 | –1700.91 |
| TS11 (C2H4O ↔ CH3CHO) | –19.26 | –10.09 | –31.09 | –21.95 | –731.56 | –742.19 |
| TS12 (C2H4O ↔ CHCH2OH) | –10.46 | –1.04 | –15.57 | –6.14 | –1084.94 | –1180.27 |
| TS13 (CHCH2OH ↔ ↔ cis-CH2CHOH) | –0.21 | 9.14 | –4.44 | 4.89 | –873.71 | –878.54 |
| TS14 (C2H4O ↔ CH2OCH2) | –22.34 | –13.35 | –30.37 | –21.29 | –595.63 | –582.46 |
| TS15 (C2H4O ↔ cis-CH3OCH) | –6.36 | 3.09 | –9.92 | –0.50 | –681.42 | –617.56 |
| TS16 (CH3CHO ↔ ↔ cis-CH2CHOH) | –43.26 | –33.90 | –45.34 | –36.01 | –2200.32 | –2181.65 |
| TS17 (CH3CHO ↔ ↔ trans-CH3OCH) | 1.79 | 10.95 | 1.00 | 10.10 | –837.76 | –847.78 |
| TS18 (CH3CHO ↔ ↔ trans-CH3COH) | –28.34 | –19.91 | –32.75 | –24.36 | –1998.78 | –2016.23 |
| TS19 (cis-CH2CHOH ↔ ↔ cis-CH3COH) | –29.12 | –19.86 | –34.56 | –25.33 | –1490.81 | –1451.04 |
| TS20 (trans-CH2CHOH ↔ ↔ trans-CH3COH) | –31.82 | –22.57 | –38.60 | –29.36 | –1459.40 | –1416.09 |
| TS21 (CH2OCH2 ↔ ↔ HCOH + CH2) | 68.89 | 77.70 | 61.49 | 70.24 | –1476.44 | –1459.73 |
| TS22 (cis-CH2CHOH ↔ ↔ trans-CH2CHOH) | –91.66 | –82.36 | –98.73 | –89.44 | –429.88 | –440.18 |
| TS23 (cis-CH3COH ↔ ↔ trans-CH3COH) | –30.55 | –22.02 | –36.76 | –28.23 | –1124.67 | –1108.06 |
| TS24 (cis-CH3OCH ↔ ↔ trans-CH3OCH) | –15.32 | –6.79 | –15.14 | –6.62 | –1007.29 | –1029.98 |
В работе установлено, что результат реакции зависит от направления приближения триплетного атома кислорода к этилену и мультиплетности системы. При этом процесс протекает с нулевой энергией активации и может приводить к образованию бирадикала СН2СН2О в возбужденном триплетном состоянии, либо к оксиду этилена С2Н4O, что было установлено ранее на основании экспериментальных исследований [53].
Оксид этилена, имеющий большое практическое значение, может участвовать в реакции изомеризации с превращением в виниловый спирт. Процесс протекает через образование промежуточного соединения по реакции
(6)
${{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}{\text{O}} \to {\text{CHC}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}} \to cis{\text{ - C}}{{{\text{H}}}_{{\text{2}}}}{\text{CHOH}}.$Энтальпия переходного состояния ΔHа этого интермедиата из C2H4O составляет 70.5 ккал/моль, а ΔHа изомеризации CHCH2OH в cis-CH2CHOH равна 14.2 ккал/моль, т.е. обратимое превращение винилового спирта в оксид этилена не является одностадийным, а проходит через промежуточное состояние CHCH2OH. Полученные нами результаты расчетов энергий всех экстремумов согласуются с данными теоретических исследований, приведенными в работах [53–56].
Локализован также интермедиат CH2OCH2, который может образоваться из оксида этилена с ΔHа = 55.7 ккал/моль. Образовавшийся бирадикал CH2OCH2 может распадаться по реакциям
(7)
${\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OC}}{{{\text{H}}}_{2}} \leftrightarrow {\text{HCOH}} + {\text{C}}{{{\text{H}}}_{{\text{2}}}},$(7a)
${\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OC}}{{{\text{H}}}_{2}} \leftrightarrow {\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} + {\text{C}}{{{\text{H}}}_{2}},$Результаты проведенных расчетов не дают однозначных выводов о протекании тех или иных реакций. Тем не менее, полученные данные дают возможность оценочно ранжировать отдельные элементарные акты в процессах горения и окисления углеводородов и оценивать вероятность различных направлений превращения химических компонентов в исследуемых системах.
ЗАКЛЮЧЕНИЕ
Проведенное квантовохимическое исследование сечений поверхности потенциальной энергии систем C2H4 + H, C2H5 + O, C2H4 + O, локализация на ней различных реакционных интермедиатов и построение энтальпийных диаграмм возможных реакций позволяет сделать следующие выводы.
Этильный радикал образуется в результате реакции присоединения атома водорода к этилену, протекающей через образование промежуточного комплексного состояния [H · C2H4]. В присутствии атомарного кислорода одним из путей превращения этильного радикала является его взаимодействие с атомом кислорода, приводящее к образованию этоксильного радикала. Анализ ППЭ показал, что основными путями превращения этоксильного радикала являются многоканальные реакции его мономолекулярного распада и изомеризации. В случае распада этоксильного радикала на ацетальдегид и атом водорода последние могут реагировать между собой по нескольким направлениям через образование трех различных ван-дер-ваальсовских комплексов. Для реакции взаимодействия этилена с атомарным кислородом на ППЭ локализован ряд ранее неизвестных интермедиатов и оценены вероятности возможных путей их превращения. Полученные данные дают возможность оценочно ранжировать отдельные элементарные акты с участием химических компонентов в процессах окислительной конверсии углеводородов.
Исследование выполнено в рамках совместного научного проекта при финансовой поддержке Комитетом по науке Республики Армения (проект 20RF-002) и Российским фондом фундаментальных исследований (проект № 20-53-05001).
Список литературы
Ushakova A., Zatsepin V., Varfolomeev M., Emelyanov D. // J. Combust. 2017. V. 2017. Article 2526596; https://doi.org/10.1155/2017/2526596
Манташян А.А. // Хим. физика. 2021. Т. 40. № 4. С. 18; https://doi.org/10.31857/S0207401X21040105
Pogosyan N.M., Pogosyan M.Dj., Arsentiev S.D., Tavadyan L.A., Strekova L.N., Arutyunov V.S. // Pet. Chem. 2020. V. 60. № 3. P. 316; https://doi.org/10.1134/S0965544120030172
Grigoryan R.R., Arsentev S.D. // Pet. Chem. 2020. V. 60. № 2. P. 187; https://doi.org/10.1134/S096554412002005X
Погосян Н.М., Погосян М.Дж., Стрекова Л.Н., Тавадян Л.А., Арутюнов В.С. // Хим. физика. 2015. Т. 34. № 3. С. 35; https://doi.org/10.7868/S0207401X15030103
Паланкоева А.С., Беляев А.А., Арутюнов В.С. // Хим. физика. 2022. Т. 41. № 6. С. 7.
Арсентьев С.Д., Тавадян Л.А., Брюков М.Г., Паланкоева А.С., Беляев А.А., Арутюнов В.С. // Хим. физика. 2022. Т. 41. № 11. С. 3.
Волохов В.М., Зюбина Т.С., Волохов А.В., Амосова Е.С., Варламов Д.А. и др. // Хим. физика. 2021. Т. 40. № 1. С. 3; https://doi.org/10.31857/S0207401X21010131
Becke A.D. // Phys. Rev. A. 1988. V. 38. P. 3098; https://doi.org/10.1103/PhysRevA.38.3098
Becke A.D. // J. Chem. Phys. 1993. V. 98. P. 5648; https://doi.org/10.1063/1.464913
Lee C., Yang W., Parr R.G. // Phys. Rev. B. 1988. V. 37. P. 785; https://doi.org/10.1103/PhysRevB.37.785
Montgomery Jr.J.A., Frisch M.J., Ochterski J.W., Petersson G.A. // J. Chem. Phys. 1999. V. 110. № 6. P. 2822; https://doi.org/10.1063/1.477924
Nyden M.R., Petersson G.A. // Ibid. 1981. V. 75. № 4. P. 1843; https://doi.org/10.1063/1.442208
Petersson G.A., Al-Laham M.A. // Ibid. 1991. V. 94. № 9. P. 6081; https://doi.org/10.1063/1.460447
Petersson G.A., Tensfeldt T.G., Montgomery J.A. // Ibid. P. 6091; https://doi.org/10.1063/1.460448
Petersson G.A., Malick D.K., Wilson W.G., Ochterski J.W., Montgomery J.A., Frisch M.J. // Ibid. 1998. V. 109. № 24. P. 10570; https://doi.org/10.1063/1.477794
Montgomery Jr.J.A., Frisch M.J., Ochterski J.W., Petersson G.A. // Ibid. 2000. V. 112. № 15. P. 6532; https://doi.org/10.1063/1.481224
Arsentev S.D., Mantashyan A.A. // React. Kinet. Catal. Lett. 1980. V. 13. № 2. P. 125; https://doi.org/10.1007/BF02074183
Mantashyan A.A., Khachatryan L.A., Niazyan O.M., Arsentev S.D. // Combustion and Flame. 1981. V. 43. P. 221; https://doi.org/10.1016/0010-2180(81)90022-5
Манташян А.А., Едигарян Н.Г., Хачатрян Л.А., Арсентьев С.Д. // Химия высоких энергий. 1989. Т. 23. № 1. С. 63.
Yang K. // J. Amer. Chem. Soc. 1962. V. 84. № 5. P. 719; https://doi.org/10.1021/JA00864A007
Азатян В.В., Налбандян А.Б., Цуй М.-Ю. // Докл. АН СССР. 1963. Т. 149. № 5. С. 1095.
Jones W.E., Macknight S.D., Teng L. // Chem. Rev. 1973. V. 73. № 5. P. 407; https://doi.org/10.1021/CR60285A001
Dupuis M., Wendoloski J.J., Takada T., Lester Jr.W.A. // J. Chem. Phys. 1982. V. 76. № 1. P. 481; https://doi.org/10.1063/1.442748
Fueno T., Takahara Y., Yamaguchi K. // Chem. Phys. Lett. 1990. V. 167. № 4. P. 291; https://doi.org/10.1016/0009-2614(90)87170-V
Smith B.J., Nguyen M.T., Bouma W.J., Radom L. // J. Amer. Chem. Soc. 1991. V. 113. P. 6452; https://doi.org/10.1021/ja00017a015
Jursic B.S. // THEOCHEM. 1999. V. 492. № 1–3. P. 85; https://doi.org/10.1016/S0166-1280(99)00123-2
West A.C., Kretchmer J.S., Sellner B., Park K., Hase W.L. et al. // J. Phys. Chem. A. 2009. V. 113. № 45. P. 12663; https://doi.org/10.1021/jp905070z
Wortmann-Saleh D., Engels B., Peyerimhoff S.D. // J. Phys. Chem. 1994. V. 98. № 38. P. 9541; https://doi.org/10.1021/J100089A029
West A.C., Lynch J.D., Sellner B., Lischka H., Hase W.L. et al. // Theor. Chem. Acc. 2012. V. 131. Article 1123; https://doi.org/10.1007/s00214-012-1123-0
West A.C., Lynch J.D., Sellner B., Lischka H., Hase W.L. et al. // Ibid. Article 1279; https://doi.org/10.1007/s00214-012-1279-7
Westleyn F., Heron J.T., Cvetanovic R.J., Hampson R.F., Mallard W.G. NIST Standard Reference Database 17, Version 3.0. Gaithersburg, MD, USA National Institute of Standards and Technology, 1991.
Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman, J.R., Scalmani G., Barone V., Petersson G.A., Nakatsuji H., Li X., Caricato M., Marenich A.V., Bloino J., Janesko B.G., Gomperts R., Mennucci B., Hratchian H.P., Ortiz J.V., Izmaylov A.F., Sonnenberg J.L., Williams-Young D., Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V.G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., Montgomery J.A. Jr., Peralta J.E., Ogliaro F., Bearpark M.J., Heyd J.J., Brothers E.N., Kudin K.N., Staroverov V.N., Keith T.A., Kobayashi R., Normand J., Raghavachari K., Rendell A.P., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Millam J.M., Klene M., Adamo C., Cammi R., Ochterski J.W., Martin R.L., Morokuma K., Farkas O., Foresman J.B., Fox D.J. Gaussian 16, Revision C.01. Wallingford. CT: Gaussian, Inc., 2016.
Dennington R., Keith T.A., Millam J.M. GaussView, Version 6.1. Shawnee Mission, KS: Semichem Inc., 2019.
Asatryan R., Davtyan A., Khachatryan L., Dellinger B. // J. Phys. Chem. A. 2005. V. 109. P. 11198; https://doi.org/10.1021/jp053047l
Asatryan R.S., Davtyan A.H., Khachatryan L.A., Dellinger B. // Organohalogen Compd. 2002. V. 56. P. 277.
Jursic. B.S. // J. Chem. Soc., Perkin Trans. 2. 1997. V. 2. Issue 3. P. 637; https://doi.org/10.1039/A603269I
Peng C., Ayala P.Y., Schlegel H.B., Frisch M.J. // J. Comput. Chem. 1996. V. 17. № 1. P. 49; https://doi.org/10.1002/(SICI)1096-987X(19960115)17: 1<49::AID-JCC5>3.0.CO;2-0
Peng C., Schlegel H.B. // Isr. J. Chem. 1993. V. 33. P. 449; https://doi.org/10.1002/IJCH.199300051
Agarwal J., Turney J.M., Schaefer H.F. // J. Phys. Chem. Lett. 2011. V. 2. № 20. P. 2587; https://doi.org/10.1021/jz201124j
Schlegel H.B., Bhalla K.C., Hase W.L. // J. Phys. Chem. 1982. V. 86. № 25. P. 4883; https://doi.org/10.1021/J100222A010
Feng Y., Niiranen J.T., Benosura A., Knyazev V.D., Gutman D. et al. // Ibid. 1993. V. 97. P. 871.
Hase W.L., Schlegel H.B., Balbyshev V., Page M. // Ibid. 1996. V. 100. № 13. P. 5354; https://doi.org/10.1021/jp9528875
Hase W.L., Schlegel H.B. // Ibid. 1982. V. 100. P. 5354; https://doi.org/10.1021/JP9528875
Jones W.E., Macknight S.D., Teng L. // Chem. Rev. 1973. V. 73. № 5. P. 407; https://doi.org/10.1021/CR60285A001
Franklin J.L., Dillard J.G., Rosenstock H.M., Herron J.T., Draxl K., Field F.H. Ionisation potentials, appearance potentials, and heats of formation of gaseous positive ions (NSRDS–NRS26). Washington: US National Bur. eau of Standards, 1969.
Ruscic B., Pinzon R.E., Morton M.L., Srinivasan N.K., Su M.-C. et al. // J. Phys. Chem. A. 2006. V. 110. № 21. P. 6592; https://doi.org/10.1021/JP056311J
Berkowitz J., Ellison G.B., Gutman D. // J. Phys. Chem. 1994. V. 98. № 11. P. 2744; https://doi.org/10.1021/j100062a009
Ruscic B., Boggs J.E., Burcat A. et al. // J. Phys. Chem. Ref. Data. 2005. V. 34. № 2. P. 573; https://doi.org/10.1063/1.1724828
Golden D.M. // J. Phys. Chem. A. 2012. V. 116. № 17. P. 4259; https://doi.org/10.1021/jp302009t
http://c3.nuigalway.ie/combustionchemistrycentre/mechanismdownloads/
Nguyen T.L., Vereecken L., Hou X.J., Nguyen M.T., Peeters J. // J. Phys. Chem. A. 2005. V. 109. № 33. P. 7489; https://doi.org/10.1021/JP052970K
Knyazev V.D., Arutyunov V.S., Vedeneev V.I. // Intern. J. Chem. Kinet. 1992. V. 24. № 6. P. 545; https://doi.org/10.1002/KIN.550240605
Li X., Jasper A.W., Zádor J., Miller J.A., Klippenstein S.J. // Proc. Combust. Inst. 2016. V. 36. № 1. P. 219; https://doi.org/10.1016/j.proci.2016.06.053
Fu B., Han Y.C., Bowman J.M., Leonori F., Balucani N. et al. // J. Chem. Phys. 2012. V. 137. № 22. P. 22A532; https://doi.org/10.1063/1.4746758
Hu W., Lendvay G., Maiti B., Schatz G.C. // J. Phys. Chem. A. 2008. V. 112. № 10. P. 2093; https://doi.org/10.1021/jp076716z
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика