

Химическая физика, 2022, T. 41, № 5, стр. 3-9
Реакция молекулярной серы в электронно-возбужденном состоянии S2(a1Δg) с сероводородом
Е. М. Лозбень 1, 2, *, А. В. Лебедев 2, М. А. Деминский 1, 2, А. А. Грановский 2, Б. В. Потапкин 1, 2
1 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
2 “Кинтех Лаб”
Москва, Россия
* E-mail: lozben88@gmail.com
Поступила в редакцию 22.10.2021
После доработки 17.12.2021
Принята к публикации 20.12.2021
- EDN: EVBRHM
- DOI: 10.31857/S0207401X22050090
Аннотация
В формализме расширенной квазивырожденной теории возмущений (XMCQDPT2) проведены расчеты поверхности потенциальной энергии для реакции ${{{\text{S}}}_{2}}({{a}^{1}}{{\Delta }_{g}}) + {{{\text{H}}}_{{\text{2}}}}{\text{S}} \to {\text{SH}} + {\text{H}}{{{\text{S}}}_{2}}.$ Найденные геометрические параметры и частоты нормальных колебаний стабильных интермедиатов и переходных состояний использованы для оценки эффективной константы скорости реакции в рамках теории переходного состояния для температурного диапазона 500–2000 К. Для константы скорости найден вид аррениусовской зависимости.
ВВЕДЕНИЕ
Сероводород (H2S) занимает важное место в промышленной химии, энергетике и атмосферной физике, поскольку является побочным продуктом добычи природного газа и газификации промышленных отходов [1–3]. Помимо того, что сероводород пагубно влияет на состояние окружающей среды и здоровье [4], он также вызывает коррозию металла [5]. Для удаления сероводорода из природных и промышленных газов ранее широко использовался процесс Клауса: ${{{\text{H}}}_{{\text{2}}}}{\text{S}} + \frac{1}{2}{{{\text{O}}}_{2}} = {\text{S}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}$ [6, 7]. Однако этот способ очистки не позволяет использовать ${{{\text{H}}}_{{\text{2}}}}{\text{S}},$ как источник водорода для энергетической промышленности. В последние годы для получения водорода из ${{{\text{H}}}_{{\text{2}}}}{\text{S}}$ исследуются и применяются методы прямого и каталитического термолиза [8–16], фотокатализа [17], плазменной диссоциации [18–21] и диссоциации в микроволновом излучении [22, 23]. Для описания всех этих процессов необходим термический кинетический механизм разложения сероводорода. Детальный химический механизм распада ${{{\text{H}}}_{{\text{2}}}}{\text{S}}$ экспериментально и теоретически исследовали в работах [8, 9, 14, 24]. Реакция
(R1)
${{{\text{S}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{S}} \to {\text{SH}} + {\text{H}}{{{\text{S}}}_{2}}$В механизме распада сероводорода, разработанном в работе [9], реакция (R1) участвует в автокатализе:
(R1)
${{{\text{S}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{S}} \to {\text{SH}} + {\text{H}}{{{\text{S}}}_{{\text{2}}}},$(R2)
${\text{H}}{{{\text{S}}}_{2}} + {\text{M}} \to {\text{H}} + {{{\text{S}}}_{2}} + {\text{M}}{\text{.}}$Результирующий процесс можно переписать в виде ${{{\text{H}}}_{{\text{2}}}}{\text{S}} + {\text{M}}\xrightarrow{{{{{\text{S}}}_{{\text{2}}}}}}{\text{H}} + {\text{SH}} + {\text{M}}{\text{.}}$ В работе [9] показано, что несмотря на малое значение констант скоростей реакций (R1) и (R2), наличие небольшого количества серы в газовой смеси приводит к ускорению разложения сероводорода.
На сегодняшний день кинетические механизмы, описывающие разложение сероводорода в широком диапазоне температур, отсутствуют. В разработанных механизмах термолиза ${{{\text{H}}}_{{\text{2}}}}{\text{S}}$ до сих пор не учитывалось влияние присутствия в газе электронно-возбужденных частиц, однако в работах [12, 25] обсуждается возможность образования в газе молекул серы ${{{\text{S}}}_{2}}({{a}^{1}}{{\Delta }_{g}})$ в первом метастабильном возбужденном состоянии при рекомбинации атомов серы при температурах выше 1000 К. Принимая во внимание потенциальную значимость автокатализа [9], следует рассмотреть участие электронно-возбужденных частиц в химии распада сероводорода.
Образование молекулярной серы в электронно-возбужденном состоянии может происходить по разным причинам, например при термическом разложении H2S, которое сопровождается образованием атомарной серы S(3P). Также это состояние возникает в среде неравновесных разрядов. Дальнейшая рекомбинация атомарной серы при относительно низких температурах приводит к образованию молекулярной серы (S2) в возбужденных электронных состояниях. Несмотря на то, что этот процесс является трехчастичным, при высоких давлениях он также может быть заметным источником молекул S2 в возбужденных электронных состояниях. К примеру, в серных лампах высокого давления излучение формируется за счет атомов серы в возбужденных электронных состояниях, а основным процессом образования возбужденных молекул является рекомбинация атомов в области низких температур при давлениях от 1 атм и выше [26, 27]. При этом первое возбужденное ${{{\text{S}}}_{2}}({{a}^{1}}{{\Delta }_{g}})$ состояние медленно распадается до основного ${{{\text{S}}}_{2}}({{X}^{3}}\Sigma ),$ поскольку этот переход формально запрещен по спину. Аналогичная ситуация имеет место в случае неравновесной плазмы, в которой кроме атомов S(1D) также присутствуют атомы S(3P).
Нередко реакции с участием молекул в возбужденном состоянии протекают быстрее, чем соответствующие реакции с основными состояниями из-за “улучшения” термохимии реакции (разница энтальпий образования ${{{\text{S}}}_{2}}({{a}^{1}}{{\Delta }_{g}})$ и ${{{\text{S}}}_{2}}({{X}^{3}}\Sigma )$ составляет около ΔH0 = 12.7 ккал/моль). Однако реальное ускорение реакции определяется не только энергией, но и наличием или отсутствием барьеров на пути реакции:
(R3)
${{{\text{S}}}_{2}}\left( {{{a}^{1}}{{\Delta }_{g}}} \right) + {{{\text{H}}}_{{\text{2}}}}{\text{S}} \leftrightarrow {\text{SH}} + {\text{H}}{{{\text{S}}}_{2}}.$Поэтому настоящая работа посвящена теоретическому исследованию механизма реакции ${{{\text{H}}}_{{\text{2}}}}{\text{S}}$ с молекулой ${{{\text{S}}}_{2}}({{a}^{1}}{{\Delta }_{g}}).$ Влияние этого процесса на общую кинетику разложения сероводорода не исследовалось, так как для этого необходимо провести детальный кинетический анализ с участием других конкурирующих каналов. Среди них, например, конкуренция химической реакции (R3) и тушения серы в возбужденном состоянии на частицах, присутствующих в газе, а также образование серы в электронных состояниях, с энергиями, превышающими энергию 1D-состояния и дальнейшее участие электронно-возбужденных частиц в химической кинетике. Также следует учитывать трехчастичную рекомбинацию атомарной серы, приводящую к образованию метасбильной молекулы ${{{\text{S}}}_{2}}({{a}^{1}}{{\Delta }_{g}}).$ Таким образом, данная работа является первым шагом в теоретическом исследовании влияния электронно-возбужденных молекул серы на кинетику разложения сероводорода.
МЕТОДЫ РАСЧЕТА
Расчет константы скорости реакции (R3) включал две стадии. На первой из них был проведен поиск стационарных точек поверхности потенциальной энергии (ППЭ) реакции и найдены положения интермедиатов и переходных состояний (transition state (TS)). Важно отметить, что рекомбинация атомарной серы происходит при относительно низких температурах (1000–2500 К), что требует от применяемого квантовохимического метода высокой точности расчета. На следующей стадии проводили оценку константы скорости реакции с применением стохастического подхода и теории переходных состояний.
Для реакции (R3) ППЭ рассчитывали с помощью пакета Firefly QC [28], который частично основан на исходном коде GAMESS [29]. Волновые функции нулевого порядка получены из расчетов многоконфигурационным методом самосогласованного поля в полном активном пространстве (CASSCF) с 16 электронами, распределенными по 12 активным орбиталям, и усреднением по четырем низшим синглетным состояниям (SA4). Затем энергии синглетных состояний корректировались в рамках расширенной квазивырожденной теории возмущений второго порядка (XMCQDPT2) [30]. Во всех расчетах был использован корреляционно-согласованный базисный набор cc-pVTZ.
Кроме ППЭ реакции (R3) была рассчитана и ППЭ реакции (R1) в приближении функционала плотности (DFT) с гибридным функционалом Беке–Ли–Янга–Парра (B3LYP) [31]. Здесь использовали базис 6-311, дополненный диффузными функциями p-, d- и f-типов. Этот выбор обеспечивает точность термохимических данных на уровне 1–2 ккал/моль. Действительно, рассчитанная энтальпия реакции (R1) ΔH298 = 32.3 ккал/моль оказалась близка к данным из других источников [8, 32]: ΔH298 = 32.3–33.2 ккал/моль. Отметим, что выбор приближения функционала плотности для расчета ППЭ реакции (R1) обусловлен тем фактом, что константа скорости реакции (R1) хорошо известна, и точность подтверждается совпадением вычисленной константы скорости с результатами ранее проведенных исследований [8], как показано в разд. “Результаты и их обсуждение”. Приближение функционала плотности уступает в точности расчета энергий молекул и переходных состояний многоконфигурационным ab initio методам, но его применение значительно сокращает время расчета и требования к вычислительной мощности. Более точный подход применен для реакции (R3), поскольку приближение функционала плотности не может обеспечить высокой точности при расчете ППЭ возбужденных электронных состояний.
Константу скорости реакции (R3) оценивали с помощью зависящего от времени управляющего уравнения с использованием программного пакета “MultiWell” [33]. Оценка проведена для давления, равного 1 атм, так как большинство экспериментальных исследований термического разложения сероводорода проводятся при атмосферном давлении. Для проверки полученных результатов и из-за отсутствия достоверных данных по константе скорости реакции (R3) дополнительно проведены расчеты с использованием канонической теории переходного состояния. В качестве параметров при расчетах константы скорости были использованы геометрия и частоты нормальных колебаний реагентов, переходных состояний и промежуточных комплексов, полученные в квантовохимических расчетах. Для безбарьерной реакции положение виртуального TS определяли с использованием модели переходного состояния Горина [34]. Константу скорости реакции (R1) рассчитывали с помощью программного комплекса Химера [35] в рамках канонической теории переходного состояния с использованием результатов квантовохимических расчетов.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Поверхность потенциальной энергии реакций (R1), (R3)
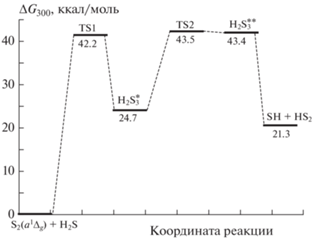
На рис. 1 изображены энергетические профили путей реакций (R1) и (R2). Для удобства анализа энергия отсчитывается от энергии реагентов реакции (R3), т.е. энергия реагентов реакции (R3) принята за 0 ккал/моль. Значения энергии Гиббса стационарных точек относительно энергии реакции (R3) представлены на рис. 2.
Рис. 1.
Стационарные точки на поверхности потенциальной энергии для реакций (R1) и (R3) и структуры соответствующих переходных состояний (TS1, TS2, TS3) и минимумов; крупные шарики – атомы серы, мелкие – атомы водорода.
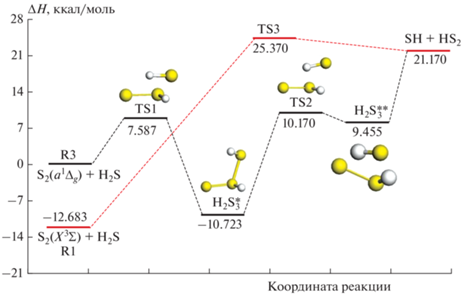
Хотя между величинами энергий реагентов и комплекса в переходном состоянии TS3 существует разница, реакцию (R1) можно рассматривать как безбарьерную. Действительно, энтальпия переходного состояния TS3 близка к энтальпии продуктов, поэтому в пределах ошибки вычислений реакция не имеет явного локального максимума вдоль реакционного пути, что соответствует модели разрыхленного переходного комплекса [36]. Разница энтальпий образования TS3 и продуктов реакции (SH + HS2) лишь немного превышает точность выбранного метода. Этот результат согласуется с выводом исследования, проведенного в работе [8]. Корректность рассчитанной ППЭ подтверждается сравнением с имеющимися термохимическими данными для реагентов и продуктов [8]:
Реакция (R3) протекает через переходное состояние TS1 с образованием в точке глобального минимума ${{{\text{H}}}_{{\text{2}}}}{\text{S}}_{3}^{*}$ с энтальпией ΔH0 = –10.723 ккал/моль. Первая ступень реакции имеет барьер ΔH0 = = 7.587 ккал/моль. Сформированная структура глобального минимума ${{{\text{H}}}_{{\text{2}}}}{\text{S}}_{3}^{*}$ обладает конфигурацией пирамидального типа. Структура молекулы ${{{\text{H}}}_{{\text{2}}}}{\text{S}}_{3}^{*}$ образована центральным атомом S с валентностью IV и двумя другими атомами S с валентностью II. Длина связи между атомами серы с разными валентностями составляет 2.189 Å. Прямое формирование продуктов невозможно, потому что сначала нужно разорвать эту связь. Поэтому происходит образование второго промежуточного состояния ${{{\text{H}}}_{{\text{2}}}}{\text{S}}_{3}^{{{\text{**}}}}.$ Реакция идет путем образования второго локального слабосвязанного минимума ${{{\text{H}}}_{{\text{2}}}}{\text{S}}_{3}^{{{\text{**}}}}$ через второе переходное состояние TS2. Разница энтальпий при температуре 0 К этих состояний составляет всего 0.715 ккал/моль. Этот минимум имеет такую топологию, что расстояние между атомами серы составляет 3.945 Å, и он готов к образованию продуктов без явного образования переходного состояния. Это возможно, поскольку ${{{\text{H}}}_{{\text{2}}}}{\text{S}}_{3}^{{{\text{**}}}}$ имеет структуру образующихся продуктов, сформированную за счет диполь-дипольного взаимодействия. Эта особенность – образование дополнительного связного состояния – важна для микрокинетического анализа, поскольку промежуточное состояние очень нестабильно по отношению к обратному процессу. Наконец, слабосвязанный минимум соответствует продуктам реакции SH и HS2. Поскольку энергия Гиббса комплекса ${{{\text{H}}}_{{\text{2}}}}{\text{S}}_{3}^{{{\text{**}}}}$ выше, чем энергия Гиббса продуктов (рис. 2), он не учитывался при расчете константы скорости.
Структуры промежуточных комплексов приведены на рис. 3а–г. Координаты атомов стационарных точек и термодинамические характеристики представлены в табл. 1–8. Частоты нормальных колебаний приведены в табл. 9, 10.
Рис. 3.
Структуры промежуточных комплексов: а – TS1, б – ${{{\text{H}}}_{{\text{2}}}}{\text{S}}_{3}^{*},$ в – TS2, г – ${{{\text{H}}}_{{\text{2}}}}{\text{S}}_{3}^{{{\text{**}}}}.$
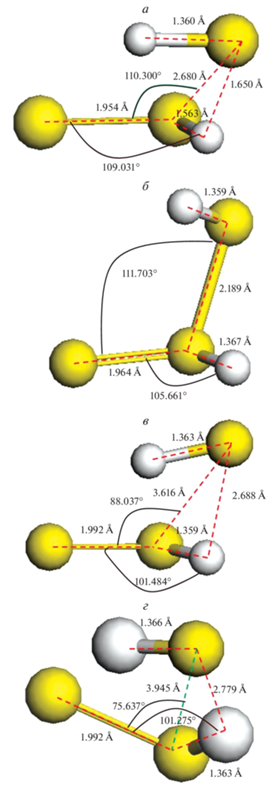
Таблица 1.
Координаты и заряд атомов в переходном состоянии TS1
| Атом | Заряд, е | X, Å | Y, Å | Z, Å |
|---|---|---|---|---|
| S | 16.0 | 0.0141 | –0.1977 | 1.0091 |
| S | 16.0 | 0.7971 | –0.5056 | –0.7548 |
| H | 1.0 | 1.0910 | 0.4501 | 1.9389 |
| S | 16.0 | 1.8334 | –0.6434 | 2.9260 |
| H | 1.0 | 2.6943 | –0.8239 | 1.8888 |
Таблица 2.
Термодинамические характеристики переходного состояния TS1
| T, K | ΔH, ккал/моль | ΔG, ккал/моль |
|---|---|---|
| 300 | 6.9 | 42.2 |
Таблица 3.
Координаты и заряд атомов в точке ${{{\mathbf{H}}}_{{\mathbf{2}}}}{\mathbf{S}}_{{\mathbf{3}}}^{{\mathbf{*}}}$ минимума
| Атом | Заряд, е | X, Å | Y, Å | Z, Å |
|---|---|---|---|---|
| S | 16.0 | 0.1632 | –0.2013 | 1.1662 |
| S | 16.0 | 0.8307 | –0.5355 | –0.6501 |
| H | 1.0 | 0.2973 | 1.1443 | 1.3669 |
| S | 16.0 | 1.6767 | –0.6547 | 2.6809 |
| H | 1.0 | 2.6660 | –0.0981 | 1.9343 |
Таблица 4.
Термодинамические характеристики ${{{\mathbf{H}}}_{{\mathbf{2}}}}{\mathbf{S}}_{{\mathbf{3}}}^{{\mathbf{*}}}$
| T, K | ΔH, ккал/моль | ΔG, ккал/моль |
|---|---|---|
| 300 | –11.7 | 24.7 |
Таблица 5.
Координаты и заряд атомов в переходном состоянии TS2
| Атом | Заряд, е | X, Å | Y, Å | Z, Å |
|---|---|---|---|---|
| S | 16.0 | –0.1854 | 0.1710 | 0.6128 |
| S | 16.0 | 1.0087 | –0.6225 | –0.7696 |
| H | 1.0 | 0.7133 | 0.7828 | 1.4277 |
| S | 16.0 | 2.3726 | –0.5589 | 3.0628 |
| H | 1.0 | 2.6279 | –0.9406 | 1.7792 |
Таблица 6.
Термодинамические характеристики переходного состояния TS2
| T, K | ΔH, ккал/моль | ΔG, ккал/моль |
|---|---|---|
| 300 | 9.9 | 43.5 |
Таблица 7.
Координаты и заряд атомов ${{{\mathbf{H}}}_{{\mathbf{2}}}}{\mathbf{S}}_{{\mathbf{3}}}^{{{\mathbf{*}}{\kern 1pt} {\mathbf{*}}}}$ в точке минимума
| Атом | Заряд, е | X, Å | Y, Å | Z, Å |
|---|---|---|---|---|
| S | 16.0 | –0.3486 | 0.2416 | 0.4604 |
| S | 16.0 | 1.0740 | –0.6328 | –0.6261 |
| H | 1.0 | 0.2092 | 0.2115 | 1.7039 |
| S | 16.0 | 2.4903 | –0.6051 | 3.0651 |
| H | 1.0 | 2.5000 | –0.8582 | 1.7228 |
Таблица 8.
Термодинамические характеристики ${{{\mathbf{H}}}_{{\mathbf{2}}}}{\mathbf{S}}_{{\mathbf{3}}}^{{{\mathbf{*}}{\kern 1pt} {\mathbf{*}}}}$
| T, K | ΔH, ккал/моль | ΔG, ккал/моль |
|---|---|---|
| 300 | 9.6 | 43.4 |
Таблица 9.
Частоты (в см–1) нормальных колебаний элементов реакции (R3)
| ${{{\text{S}}}_{2}}({{a}^{1}}{{\Delta }_{g}})$ | ${{{\text{H}}}_{{\text{2}}}}{\text{S}}_{3}^{*}$ | ${\text{TS1}}_{3}^{*}$ | ${{{\text{H}}}_{{\text{2}}}}{\text{S}}_{3}^{*}$ |
|---|---|---|---|
| 669.56 | 1136.24 | 1035.29*i | 152.51 |
| 2624.94 | 116.86 | 240.54 | |
| 2657.80 | 178.27 | 368.73 | |
| 244.20 | 586.15 | ||
| 565.61 | 702.11 | ||
| 633.20 | 800.93 | ||
| 1011.35 | 890.00 | ||
| 1376.35 | 2309.20 | ||
| 2585.59 | 2584.20 |
Таблица 10.
Частоты (в см–1) нормальных колебаний элементов реакции (R3)
| ${\text{TS2}}_{3}^{*}$ | ${{{\text{H}}}_{{\text{2}}}}{\text{S}}_{3}^{{*{\kern 1pt} *}}$ | ${\text{SH}}_{3}^{*}$ | ${\text{HS}}_{2}^{*}$ |
|---|---|---|---|
| 158.32*i | 66.09 | 2590.06 | 573.84 |
| 59.81 | 82.19 | 873.41 | |
| 86.08 | 162.49 | 2487.76 | |
| 152.04 | 176.90 | ||
| 233.52 | 231.87 | ||
| 573.53 | 579.66 | ||
| 904.73 | 894.73 | ||
| 2501.15 | 2458.51 | ||
| 2569.72 | 2540.48 |
Расчет константы скорости
Результирующий процесс (R3) представлен последовательностью двух реакций (R3*) с прямой и обратной константами скорости k1, –1 и k2, –2: первая реакция – это образование стабильного промежуточного комплекса. ${{{\text{H}}}_{{\text{2}}}}{\text{S}}_{3}^{{\text{*}}}$ с явным жестким переходным состоянием TS1; вторая реакция – распад стабильного промежуточного комплекса на продукты без явного барьера, как это показано на рис. 4.
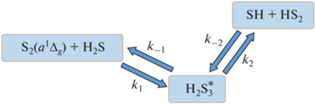
В первом процессе система проходит потенциальный барьер высотой ΔH0 = 7.587 ккал/моль через переходное состояние TS1. Диссоциация промежуточного комплекса в область продуктов является эндотермическим процессом с ΔH0 = 31.893 ккал/моль. Однако комплекс ${{{\text{H}}}_{{\text{2}}}}{\text{S}}_{3}^{{\text{*}}}$(v) образуется в колебательно-возбужденном состоянии, и реакция идет через химически активированный комплекс. Это означает, что реальное значение ΔH(TS1) может быть намного меньше, а скорость распада определяется процессом столкновительно-колебательной релаксации ${{{\text{H}}}_{{\text{2}}}}{\text{S}}_{3}^{{\text{*}}}{\text{:}}$
Эффективная константа скорости сложной химической реакции (R3) оценена методом квазистационарных концентраций Боденштейна (M. Bodenstein):
Как упоминалось ранее, константа скорости реакции (R1) была рассчитана с использованием канонической теории переходного состояния. На рис. 5 представлены результаты приведенных выше расчетов константы скорости реакции молекулярной серы в основном состоянии с сероводородом (R1), полученные в этой работе и работе [8], а также константа скорости реакции молекулярной серы в возбужденном состоянии с сероводородом (R3). Участие электронно-возбужденного состояния двухатомной молекулы S2 приводит к существенному ускорению реакции. Отметим, что из-за сложного характера ППЭ (наличие стабильного промежуточного комплекса, наличие барьеров для разложения комплекса реагентами) константа скорости реакции (R3) не может быть оценена простым уменьшением энергии активации на величину энергии возбуждения: ∆H0 = –H0(${{{\text{S}}}_{2}}({{a}^{1}}{{\Delta }_{g}})$) – ‒ ∆H0(${{{\text{S}}}_{2}}({{X}^{3}}\Sigma )$) = 12.68 ккал/моль.
Рис. 5.
Температурные зависимости констант скорости для реакций (R1) и (R3): сплошная тонкая линия – результат для реакции (R3) из канонической теории переходного состояния; звездочки – константа для реакции (R3) из решения управляющего уравнения (заштрихованная область – оценка погрешности); штриховая линия – константа скорости реакции (R1), полученная в этой работе; сплошная жирная линия – константа для реакции (R1), полученная в исследовании [8].
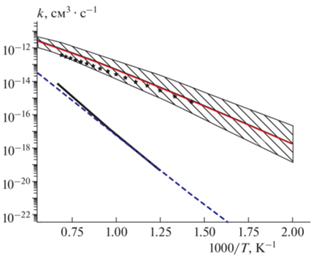
Как и ожидалось, константа скорости реакции (R3) намного выше, чем константа скорости реакции (R1). Для характерной температуры T = = 1000 K константы различаются более чем на четыре порядка. Повышение температуры приводит к “сближению” значений констант скоростей реакции с участием молекул в основном и возбужденном состояниях за счет влияния тепловой энергии в реакции. Аррениусовский вид констант скорости реакций описывается следующими зависимостями:
ЗАКЛЮЧЕНИЕ
Константа скорости реакции сероводорода с серой в электронно-возбужденном состоянии была рассчитана из первых принципов (ab initio). Реакция имеет сложную поверхность потенциальной энергии со стабильными промежуточными состояниями. Эти особенности, наряду с участием в реакции серы в возбужденном метастабильном состоянии, ${{{\text{S}}}_{2}}({{a}^{1}}{{\Delta }_{g}}),$ приводят к значительному (на несколько порядков) увеличению константы скорости по сравнению с реакцией молекулы серы в основном состоянии. Полученный результат важен для анализа кинетики разложения сероводорода в разрядах различного типа, исследованиях каталитических процессов в которых образуется электронно-возбужденная двухатомная молекула серы ${{{\text{S}}}_{2}}({{a}^{1}}{{\Delta }_{g}}).$
Приведенные в статье результаты квантомеханических расчетов были выполнены Александром Александровичем Грановским – известным российским квантовым химиком, создателем программного пакета ab initio расчетов Firefly. Александр Александрович очень много сделал для развития высокоэффективных алгоритмов квантовохимических методов. Авторы глубоко сожалеют о том, что Александра Александровича более нет с нами, данная публикация была бы невозможной без его определяющего участия.
Список литературы
Кислов В.М., Цветкова Ю.Ю., Глазов С.В. и др. // Хим. физика. 2020. Т. 39. № 8. С. 64.
Федяева O.Н., Востриков A.A. // Сверхкритические флюиды: теория и практика. 2019. Т. 14. № 3. С. 77.
Кислов В. M., Цветкова Ю. Ю., Цветков M.В. и др. // Хим. физика. 2021. Т. 40. № 8. С. 19.
Daldal H., Beder B., Serin B., Sungurtekin H. // Clin. Toxicol. 2010. V. 48. Issue 7. P. 755.
Kahyarian A., Nesic S. // Electrochim. Acta. 2018. V. 297. P. 676.
Zaman J., Chakma A. // Fuel Process. Technol. 1995. V. 41. P. 159.
Barba D., Cammarota F., Vaiano V. et al. // Fuel. 2017. V. 198. P. 68.
Sendt K., Jazbec M., Haynes B.S. // Proc. Combust. Inst. 2002. V. 29. P. 2439.
Binoist M. et al. // Ind. Eng. Chem. Res. 2003. V. 42. Issue 17. P. 3943.
Mohamadi S., Bashiri H. // Intern. J. Chem. Kinet. 2019. V. 52. Issue 1. P. 16.
Kraia T., Kaklidis N., Konsolakis M., Marnellos G.E. // Intern. J. Hydrogen Energy. 2019. V 44. P. 9753.
Startsev A.N. // Kinet. Catal. 2016. V. 57. Issue 4. P. 511.
Wang B., Zhang S., Zhang L. Ye. X. et al. // ACS Omega. 2020. V. 5. Issue 22. P. 13324.
Hawboldt K.A., Monnery W.D., Svrcek W.Y. // Chem. Eng. Sci. 2000. V. 55. P. 957.
Startsev A.N., Kruglyakova O.V., Chesalov Yu.A. // Top. Catal. 2013. V. 56. P. 969.
Zakharov I.I., Startsev A.N., Voroshina O.V. et al. // Rus. J. Phys. Chem. B. 2006. V. 80. № 9. P. 1403.
Bai X., Cao Y., Wu W. // Renew. Energy. 2011. V. 36. Issue 9. P. 2589.
Reddy E.L., Karuppiah J., Biju V.M., Subrahmanyam C. // Intern. J. Energy Res. 2012. V. 37. P. 1280.
Reddy E.L., Biju B.M., Subrahmanyam C. // Intern. J. Hydrogen Energy 2012. V. 37. P. 8217.
John S., Hamann J.S., Muknahallipatna S.S. et al. // Chem. Eng. Sci. 2009. V. 64. P. 4826.
Gutsol K. High-Conversion Plasma Dissociation of Hydrogen Sulfid. A Thesis Submitted to the Faculty of Drexel University. 2014.
Luo M., Zhou J., Xu W., Chen J. et al. // Fuel. 2020. V. 281. Article 118729.
Zhou J. et al. // Sci. Rep. 2016. V. 6. Article 25149.
Cong T.Y., Raj A., Chanaphet J. et al. // Intern. J. Hydrogen Energy. 2016. V. 41. Issue 15. P. 6662.
Startsev A.N., Bulgakov N.N., Pazankin S.Ph. et al. // J. Sulfur Chem. 2015. V. 36. Issue 3. P. 234.
Johnston C.W. et al. // J. Phys. D: Appl. Phys. 2004. V. 37. Issue 2. P. 211
Johnston C.W. et al. // Ibid. 2002. V. 35. Issue 4. P. 342
Granovsky A.A. Firefly computational chemistry program. version 8.0.0; http://classic.chem.msu.su/gran/firefly/index.html
Baldridge K., Boatz J., Force A. et al. // J. Comput. Chem. 1993. V. 14. Issue 11. P. 1347.
Granovsky A.A. // J. Chem. Phys. 2011. V. 134. Article 214113.
Kim K., Jordan K.D. // J. Phys. Chem. 1994. V. 98. Issue 40. P. 10089.
NIST Chemistry WebBook. NIST Standard Reference Database Number 69 / Eds. Linstrom P.J., Mallard W.G. Gaithersburg USA: Natl Inst. Stand. Technol., 2011.
Barker J.R. // Inern. J. Chem. Kinet. 2001. V. 33. Issue 4. P. 232.
Smith G.P., Golden D.M. // Ibid. 1978. V. 10. P. 489.
Novoselov K.P., Shirabaikin D.S., Umanskii S.Y. et al. // J. Comput. Chem. 2002. V. 23. Issue 14. P. 1375.
Уманский С.Я. Теория элементарных химических реакций. Долгопрудный: Издат. Дом “Интеллект”, 2009.
Neufeld P.D., Janzen A.R., Aziz R.A. // J. Chem. Phys. 1972. V. 57. P. 1100; https://doi.org/10.1063/1.1678363
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика